

The role of a topologically conserved isoleucine in the structure of glutathione transferase was investigated by replacing the Ile71 residue in human GSTA1-1 by alanine or valine.
Keywords: glutathione transferases
Abstract
The common fold shared by members of the glutathione-transferase (GST) family has a topologically conserved isoleucine residue at the N-terminus of helix 3 which is involved in the packing of helix 3 against two β-strands in domain 1. The role of the isoleucine residue in the structure, function and stability of GST was investigated by replacing the Ile71 residue in human GSTA1-1 by alanine or valine. The X-ray structures of the I71A and I71V mutants resolved at 1.75 and 2.51 Å, respectively, revealed that the mutations do not alter the overall structure of the protein compared with the wild type. Urea-induced equilibrium unfolding studies using circular dichroism and tryptophan fluorescence suggest that the mutation of Ile71 to alanine or valine reduces the stability of the protein. A functional assay with 1-chloro-2,4-dinitrobenzene shows that the mutation does not significantly alter the function of the protein relative to the wild type. Overall, the results suggest that conservation of the topologically conserved Ile71 maintains the structural stability of the protein but does not play a significant role in catalysis and substrate binding.
1. Introduction
Glutathione transferases (GSTs; EC 2.5.1.18) are a ubiquitous superfamily of enzymes that are found in eukaryotes and prokaryotes. They are primarily involved in the defence of the cellular milieu against an array of reactive exogenous and endogenous compounds (Armstrong, 1997 ▶). Their key function is to mediate the catalytic conjugation of glutathione (γ-glutamyl-cysteinyl-glycine; GSH) to an electrophilic substrate, thus increasing the hydrophilicity and hence the excretion of the compound from the cell. In addition to their role as detoxifying enzymes, GSTs are involved in a broad range of diverse cellular processes including transport and storage (Litwack et al., 1971 ▶), thiol transfer (Eklund et al., 2007 ▶), stress kinase regulation (Desmots et al., 2005 ▶), prostaglandin synthesis (Kanaoka et al., 1997 ▶) and peroxidase activity (Board et al., 1997 ▶). GSTs are widely distributed in several tissues in higher eukaryotes as either cytosolic, microsomal or mitochondrial GSTs (Mannervik et al., 1985 ▶; Sheehan et al., 2001 ▶; Frova, 2006 ▶).
The cytosolic GSTs have been well characterized and form a number of species-independent gene classes (Mannervik et al., 1992 ▶; Pemble & Taylor, 1992 ▶; Board et al., 1997 ▶; Sheehan et al., 2001 ▶; Frova, 2006 ▶). Cytosolic GSTs are dimers, with each subunit comprising of an N-terminal domain with a thioredoxin-like fold (βαβαββα) and a larger C-terminal domain which is predominantly α-helical (Dirr et al., 1994 ▶). There is one active site per subunit, which is made up of the glutathione-binding site (G-site) and an adjacent hydrophobic substrate-binding site (H-site). The conformation of the H-site enables GSTs to bind to a variety of electrophilic substrates (Mannervik et al., 1985 ▶; Oakley et al., 1999 ▶).
Most GSTs share a common secondary-structure topology despite the limited sequence identity between the various gene classes (Sheehan et al., 2001 ▶; Frova, 2006 ▶). Therefore, the functional diversity within the GST superfamily arises from each class showing unique physicochemical features within the active site (Dirr et al., 1994 ▶). Structural alignments of GSTs have shown that only about 5% of the residues are strictly conserved (Sheehan et al., 2001 ▶; Cromer et al., 2002 ▶; Frova, 2006 ▶). Ile71 in helix 3 of human GSTA1-1 (hGSTA1-1) is topologically a highly conserved residue in the core of the N-terminal domain of the dimeric GSTs as well as in the dimeric and monomeric GST-like proteins Grx2, Urep2 and CLICs, with the exception of omega GST, which has a threonine residue (Fig. 1 ▶ a). The topologically conserved isoleucine (and isosteric threonine) is involved in the packing of helix 1 and strands 3 and 4 against helix 3, as shown for hGSTA1-1 in Fig. 1 ▶(b), suggesting that it might play a significant role in maintaining the stability of the tertiary structure of the N-terminal domain and, given its proximity to the active site which is located above helices 1 and 3 and to strands 3 and 4, the functionality of the enzyme. In this study, we investigated the effect of the replacement of the topologically conserved Ile71 residue in hGSTA1-1 by alanine or valine on the structure, stability and catalytic function of the enzyme.
