

Crystal and solution structures of Rv1848 protein and their implications in the biological assembly of Mtb urease is presented.
Keywords: Mycobacterium tuberculosis, urease, structural genomics
Abstract
The crystal structure of the urease γ subunit (UreA) from Mycobacterium tuberculosis, Rv1848, has been determined at 1.8 Å resolution. The asymmetric unit contains three copies of Rv1848 arranged into a homotrimer that is similar to the UreA trimer in the structure of urease from Klebsiella aerogenes. Small-angle X-ray scattering experiments indicate that the Rv1848 protein also forms trimers in solution. The observed homotrimer and the organization of urease genes within the M. tuberculosis genome suggest that M. tuberculosis urease has the (αβγ)3 composition observed for other bacterial ureases. The γ subunit may be of primary importance for the formation of the urease quaternary structure.
1. Introduction
Urease, a nickel-containing enzyme that catalyzes the hydrolysis of urea to form ammonia and carbamate (which subsequently decomposes with water to form ammonia and carbon dioxide), is found in a broad range of plants and bacteria. Urease has been implicated as a virulence factor in diseases of the human gastrointestinal and urinary tracts (Mobley & Hausinger, 1989 ▶; Jones & Mobley, 1989 ▶; Eaton et al., 1991 ▶). Urease is also thought to be important for the survival of Mycobacterium tuberculosis (Mtb) in lung tissue, possibly via pH modulation and nitrogen biosynthesis (Clemens et al., 1995 ▶; Gordon et al., 1980 ▶). The subunit composition of urease shows variations from monomeric, as in jack bean urease (Takishima et al., 1988 ▶), to heterodimeric, as in Helicobacter pylori urease, in which the β and γ subunits are tethered together (Ha et al., 2001 ▶), and heterotrimeric, as in Klebsiella aerogenes urease (Jabri et al., 1995 ▶) and Bacillus pasteurii urease (Benini et al., 1999 ▶). Prior to determination of the K. aerogenes urease structure, various other subunit compositions had been suggested based on gel-filtration results (Todd & Hausinger, 1987 ▶); the trimer-of-trimers assembly is commonly believed to exist but has not been confirmed.
In multi-subunit urease complexes the α subunit contains the nickel-binding active-site residues and the β subunit is involved in directing nickel-ion incorporation by interacting with accessory metallochaperones, while the γ subunit has no known enzymatic function. Urease follows normal Michaelis–Menten kinetics, showing no known allosteric or cooperative effects (Mobley et al., 1995 ▶).
The Mtb urease is encoded within an operon in the Mtb genome. Sequence analysis indicates the urease operon consists of α (UreC), β (UreB) and γ (UreA) subunits and three accessory proteins UreD, UreF and UreG that are used in nickel incorporation (Fig. 1 ▶). This operon composition suggests the formation of a heterotrimeric urease complex akin to the structure of K. aerogenes urease. The active Mtb urease complex has several unique properties. These include a K m of 0.3 mM, which is comparatively lower than those of other ureases and is believed to be a consequence of the ammonia-deficient cellular environment that Mtb resides in, and exceptional temperature and chemical stability, with normal activity seen up to 333 K as well as in 4 M urea (Clemens et al., 1995 ▶).
Figure 1.

Urease operon arrangement in M. tuberculosis, consisting of subunit γ (Rv1848), subunit β (Rv1849) and subunit α (Rv1850) and accessory proteins UreF (Rv1851), UreG (Rv1852) and UreD (Rv1853) that are believed to be involved in nickel incorporation (http://www.doe-mbi.ucla.edu/TB/PUBLIC/qs/qsearch.php?orf=Rv1848&checkorf=1).
