

Two crystal forms of an Mn2+-dependent phosphopentomutase were identified from chemically distinct conditions by sparse-matrix screening with and without the inclusion of 50 mM Mn2+. The crystals identified in the presence of Mn2+ were of dramatically better diffraction quality than those identified in the absence of added Mn2+.
Keywords: phosphopentomutase; alkaline phosphatase superfamily; ribose 5-phosphate, ribose 1-phosphate; sparse-matrix screening; metalloproteins; metalloenzymes
Abstract
Phosphopentomutases (PPMs) interconvert d-ribose 5-phosphate and α-d-ribose 1-phosphate to link glucose and nucleotide metabolism. PPM from Bacillus cereus was overexpressed in Escherichia coli, purified to homogeneity and crystallized. Bacterial PPMs are predicted to contain a di-metal reaction center, but the catalytically relevant metal has not previously been identified. Sparse-matrix crystallization screening was performed in the presence or absence of 50 mM MnCl2. This strategy resulted in the formation of two crystal forms from two chemically distinct conditions. The crystals that formed with 50 mM MnCl2 were more easily manipulated and diffracted to higher resolution. These results suggest that even if the catalytically relevant metal is not known, the crystallization of putative metalloproteins may still benefit from supplementation of the crystallization screens with potential catalytic metals.
1. Introduction
The pentose-phosphate pathway is an alternative metabolic process to glycolysis that can proceed through an oxidative or a non-oxidative phase. During the non-oxidative phase, ribulose 5-phosphate is converted to numerous important cellular building blocks (Kanehisa et al., 2002 ▶). One important product is d-ribose 5-phosphate (ribose 5-phosphate), which is used in de novo nucleotide biosynthesis after its conversion to α-d-ribose 1-phosphate by phosphopentomutase (PPM; Tozzi et al., 2006 ▶). Interest in prokaryotic PPMs has recently been stimulated by the observation that targeted deletion of the gene encoding PPM in the pathogen Francisella tularensis dramatically decreased its virulence (Horzempa et al., 2008 ▶). Therefore, PPMs may become a novel antibiotic target and an enhanced understanding of the architecture of the active site would greatly facilitate antibiotic development. While the coordinates for a PPM from Streptococcus mutans are available (PDB code 2i09; New York SGX Research Center for Structural Genomics, unpublished work), no analysis of this structure has been reported in the literature.
Based on sequence similarity, PPMs have been identified as alkaline phosphatase superfamily members and contain a metal-binding domain that coordinates two divalent cations (Galperin et al., 1998 ▶). The metal requirement for Bacillus cereus PPM has not previously been reported. The characterization of a PPM from Escherichia coli revealed that it is catalytically active when incorporated with either Co2+ or Mn2+ (Hammer-Jespersen & Munch-Petersen, 1970 ▶).
It has been hypothesized that as many as one in three proteins bind metals (Tainer et al., 1991 ▶) and an increasing number of these structures are being approached by high-throughput methods. Unfortunately, the heterologous overexpression of metalloenzymes in E. coli can sometimes result in protein with under-incorporated or mis-incorporated metal centers. For example, during the expression of the Mn2+-containing bacteriophage lambda protein phosphatase, the intracellular concentration of Mn2+ increased but was not sufficient to saturate all metal-binding sites. Supplementation with 1 mM Mn2+ led to full activation of the protein (Reiter et al., 2002 ▶). Simple modification of currently accepted techniques for the expression and crystallization of metalloproteins may improve the success rate of structure determination.
In an effort to further understand the structure and mechanism of PPMs, we have cloned, overexpressed and crystallized PPM from B. cereus. The catalytically relevant metal for the PPM active site was unknown at the time of crystallization and standard high-throughput screening techniques were modified such that crystallization trials were performed both with and without the addition of 50 mM MnCl2, yielding substantially different results. The method by which the crystallization screening was performed is simple and can easily be applied to high-throughput crystallization of other metalloproteins.
2. Materials and methods
2.1. Materials
Unless otherwise stated, all materials were of the highest grade obtained from Sigma–Aldrich.