Figure 1.

(a) Structure-based sequence alignment of GST proteins corresponding to the region containing strand β4 and helix α3. The residue in bold indicates the topologically conserved isoleucine in helix α3. Alpha to zeta represent different gene classes of dimeric GSTs; Clic1 (chloride intracellular channel 1) and Grx2 (glutaredoxin 2) are monomeric GST homologues and Ure2p (a prion protein in yeast) is a dimeric GST homologue. The PDB codes are shown in parentheses. The alignment was performed with 3DCoffee (O’Sullivan et al., 2004 ▶). (b) Ribbon structure of domain 1 in human GSTA1-1 (PDB code 1k3l), showing the location of Ile71 in the core of domain 1. The image was generated with PyMOL (DeLano, 2002 ▶).
2. Experimental methods
2.1. Mutagenesis, expression and purification
The pKHA1 plasmid encoding an open reading frame cDNA sequence for wild-type hGSTA1-1 (GenBank ID No. AAB20973.1; Rozen et al., 1992 ▶) containing no purification tag was a gift from Professor B. Mannervik (Uppsala University, Uppsala, Sweden; Stenberg et al., 1992 ▶). The pKHA1 plasmid was used as a template for site-directed mutagenesis to create the I71V and I71A mutants. The I71V mutant was generated with the QuikChange method (Stratagene, La Jolla, California, USA), while the GeneEditor method (Promega, Madison, Wisconsin, USA) was used to generate the I71A mutant. The replacement of the leucine TTA codon by the valine CTG and alanine TCG codons was confirmed by sequencing (Inqaba Biotechnical Industries Pty Ltd, Pretoria, South Africa). Recombinant I71A and I71V hGSTA1-1 were overexpressed in Escherichia coli strain BL21 (DE3) pLysS and purified as described previously (Stenberg et al., 1992 ▶).
2.2. Crystallization, X-ray detection and data processing
Crystals of recombinant I71A and I71V hGSTA1-1 were grown as described previously (Gildenhuys et al., 2010 ▶). Briefly, crystals of both mutant proteins were grown by the hanging-drop vapour-diffusion method at 293 K using a 24-well microplate. Each hanging drop (4, 6 or 8 µl) was comprised of equal volumes of protein stock solution and reservoir buffer. The stock protein concentration was 10 mg ml−1 (I71A hGSTA1-1) or 15 mg ml−1 (I71V hGSTA1-1) in 0.1 M Tris–HCl pH 7.5 containing 10 mM DTT, 2.5 mM S-hexylglutathione and 0.02% sodium azide. The reservoir buffer was PEG 4000 [19%(w/v)] in 0.1 M Tris–HCl pH 7.5, 10 mM DTT and 0.02% sodium azide. A paraffin–silicon oil mixture (1:1 ratio) was placed on top of the reservoir buffer in the wells for the I71A hGSTA1-1 crystallizations to induce favourable conditions for the growth of large crystals by slowing evaporation and supersaturation in the wells (Chayen et al., 1990 ▶). The crystals were harvested, briefly soaked in the reservoir buffer and mounted on a cryoloop. X-ray diffraction data for I71A hGSTA1-1 were collected on a Bruker X8 Proteum system with a Microstar copper rotating-anode generator with Montel 200 optics, a PLATINUM 135 CCD detector and an Oxford Cryostream Plus system and the diffraction data for I71V hGSTA1-1 crystals were collected on a Rigaku RUH3R copper rotating-anode X-ray source with a Rigaku R-AXIS IV+ image-plate camera, an X-stream 2000 low-temperature system and an AXCO PX50 glass capillary optics system. Crystals were cooled to 113 K in a stream of nitrogen during data collection and images were collected covering an oscillation angle of 0.5° per image. The data sets were processed using APEX and SAINT software (Bruker AXS Inc., Madison, Wisconsin, USA) for I71A hGSTA1-1 and HKL-2000 (Otwinowski & Minor, 1997 ▶) for I71V hGSTA1-1.