Owing to the potential virulence-enhancing effects seen in other microbial ureases (Eaton et al., 1991 ▶; Jones et al., 1990 ▶; Tsuda et al., 1994 ▶; Cox et al., 2000 ▶), Mtb urease remains an enticing target in the war against tuberculosis (TB), which saw 9.27 million new cases and 1.8 million deaths in 2007 (World Health Organization, 2009 ▶). In an effort to elicit the structures of potential TB drug targets in order to enable structure-based rational drug design, the Mycobacterium tuberculosis Structural Genomic Consortium (TBSGC) has targeted Mtb urease and its subunits for structure determination. Here, we report the crystal structure of Rv1848 (the γ subunit; UreA).
2. Materials and methods
2.1. Cloning and expression
Cloning and overexpression of Rv1848 protein were carried out at the TBSGC high-throughput protein-production facility located at the Los Alamos National Laboratory, USA. A 0.3 kbp DNA fragment encoding the Mtb Rv1848 (UreA) gene was amplified by conventional PCR using Mtb H37Rv genomic DNA as the template. The amplified DNA fragment was digested and subcloned into a modified pET28b vector encoding an N-terminal six-His tag and a thrombin cleavage site upstream of the NdeI site. The expressed protein has the N-terminal tag MGSSHHHHHHSSGLVPRGSH and two additional amino acids (GS) at the C-terminus.
Escherichia coli BL21 (DE3) cells were transformed with the UreA-modified pET28b/6His vector and grown to exponential phase at 310 K in 3 ml Luria–Bertani broth containing 30 µg ml−1 kanamycin. The seed culture was transferred to 500 ml Terrific broth and expression was induced with 0.5 mM isopropyl β-d-1-thiogalactopyranoside at an OD600 of approximately 0.5. Growth was continued at 293 K for approximately 20 h until the OD600 reached approximately 15 (as measured from dilutions). The cells were harvested and stored at 193 K.
2.2. Protein purification
The cell pellets were lysed by sonication in 10 ml buffer A (20 mM Tris–HCl pH 8.0, 100 mM NaCl) per gram of cells for 10 min in 30 s pulses at 283 K. After ultracentrifugation for 30 min at 38 000 rev min−1, the supernatant was filtered through a 0.2 µm pore membrane and loaded onto a 5 ml Talon Superflow affinity column (Clontech) equilibrated with buffer A. After washing with 50 ml buffer A, the His-tagged Rv1848 was eluted from the cobalt-affinity column using buffer B (20 mM Tris pH 8.0, 500 mM NaCl, 300 mM imidazole). The eluent was dialyzed against buffer C (20 mM Tris pH 8.0, 100 mM NaCl, 10 mM β-mercaptoethanol) and purified by gel filtration on a Superdex-75 column (Amersham Pharmacia Biotech) using buffer C for equilibration and elution. The peak fractions (monitored by OD280) were analyzed by SDS–PAGE and the pooled protein fractions were concentrated using a Centriprep YM-3 (Millipore). The protein was 98% pure as estimated by SDS–PAGE and matrix-assisted laser desorption/ionization-time of flight mass spectrometry (Applied Biosystems).
2.3. Crystallization
The Rv1848 protein was crystallized at the TBSGC’s protein-crystallization facility at the Lawrence Livermore National Laboratory using the sitting-drop vapor-diffusion method. Initial random screens generated by CRYSTOOL were used to identify successful crystallization conditions. Diffraction-quality crystals were obtained by mixing 250 nl protein stock with 250 nl reservoir solution. The reservoir contained 1.1% 2-butanol, 23.76% PEG 750 MME, 0.16% glycerol and 0.1 M sodium cacodylate pH 5.5. No additional cryoprotectant was necessary; crystals were mounted in loops and plunged into liquid nitrogen straight from the crystallization drop.