2.2. Cloning, expression and purification
Genomic DNA for B. cereus ATCC 14579D was purchased from ATCC. DNA encoding PPM (accession No. Q818Z9.1) was amplified by PCR using 5′-GCTAGCAATAAATATAAACGTATATTCCTAGTCG-3′ as the sense primer and 5′-CTCGAGCTATTTCTTTAGCTCGTTTAAG-3′ as the antisense primer. PPM was ligated into the plasmid pET28a(+) (Novagen) between the NheI and XhoI sites to include an amino-terminal hexahistidine tag and thrombin-cleavage site. The resulting plasmid was electroporated into E. coli strain BL21-Gold (DE3) (Stratagene) using a Gibco BRL Cell Porator E. coli Pulser (Life Technologies) at 2.5 kV. Cells were grown in LB supplemented with 100 mg l−1 kanamycin at 310 K with continuous shaking at 250 rev min−1 until the A 600 was between 0.4 and 0.6. Protein expression was induced by the addition of isopropyl β-d-1-thiogalactopyranoside to 0.3 mM and was continued with shaking at 310 K for 4 h. Cells were harvested by centrifugation at 5000g for 15 min at 277 K and were resuspended in 25 ml buffer A (50 mM Tris–HCl pH 7.5, 200 mM NaCl, 10 mM imidazole) enriched with 1 µg ml−1 DNase I, 1 µg ml−1 leupeptin and 1 µg ml−1 pepstatin A. Cells were lysed by sonication on ice for 10 min (60 Sonic Dismembrator, Fischer Scientific) and then clarified by centrifugation at 30 000g for 1 h at 277 K in an SS-34 rotor (Sorvall). The supernatant was filtered through a 0.45 µm filter prior to chromatography at 277 K. The filtered lysate was passed over a HisTrap HP column (GE) equilibrated with buffer A. Nonspecifically bound protein was removed by washing with ten column volumes of buffer A supplemented with 50 mM imidazole. Purified protein was eluted by adding buffer A supplemented with 300 mM imidazole. The eluant was concentrated to 500 µl using an Amicon Ultra 30 000 molecular-weight cutoff centrifugal filter (Millipore) spun in a swinging-bucket rotor at 5000g at 277 K. The hexahistidine tag was removed by exchanging into buffer B (50 mM Tris–HCl pH 7.5 and 200 mM NaCl) and cleaving overnight with 50 units of thrombin. The resultant protein contained three additional residues, Gly-Ser-His, at the N-terminus. PPM was separated from protein aggregates and the cleaved peptide tag by size-exclusion chromatography using a Superdex 200 10/300 GL column (GE) equilibrated with 25 mM Tris–HCl pH 7.5 (for crystals without manganese) or 25 mM Tris–HCl pH 7.5 and 1 mM MnCl2 (for crystals with manganese). The protein was concentrated to between 50 and 75 mg ml−1 as determined by the Bio-Rad Protein Assay (Bio-Rad) and was stored at 277 K prior to use.
2.3. Crystallization
Initial crystallization conditions were identified by sparse-matrix screening with Index, Crystal Screen 1 and Crystal Screen 2 (Hampton Research) and the Wizard I and Wizard II screens (Emerald BioSystems). For these screens, a Mosquito NanoDrop liquid-handling robot (TTP Labtech) was used to set up 200 nl drops of protein solution mixed in a 1:1 ratio with reservoir solution. Drops were equilibrated over 100 µl reservoir solution using the sitting-drop vapor-diffusion method at 291 K. For optimization of initial crystallization leads, the conditions were replicated in 24-well Linbro plates using the hanging-drop vapor-diffusion method and 1 µl protein plus 1 µl reservoir solution equilibrated over 1 ml reservoir solution. An initial crystallization condition for PPM in the absence of Mn2+ was identified from a 10 mg ml−1 protein mixture in 25 mM Tris–HCl pH 7.5. Optimized crystals grew from protein equilibrated against 33–36% polyethylene glycol 6000, 100 mM 2-(N-morpholino)ethanesulfonic acid (MES) pH 6.5, 100–300 mM ammonium sulfate and appeared in 2–4 d at 291 K. Crystals were flash-cooled in liquid nitrogen in reservoir solution without additional cryoprotectant prior to data collection.