The structures of both mutants were solved by molecular replacement using MOLREP (Vagin & Teplyakov, 2000 ▶) as implemented in the CCP4 suite of programs (Collaborative Computational Project, Number 4, 1994 ▶), using wild-type hGSTA1-1 (PDB code 1k3l; Le Trong et al., 2002 ▶) as the search model. Model refinement was perfomed with REFMAC5 (Murshudov et al., 1997 ▶) and model building was performed with Coot (Emsley & Cowtan, 2004 ▶). After refinement of the ligand-free proteins, the model of S-hexylglutathione from 1k3l was built into the mutant structure and solvent molecules were added using Coot (Emsley & Cowtan, 2004 ▶). The data-collection and refinement statistics are given in Table 1 ▶. Stereochemical validation of the model was performed using PROCHECK (Laskowski et al., 1996 ▶) and MolProbity (Chen et al., 2010 ▶). PyMOL (DeLano, 2002 ▶) was used to generate images of the structures.
Table 1. Data-collection and refinement statistics.
Values in parentheses are for the outer shell.
| I71A hGSTA1-1 | I71V hGSTA1-1 | |
|---|---|---|
| Wavelength (Å) | 1.5418 | 1.5418 |
| Space group | C2 (C121) | C2 (C121) |
| Unit-cell parameters (Å, °) | a = 99.67, b = 93.93, c = 51.50, β = 92.94 | a = 98.41, b = 93.58, c = 50.72, β = 93.05 |
| Wilson plot B factor (Å2) | 10.5 | 38.1 |
| Solvent content (%) | 47.89 | 45.87 |
| Resolution range (Å) | 68.32–1.75 (1.84–1.75) | 67.30–2.51 (2.57–2.51) |
| No. of observed reflections | 47499 | 20428 |
| No. of unique reflections | 45093 | 14904 |
| Completeness | 99.4 | 97.2 |
| 〈I/σ(I)〉 | 4.8 (3.57) | 9.63 (2.25) |
| Rmerge† | 0.090 (0.405) | 0.095 (0.215) |
| Final overall R factor | 0.211 | 0.211 |
| Rwork‡ | 0.207 (0.492) | 0.206 (0.279) |
| Rfree‡ | 0.281 (0.456) | 0.298 (0.322) |
| No. of protein atoms | 3572 | 3576 |
| No. of ligand atoms | 52 | 52 |
| Matthews coefficient VM (Å3 Da−1) | 2.36 | 2.27 |
| Solvent content (%) | 47.9 | 45.9 |
| Total No. of atoms | 4469 | 3726 |
| Average B value (Å2) | 15.814 | 32.23 |
| R.m.s.d. in bond length (Å) | 0.0229 | 0.07 |
| R.m.s.d. in bond angles (°) | 1.930 | 1.716 |
| Ramachandran statistics | ||
| Most allowed (%) | 97.25 | 94.95 |
| Allowed (%) | 2.29 | 3.44 |
| Asymmetric unit content | Dimer | Dimer |
| PDB code | 3ktl | 2r6k |
R
merge = 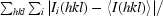
 , where I(hkl) is the intensity of reflection hkl,
, where I(hkl) is the intensity of reflection hkl,  is the sum over all reflections and
is the sum over all reflections and 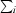 is the sum over i measurements of reflection hkl.
is the sum over i measurements of reflection hkl.
R free is calculated for a randomly chosen 5% of reflections which were not used for refinement of the structure and R work is calculated for the remaining reflections.
2.3. Enzyme-activity studies
Enzyme activity was determined at 293 K with 1 mM 1-chloro-2,4-dinitrobenzene (CDNB) and 1 mM GSH in 0.1 M sodium phosphate, 1 mM EDTA pH 6.5 by monitoring the reaction at 340 nm (Habig & Jakoby, 1981 ▶). All rates were corrected for non-enzymatic rates.