2.4. Data collection and structure solution
A 1.8 Å resolution data set was collected on beamline 5.0.1 at the Advanced Light Source (ALS). Rv1848 crystals belonged to space group P6, with unit-cell parameters a = b = 109.99, c = 40.87 Å. The data were processed with HKL-2000 (Otwinowski & Minor, 1997 ▶) with an R merge of 4.3% and a completeness of 95.8% (79.4% in the highest resolution shell). Cell-content analysis gave a Matthews coefficient of 2.15 Å3 Da−1 and a solvent content of 42.7% with three molecules per asymmetric unit. The structure was determined by the molecular-replacement method using the Phaser program (McCoy et al., 2007 ▶). The Cα chain trace of the K. aerogenes urease structure γ subunit (PDB code 2kau chain A; 65% sequence identity; Jabri et al., 1995 ▶) was used as the search model. Refinement was carried out using REFMAC (Murshudov et al., 1997 ▶) and model building and visualization were carried out with Coot (Emsley & Cowtan, 2004 ▶). The final R work and R free values were 18.6% and 22.9%, respectively, with acceptable protein stereochemistry. Table 1 ▶ shows a more complete listing of the data-collection and refinement statistics. The final model coordinates and structure factors of Rv1848 have been deposited in the PDB with code 2fvh.
Table 1. Data-collection and refinement statistics.
Values in parentheses are for the outermost shell.
| Data-collection statistics | |
| Wavelength (Å) | 1.0 |
| Resolution range (Å) | 50–1.80 (1.86–1.80) |
| Space group | P6 |
| Unit-cell parameters (Å) | a = b = 109.99, c = 40.87 |
| Total reflections | 28667 |
| Completeness (%) | 95.8 (79.4) |
| Multiplicity | 3.1 (1.8) |
| Rmerge† (%) | 4.3 (28.9) |
| Rp.i.m.‡ | 0.023 (0.318) |
| Rmeas/Rr.i.m.‡ | 0.048 (0.491) |
| Wilson B factor | 25.6 |
| Average I/σ(I) | 28 (2.2) |
| Refinement statistics | |
| Resolution (Å) | 27.02–1.8 (1.85–1.8) |
| Completeness (%) | 97.3 (88.9) |
| Working reflections | 24482 (1648) |
| Test reflections | 1311 (92) |
| Rwork§ (%) | 18.4 (22.0) |
| Rfree§ (%) | 22.9 (30.1) |
| No. of waters | 164 |
| R.m.s. deviations from ideal geometry | |
| Bonds (Å) | 0.015 |
| Angles (°) | 1.6 |
| Average B factor (Å2) | 29.7 |
| Ramachandran analysis by MolProbity¶ | |
| Residues in favored regions (%) | 99.31 |
| Residues in allowed regions (%) | 100 |
R
merge = 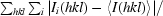
 , where I
i(hkl) is the intensity of the ith observation and 〈I(hkl)〉 is the mean intensity of the reflections. The values are for unmerged Friedel pairs.
, where I
i(hkl) is the intensity of the ith observation and 〈I(hkl)〉 is the mean intensity of the reflections. The values are for unmerged Friedel pairs.
R p.i.m. [precision-indicating (multiplicity-weighted) R merge] and R meas/R r.i.m. [redundancy-independent(multiplicity-weighted) R merge] as ouptut by the program SCALA (Evans, 2006 ▶; Weiss, 2001 ▶).
The crystallographic R factor R = 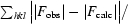
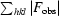 ; R
free =
, where all reflections belong to a test set of randomly selected data.
; R
free =
, where all reflections belong to a test set of randomly selected data.
Chen et al. (2010 ▶).
2.5. SAXS data collection and processing
Small-angle X-ray scattering (SAXS) data were collected on ALS beamline 12.3.1. Rv1848 was diluted with buffer C (20 mM Tris pH 8.0, 100 mM NaCl and 10 mM β-mercaptoethanol) to give 20 µl samples at final concentrations of 2.73, 1.36 and 0.68 mg ml−1. For each protein sample and a buffer blank, SAXS data were collected using a 0.5 s, a 5 s and a second 0.5 s exposure. Buffer-subtracted files were analyzed using PRIMUS (Konarev et al., 2003 ▶) and the GNOM (Svergun, 1992 ▶) P(r) output file with d max = 62 was used to calculate electron-density envelopes. Each envelope is the product of 16 GASBOR (Svergun et al., 2001 ▶) runs (P1 symmetry and 300 residues) averaged with DAMMAVER (Volkov & Svergun, 2003 ▶).
3. Results and discussion
3.1. Overall structure
The crystal structure of the Mtb Rv1848 protein (UreA; Fig. 2 ▶) closely resembles the γ subunit of K. aerogenes UreA (PDB code 2kau chain A). Each Rv1848 monomer consists of an α/β structure containing a two-stranded antiparallel β-sheet two-layer sandwich with helices 2–5. The N-terminal helix 1 (amino acids 4–26) is at an approximate angle of 60° from helix 2 in the two-layer α/β sandwich. Fig. 3 ▶ shows sequence alignments and structural overlays of Rv1848 and the known urease complex γ subunits.
Figure 2.

Crystal structure of the Rv1848 homotrimer looking down the threefold axis (PDB code 2fvh). This figure was produced using PyMOL (DeLano, 2002 ▶).
Figure 3.

(a) Structural overlay of urease γ-subunit monomers using PyMOL: M. tuberculosis (PDB code 2fvh; green), H. pylori (PDB code 1e9y; cyan; Ha et al., 2001 ▶), in which the tethered domain corresponding to the β subunit has been removed from the picture, B. pasteurii (PDB code 1ubp; yellow; Benini et al., 1999 ▶) and K. aerogenes (PDB code 2kau; grey; Jabri et al., 1995 ▶). (b) Multiple sequence alignment of known urease γ subunits using MULTALIN (Corpet, 1988 ▶). The N-terminal 6×His tag from 2fvh and the tethered urease β subunit from 1e9y were removed from the alignment for clarity. (c) Chain A of Rv1848 color-coded by sequence-alignment results. Secondary-structure elements are labeled.
3.2. Quaternary structure
The Rv1848 protein forms a homotrimer in the asymmetric unit of the crystal. This trimer forms a disk with a diameter of approximately 60 Å and a maximum height of approximately 30 Å at the threefold centre of symmetry. Stabilized by the interactions of helix 1 and 2, 20% of the protein’s residues are involved in homotrimer formation, beginning with hydrophobic interactions at the N-terminal portion of helix 1 and transitioning at Tyr15 to a large network of electrostatic and ionic intersubunit interactions (Fig. 4 ▶). The core of the homotrimer threefold is stabilized by the hydrophobic interactions of the three Leu12 side chains, the aromatic ring interactions of Tyr15, the hydroxyl of the Tyr15 side chain interacting with the side chain of Ser14 from a neighboring chain and the ionic network of Glu18 side chains. In accordance with the interactions seen at the homotrimer threefold, inspecting the calculated electrostatic potentials of the γ-subunit monomer shows the exposed surface of helix 1 to be predominantly hydrophobic in nature, ending with the positive potential provided by Arg22, Arg23 and Arg26 corresponding to the external solvent surface of the protein (Fig. 5 ▶). Overall, the homotrimer maintains a more hydrophobic underside that presumably maintains interactions with the α subunits in the proposed (αβγ)3 trimer of trimers, as seen in the K. aerogenes structure, while the external solvent-exposed surface has a slight negative charge.
Figure 4.

Rv1848 homotrimer subunit interactions colored by type of interaction. The homotrimer is colored by chain and shown in cartoon representation. All amino acids involved in intersubunit interactions (main-chain or side-chain interactions) are shown in stick form, with hydrophobic and aromatic interactions in yellow and hydrogen bonding and ionic interactions in blue. This analysis was carried out with the Protein Interactions Calculator (Tina et al., 2007 ▶).
Figure 5.

(a) Side view of the Rv1848 monomer along helix 1 with the surface colored by electrostatic potential and the backbone ribbon in green. (b) Top and bottom surface of the Rv1848 homotrimer colored by electrostatic potential. The homotrimer orientation is based on the K. aerogenes 2kau structure. The potentials were calculated using APBS (Baker et al., 2001 ▶) from an input file generated from the PDB2PQR web server (Dolinsky et al., 2004 ▶, 2007 ▶) and were visualized using CHIMERA (Pettersen et al., 2004 ▶) with a scale of red = −10 V and blue = 10 V.