2.4. Urea-induced equilibrium unfolding
Equilibrium unfolding studies were conducted as described previously (Wallace et al., 1998 ▶). The protein concentration was 2 µM in 20 mM sodium phosphate buffer pH 6.5 containing 1 mM EDTA and 0.02% sodium azide and the concentration of urea was between 0 and 8 M. Structural changes were monitored by far-UV CD on a Jasco model 810 CD spectropolarimeter at 222 nm and by tryptophan fluorescence on a Perkin–Elmer LS50B luminescence spectrometer with the excitation wavelength at 295 nm. The unfolding data were analysed according to a two-state model with only folded dimeric (N2) and unfolded monomeric (U) states (Pace, 1986 ▶) using the global fitting program SAVUKA v.6.2.26 (Beechem, 1992 ▶; Zitzewitz et al., 1995 ▶; Bilsel et al., 1999 ▶).
3. Results and discussion
In this study, the structures of human I71A and I71V GSTA1-1 mutants in complex with S-hexylglutathione were determined at 1.75 and 2.51 Å resolution, respectively; the data-collection and refinement statistics are shown in Table 1 ▶. The Matthews coefficient (V M; Matthews, 1968 ▶) for both crystals was calculated using the CCP4 suite of programs (Collaborative Computational Project, Number 4, 1994 ▶). V M values of 2.36 Å3 Da−1 for the I71A mutant and 2.27 Å3 Da−1 for the I71V mutant were obtained, with associated solvent contents of 47.9% and 45.9%, respectively, indicating that both crystals contained two molecules in the asymmetric unit. The electron densities of the final models are well defined for residues 2–222 in chain A and for residues 4–222 in chain B. The electron density for residue 71 in chains A and B is consistent with an alanine and a valine residue in the I71A and I71V GSTA1-1 mutants, respectively. Electron density defining the locations and the conformations of S-hexylglutathione bound to chains A and B were clear, except for the terminal regions of the hexyl moieties owing to their flexibility, as observed in other structures (Gildenhuys et al., 2010 ▶). The replacement of Ile71 by either an alanine or a valine does not alter the backbone structure, as indicated by the Cα r.m.s.d. values of 0.24 Å between wild-type and I71A GSTA1-1 and of 0.31 Å between wild-type and I71V GSTA1-1. In wild-type hGSTA1-1 the structurally conserved Ile71 forms van der Waals interactions within the core of the N-terminal domain with its side chain tightly packed against Thr19 (α1), Pro56 (loop connecting α2 and β3), Val58 (β3) and Leu65 (β4) (Fig. 2 ▶ a). While the conformation of the domain is preserved by the replacement of Ile71 by alanine or valine (Figs. 2 ▶ b and 2 ▶ c), the cavity (193 Å3 for chain A; 192 Å3 for chain B) created by the I71A mutation results in the inclusion of two water molecules into the core of the domain (Fig. 2 ▶ b). These water molecules are hydrogen bonded to one another and to the protein and improve the packing density of the mutant.
Figure 2.

Stereo diagrams of (a) wild-type, (b) I71A and (c) I71V hGSTA1-1 showing the structural elements around residue 71 in helix 3. Secondary structures are shown as ribbons and amino-acid side chains are shown as sticks. The two water molecules included in the core of I71A hGSTA1-1 (b) are shown as spheres.
The specific activity of the mutant enzymes in conjugating CDNB to GSH is only slightly reduced to 97% (for I71V GSTA1-1) and to 86% (for I71A GSTA1-1) of the wild-type activity. This is consistent with the fact that although the replacement of Ile71 by alanine creates a cavity in domain 1 just below the active site, the replacement of Ile71 by either an alanine or a valine does not significantly alter the structure of the active site. Furthermore, the conformations of and interactions with the glutathione moiety bound to both mutants are essentially the same as those observed for the moiety in the wild-type complex.