3.3. Oligomeric state and biological relevance
The crystal structure of Rv1848 is the first γ-subunit-only structure of the (αβγ)3-type ureases reported to date. The formation of the Rv1848 homotrimer leads to a reduction of approximately 4300 Å2 in accessible surface area, indicating that the intersubunit interactions in the Rv1848 homotrimer are highly specific (Bahadur et al., 2004 ▶; Ponstingl et al., 2000 ▶). Similar to the γ subunits in other (αβγ)3 urease structures, Rv1848 also forms a homotrimer (Fig. 2 ▶) and shares a striking resemblance to the quaternary structures of those in the holoenzymes. When superposed (Fig. 6 ▶), the Rv1848 homotrimer and the trimer of γ subunits in the K. aerogenes (αβγ)3 urease structure have a main-chain r.m.s.d. of 1.13 Å. Together with nickel analysis, which shows six molecules per complex (Clemens et al., 1995 ▶), this suggests that the Mtb urease complex may also adopt the trimer-of-trimers formation seen in K. aerogenes urease as well as in related species. To further confirm that the Rv1848 homotrimer in the crystal structure was not a crystallization artifact, an SAXS experiment was performed with the Rv1848 protein in its solution state. Fig. 7 ▶ shows the electron-density envelope of the average result of 16 individual calculations overlaid onto the crystal structure. This suggests that Rv1848 forms a homotrimer in solution and that it may be biologically relevant.
Figure 6.

Overlay of the γ subunits from M. tuberculosis (PDB code 2fvh; green), K. aerogenes (PDB code 2kau; cyan) with an r.m.s.d. of 1.13 Å and B. pasteurii (PDB code 1ubp; magenta) with an r.m.s.d. of 0.968 Å superimposed onto the K. aerogenes urease trimer of trimers with semi-transparent orange (α) and yellow (β) surfaces, highlighting the centralized location of the γ subunits in the urease complexes.
Figure 7.

Top and side view of the overlay of 2fvh with the electron-density envelope using CHIMERA. The envelope is calculated from an average of 16 GASBOR runs with P1 symmetry and 300 residues.
3.4. Functional implications
Since UreA has no known enzymatic activity, it is possible that the γ-subunit trimer located at the center of the urease trimer serves as a scaffold for the urease-trimer complex. Based on the tight interactions of the γ subunits, we further speculate that γ-subunit trimerization might be an important driving force in the formation of the trimer-of-trimers complex previously seen in bacterial urease crystal structures. It is well understood that the α subunit of the urease complex contains the active-site and nickel-binding residues. In H. pylori the γ subunit is tethered to the β subunit but the urease complex still maintains threefold symmetry, with the γ subunit in the center and the β subunit at the vertices contacting two adjacent α subunits. Research in H. pylori implicates trimerization of the C-terminal portion of the β subunit as the structural stabilization required in the formation of a supramolecular urease complex that is believed to help in low-pH environments (Ha et al., 2001 ▶). Recent research on the K. aerogenes urease complex demonstrated that the shifting in position of the β subunit is necessary for the accessibility of the active site to nickel and carbon dioxide (Quiroz-Valenzuela et al., 2008 ▶). Combined together, this seems to suggest that while the α and β subunits are involved in the catalytic activity of the urease complex, the γ subunit might drive the formation or act as a scaffold in the urease trimer of trimers that is necessary to sustain high-heat and/or harsh chemical environments.
3.5. Implications for protein complexes in structural genomics
Protein complexes represent a significant challenge to structural genomics. Current production pipelines are designed to handle single high-expression soluble proteins instead of multi-subunit complexes. Targeting single members of a protein complex can be advantageous since individual components tend to crystallize more readily and diffract better than the full complex. With the re-emergence of SAXS, it is becoming a viable option to determine the structures of the individual pieces and then reassemble them into an SAXS electron-density envelope. This jigsaw-puzzle approach of solving complex assembly is more in tune with the established production pipelines at structural genomics centers.
There are inherent limitations in terms of assembling complexes from individual subunit structures and missing pieces could lead to arrangement uncertainty in the final model. Conversely, there are also cases in which subunit structures provide information beyond that elucidated from the complex structure alone. Rv1848 is a good example of using the advantages of structural genomics pipelines and combining complementary methods to obtain functional information from a single piece of a protein complex that would not otherwise have been discovered.
4. Conclusions
The structure of the urease γ subunit of M. tuberculosis, Rv1848, has been determined at 1.8 Å resolution and shows homology to other known γ-subunit monomers in urease complexes. Electrostatic mapping suggests that Rv1848 homotrimer formation is driven by the burial of hydrophobic residues along helix 1 at the threefold center. The crystallographic asymmetric unit and SAXS solution experiments demonstrate that Rv1848 forms a homotrimer in solution that has not previously been observed outside of urease complex formation. Based on sequence homology, gene placement and biochemical analysis, it is expected that the Mtb urease complex also adopts the trimer-of-trimers arrangement. The in-solution auto-assembly seen with Rv1848 and the non-enzymatic nature of UreA leads to the possibility of homotrimerization of the γ subunit being the driving force behind the formation of the urease trimer-of-trimers complex proposed for M. tuberculosis and seen in K. aerogenes and B. pasteurii. The need for oligomerization beyond catalytic efficiency may play a role in the extreme tolerance of Mtb urease to environmental challenges.
Supplementary Material
PDB reference: Rv1848, 2fvh
Supplementary material file. DOI: 10.1107/S1744309110019536/gx5165sup1.pdf
Supplementary material file. DOI: 10.1107/S1744309110019536/gx5165sup2.pdf
Acknowledgments
The authors would like to thank the staff at beamlines 5.0.2 (BL5.0.2) and 12.3.1 (BL12.3.1) at the ALS for technical support. Beamline 5.0.2 is managed by the Berkeley Center for Structural Biology, supported in part by the National Institutes of Health, National Institute of General Medical Sciences (NIGMS). Beamline 12.3.1 is supported in part by the US Department of Energy (DOE) program Integrated Diffraction Analysis Technologies (IDAT) and the DOE program Molecular Assemblies Genes and Genomics Integrated Efficiently (MAGGIE) under Contract No. DE-AC02-05CH11231 with the DOE. The ALS is supported by the Director, Office of Science, Office of Basic Energy Sciences of the DOE under Contract No. DE-AC02-05CH11231. This work was supported by the NIGMS Protein Structure Initiative program (NIH U54 GM074946).
References
- Bahadur, R. P., Chakrabarti, P., Rodier, F. & Janin, J. (2004). J. Mol. Biol.336, 943–955. [DOI] [PubMed]
- Baker, N. A., Sept, D., Joseph, S., Holst, M. J. & McCammon, J. A. (2001). Proc. Natl Acad. Sci. USA, 98, 10037–10041. [DOI] [PMC free article] [PubMed]
- Benini, S., Rypniewski, W. R., Wilson, K. S., Miletti, S., Ciurli, S. & Mangani, S. (1999). Structure, 7, 205–216. [DOI] [PubMed]
- Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S. & Richardson, D. C. (2010). Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed]
- Clemens, D. L., Lee, B. Y. & Horwitz, M. A. (1995). J. Bacteriol.177, 5644–5652. [DOI] [PMC free article] [PubMed]
- Corpet, F. (1988). Nucleic Acids Res.16, 10881–10890. [DOI] [PMC free article] [PubMed]
- Cox, G. M., Mukherjee, J., Cole, G. T., Casadevall, A. & Perfect, J. R. (2000). Infect. Immun.68, 443–448. [DOI] [PMC free article] [PubMed]
- DeLano, W. L. (2002). The PYMOL Molecular Viewer. http://www.pymol.org.