Urea-induced unfolding of I71A GSTA1-1 (Fig. 3 ▶ a) and I71V GSTA1-1 (Fig. 3 ▶ b) produced single sigmoidal transitions for both secondary (far-UV CD) and tertiary (fluorescence) structural changes. This two-state cooperative unfolding behaviour has previously been observed for wild-type GSTA1-1 and involves the folded dimer and unfolded monomers (Fig. 3 ▶; Wallace et al., 1998 ▶). However, the shift of the C m value or midpoint of the unfolding transition from 4.3 M urea for wild-type GSTA1-1 to lower values for both mutants (Fig. 3 ▶) indicates that both mutations destabilize hGSTA1-1. This is confirmed by a significant reduction in the value of ΔG H2 O (the energy difference between the folded and unfolded states) from 86.60 ± 0.74 kJ mol−1 for the wild type to 68.65 ± 1.34 kJ mol−1 for I71V GSTA1-1 and 62.33 ± 2.26 kJ mol−1 for I71A GSTA1-1. Furthermore, the cooperativity of unfolding (i.e. the slope of the unfolding curves) is diminished by both mutations. The m values obtained from the global fits to the unfolding data in Fig. 3 ▶ are 12.93 ± 0.67, 9.67 ± 0.33 and 7.95 ± 0.50 kJ mol−1 M −1 for wild-type, I71V and I71A GSTA1-1, respectively. The substantially reduced m values for the mutants suggest that their unfolding process is not truly two-state involving only folded dimer and unfolded monomers, but that an intermediate state or states might become populated during their unfolding as a result of a destabilized N-terminal domain (Soulages, 1998 ▶). Ile71 is located in a tightly packed environment in the core of the N-terminal domain and while the mutations do not perturb the structure of hGSTA1-1, both mutations create a cavity that diminished the packing density and in turn reduces the number of stabilizing van der Waals interactions within the core. In the I71A mutant, however, there are two highly ordered water molecules that are included in the larger hydrophobic cavity and therefore contribute to the packing density (Fig. 2 ▶ b), thus maintaining the structural integrity of the protein.
Figure 3.

Urea-induced unfolding transitions for (a) I71A and (b) I71V hGSTA1-1 determined by circular dichroism at 222 nm (filled circles) and tryptophan fluorescence (open circles). The unfolding transition for wild-type hGSTA1-1 (Wallace et al., 1998 ▶) is shown by the dotted curves in (a) and (b). The protein concentration was 2 µM in 20 mM sodium phosphate buffer pH 6.5, 1 mM EDTA, 0.02% sodium azide. The solid lines are the global regression fits to a two-state N2↔2U model.
In conclusion, the present study demonstrates that the isoleucine residue that is topologically conserved in the core of the N-terminal domain of GSTs and GST-like proteins contributes significantly to the stability of the GST fold but not to structure and function.
Supplementary Material
PDB reference: glutathione transferase, I17V mutant, 2r6k
PDB reference: I17A mutant, 3ktl
Acknowledgments
This work was supported by the University of the Witwatersrand, South African National Research Foundation Grants 60810, 65510 and 68898 (to HWD) and South African Research Chairs Initiative of the Department of Science and Technology and National Research Foundation Grant 64788 (to HWD). Any opinion, findings and conclusions or recommendations expressed in this material are those of the author(s) and therefore the NRF and DST do not accept any liability with regard thereto.
References
- Armstrong, R. N. (1997). Chem. Res. Toxicol.10, 2–18. [DOI] [PubMed]
- Beechem, J. M. (1992). Methods Enzymol.210, 37–54. [DOI] [PubMed]
- Bilsel, O., Zitzewitz, J. A., Bowers, K. E. & Matthews, C. R. (1999). Biochemistry, 38, 1018–1029. [DOI] [PubMed]
- Board, P. G., Baker, R. T., Chelvanayagam, G. & Jermiin, L. S. (1997). Biochem. J.328, 929–935. [DOI] [PMC free article] [PubMed]
- Chayen, N. E., Shaw Stewart, P. D., Maeder, D. L. & Blow, D. M. (1990). J. Appl. Cryst.23, 297–302.
- Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S. & Richardson, D. C. (2010). Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Cromer, B. A., Morton, C. J., Board, P. G. & Parker, M. W. (2002). Eur. Biophys. J.31, 356–364. [DOI] [PubMed]
- DeLano, W. L. (2002). The PyMOL Molecular Viewer. http://www.pymol.org.