- Dolinsky, T. J., Czodrowski, P., Li, H., Nielsen, J. E., Jensen, J. H., Klebe, G. & Baker, N. A. (2007). Nucleic Acids Res.35, W522–W525. [DOI] [PMC free article] [PubMed]
- Dolinsky, T. J., Nielsen, J. E., McCammon, J. A. & Baker, N. A. (2004). Nucleic Acids Res.32, W665–W667. [DOI] [PMC free article] [PubMed]
- Eaton, K. A., Brooks, C. L., Morgan, D. R. & Krakowka, S. (1991). Infect. Immun.59, 2470–2475. [DOI] [PMC free article] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Evans, P. (2006). Acta Cryst. D62, 72–82. [DOI] [PubMed]
- Gordon, A. H., Hart, P. D. & Young, M. R. (1980). Nature (London), 286, 79–80. [DOI] [PubMed]
- Ha, N.-C., Oh, S.-T., Sung, J. Y., Cha, K. A., Lee, M. H. & Oh, B.-H. (2001). Nature Struct. Biol.8, 505–509. [DOI] [PubMed]
- Jabri, E., Carr, M. B., Hausinger, R. P. & Karplus, P. A. (1995). Science, 268, 998–1004. [PubMed]
- Jones, B. D., Lockatell, C. V., Johnson, D. E., Warren, J. W. & Mobley, H. L. (1990). Infect. Immun.58, 1120–1123. [DOI] [PMC free article] [PubMed]
- Jones, B. D. & Mobley, H. L. (1989). J. Bacteriol.171, 6414–6422. [DOI] [PMC free article] [PubMed]
- Konarev, P. V., Volkov, V. V., Sokolova, A. V., Koch, M. H. J. & Svergun, D. I. (2003). J. Appl. Cryst.36, 1277–1282.
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst.40, 658–674. [DOI] [PMC free article] [PubMed]
- Mobley, H. L. & Hausinger, R. P. (1989). Microbiol. Rev.53, 85–108. [DOI] [PMC free article] [PubMed]
- Mobley, H. L., Island, M. D. & Hausinger, R. P. (1995). Microbiol. Rev.59, 451–480. [DOI] [PMC free article] [PubMed]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C. & Ferrin, T. E. (2004). J. Comput. Chem.25, 1605–1612. [DOI] [PubMed]
- Ponstingl, H., Henrick, K. & Thornton, J. M. (2000). Proteins, 41, 47–57. [DOI] [PubMed]
- Quiroz-Valenzuela, S., Sukuru, S. C., Hausinger, R. P., Kuhn, L. A. & Heller, W. T. (2008). Arch. Biochem. Biophys.480, 51–57. [DOI] [PMC free article] [PubMed]
- Svergun, D. I. (1992). J. Appl. Cryst.25, 495–503.
- Svergun, D. I., Petoukhov, M. V. & Koch, M. H. (2001). Biophys. J.80, 2946–2953. [DOI] [PMC free article] [PubMed]
- Takishima, K., Suga, T. & Mamiya, G. (1988). Eur. J. Biochem.175, 151–165. [DOI] [PubMed]
- Tina, K. G., Bhadra, R. & Srinivasan, N. (2007). Nucleic Acids. Res.35, W473–W476. [DOI] [PMC free article] [PubMed]
- Todd, M. J. & Hausinger, R. P. (1987). J. Biol. Chem.262, 5963–5967. [PubMed]
- Tsuda, M., Karita, M., Morshed, M. G., Okita, K. & Nakazawa, T. (1994). Infect. Immun.62, 3586–3589. [DOI] [PMC free article] [PubMed]
- Volkov, V. V. & Svergun, D. I. (2003). J. Appl. Cryst.36, 860–864.
- Weiss, M. S. (2001). J. Appl. Cryst.34, 130–135.
- World Health Organization (2009). Global Tuberculosis Control – Epidemiology, Strategy, Financing. WHO/HTM/TB/2009.411. Geneva: World Health Organization.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: Rv1848, 2fvh
Supplementary material file. DOI: 10.1107/S1744309110019536/gx5165sup1.pdf
Supplementary material file. DOI: 10.1107/S1744309110019536/gx5165sup2.pdf