- Desmots, F., Loyer, P., Rissel, M., Guillouzo, A. & Morel, F. (2005). FEBS Lett.579, 5691–5696. [DOI] [PubMed]
- Dirr, H., Reinemer, P. & Huber, R. (1994). Eur. J. Biochem.220, 645–661. [DOI] [PubMed]
- Eklund, B. I., Gunnarsdottir, S., Elfarra, A. A. & Mannervik, B. (2007). Biochem. Pharmacol.73, 1829–1841. [DOI] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Frova, C. (2006). Biomol. Eng.23, 149–169. [DOI] [PubMed]
- Gildenhuys, S., Dobreva, M., Kinsley, N., Sayed, Y., Burke, J., Pelly, S., Gordon, G. P., Sayed, M., Sewell, T. & Dirr, H. W. (2010). Biophys. Chem.146, 118–125. [DOI] [PubMed]
- Habig, W. H. & Jakoby, W. B. (1981). Methods Enzymol.77, 398–405. [DOI] [PubMed]
- Kanaoka, Y., Ago, H., Inagaki, E., Nanayama, T., Miyano, M., Kikuno, R., Fujii, Y., Eguchi, N., Toh, H., Urade, Y. & Hayaishi, O. (1997). Cell, 90, 1085–1095. [DOI] [PubMed]
- Laskowski, R. A., Rullmannn, J. A., MacArthur, M. W., Kaptein, R. & Thornton, J. M. (1996). J. Biomol. NMR, 8, 477–486. [DOI] [PubMed]
- Le Trong, I., Stenkamp, R. E., Ibarra, C., Atkins, W. M. & Adman, E. T. (2002). Proteins, 48, 618–627. [DOI] [PubMed]
- Litwack, G., Ketterer, B. & Arias, I. M. (1971). Nature (London), 234, 466–467. [DOI] [PubMed]
- Mannervik, B., Alin, P., Guthenberg, C., Jensson, H., Tahir, M. K., Warholm, M. & Jornvall, H. (1985). Proc. Natl Acad. Sci. USA, 82, 7202–7206. [DOI] [PMC free article] [PubMed]
- Mannervik, B., Awasthi, Y. C., Board, P. G., Hayes, J. D., Di Ilio, C., Ketterer, B., Listowsky, I., Morgenstern, R., Muramatsu, M., Pearson, W. R., Pickett, C. B., Sato, K., Widerstein, M. & Wolf, C. R. (1992). Biochem. J.282, 305–306. [DOI] [PMC free article] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed]
- Oakley, A. J., Lo Bello, M., Nuccetelli, M., Mazzetti, A. P. & Parker, M. W. (1999). J. Mol. Biol.291, 913–926. [DOI] [PubMed]
- O’Sullivan, O., Suhre, K., Abergel, C., Higgins, D. G. & Notredame, C. (2004). J. Mol. Biol.340, 385–395. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Pace, C. N. (1986). Methods Enzymol.131, 266–280. [DOI] [PubMed]
- Pemble, S. E. & Taylor, J. B. (1992). Biochem. J.287, 957–963. [DOI] [PMC free article] [PubMed]
- Rozen, F., Nguyen, T. & Pickett, C. B. (1992). Arch. Biochem. Biophys.292, 589–593. [DOI] [PubMed]
- Sheehan, D., Meade, G., Foley, V. M. & Dowd, C. A. (2001). Biochem. J.360, 1–16. [DOI] [PMC free article] [PubMed]
- Soulages, J. L. (1998). Biophys. J.75, 484–492. [DOI] [PMC free article] [PubMed]
- Stenberg, G., Bjornestedt, R. & Mannervik, B. (1992). Protein Expr. Purif.3, 80–84. [DOI] [PubMed]
- Vagin, A. & Teplyakov, A. (2000). Acta Cryst. D56, 1622–1624. [DOI] [PubMed]
- Wallace, L. A., Sluis-Cremer, N. & Dirr, H. W. (1998). Biochemistry, 37, 5320–5328. [DOI] [PubMed]
- Zitzewitz, J. A., Bilsel, O., Luo, J., Jones, B. E. & Matthews, C. R. (1995). Biochemistry, 34, 12812–12819. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: glutathione transferase, I17V mutant, 2r6k
PDB reference: I17A mutant, 3ktl