A second crystallization condition for PPM was identified by re-screening 12 mg ml−1 B. cereus PPM in a buffer containing 50 mM MnCl2 and 25 mM Tris–HCl pH 7.5. 50 mM MnCl2 was only included in the protein buffer during sparse-matrix screening; all other crystallizations were performed with 1 mM MnCl2 in the protein buffer and 50 mM in the reservoir solution. Inclusion of MnCl2 in the protein buffer itself during the screening process prevented the labor-intensive process of adding MnCl2 to a final concentration of 50 mM in all of the sparse-matrix screening solutions. After initial conditions with manganese had been identified, the protein was stored in buffer containing 1 mM MnCl2, which increased the stability of the protein. All further protein crystallizations were determined from protein stored in 20 mM Tris–HCl pH 7.5 and 1 mM MnCl2. Optimized protein crystals grew from a 12 mg ml−1 protein mixture in 25 mM Tris–HCl pH 7.5 and 1 mM MnCl2 equilibrated against a reservoir solution containing 100 mM 2,2-bis(hydroxymethyl)-2,2′,2′′-nitrilotriethanol (bis-tris) pH 5.5, 50 mM MnCl2, 13–16% PEG 3350 and 25–100 mM ammonium acetate and appeared after 3–5 d at 291 K. Prior to data collection, crystals were cryoprotected by briefly passing them through a solution containing all of the crystallization components along with 30% glycerol and were flash-cooled in liquid nitrogen.
2.4. Data collection and processing
Crystal quality was assessed by analysis of X-ray diffraction at the Advanced Photon Source (Argonne, Illinois, USA) on the Life Science Collaborative Access Team (LS-CAT) beamlines 21-ID-D/F/G and at the Stanford Synchrotron Radiation Lightsource beamline 9-2. X-ray diffraction data sets for the best diffracting crystals were collected at 100 K on LS-CAT beamline 21-ID-G using a wavelength of 0.979 Å and a MAR 225 CCD detector. A data set consisting of 120 frames was collected from the orthorhombic PPM crystals grown in the absence of Mn2+ by rotating 1° over 9 s per frame with the detector 300 mm from the crystal. A data set consisting of 180 frames was collected from the monoclinic crystals of PPM grown in the presence of 50 mM Mn2+ by rotating 1° over 4 s per frame with the detector 250 mm from the crystal. All data were processed and scaled using the HKL-2000 suite of programs (Otwinowski & Minor, 1997 ▶).
3. Results and discussion
3.1. Expression and purification
Heterologously expressed B. cereus PPM was purified using two chromatography steps: Ni affinity and size exclusion. Cell growth, induction and affinity purification were performed in a single day. The protein was nearly pure after a single Ni-affinity purification step. After overnight incubation in the presence of thrombin, nearly all of the hexahistidine tag was cleaved from the protein and a single pass over a size-exclusion column was sufficient to prepare the protein for crystallization trials (Fig. 1 ▶). Typical yields from a 1 l cell culture ranged between 15 and 20 mg purified protein. Prior to initiating crystallization trials, the catalytic metal or metals utilized by B. cereus PPM had not yet been determined. Based on homology to E. coli PPM (Hammer-Jespersen & Munch-Petersen, 1970 ▶), it was hypothesized that Mn2+ could be one possibility. To investigate whether additional Mn2+ provided a benefit to crystallization, sparse-matrix crystallization screening was performed both with and without simple supplementation of 50 mM MnCl2 to the protein buffer.
Figure 1.

Purification of B. cereus PPM. 3 µg total protein was loaded onto a 12% SDS–PAGE gel. Lane MW contains molecular-weight markers (labelled in kDa), lane 1 contains the eluant from the 300 mM imidazole wash from the Ni–NTA affinity column, lane 2 contains the sample after incubation with thrombin and lane 3 contains the purified protein following size-exclusion chromatography. The arrows indicate the position of full-length PPM with a hexahistidine tag (46 kDa) and full-length PPM without the His6 affinity tag (44 kDa).
3.2. Crystallization and data collection of PPM without manganese
Sparse-matrix screening of PPM without the addition of Mn2+ to the protein resulted in the growth of crystals with a needle-like morphology that grew from a single nucleation site (Fig. 2 ▶ a). However, once exposed to the air the 2 µl drop evaporated in less than 1 min, leaving little time to manipulate the crystals. While diffraction spots were observed to 2 Å resolution (Fig. 2 ▶ b), these spots were oblate and split and the high-resolution reflections could not be processed. A data set merging to 2.5 Å resolution was collected that was 97.1% complete overall and 97.8% complete in the highest resolution shell (Table 1 ▶). These crystals formed in the orthorhombic space group P212121, with unit-cell parameters a = 68.2, b = 75.7, c = 96.1 Å, α = β = γ = 90°. Specific volume calculations based on the unit-cell parameters and the molecular weight of 44 kDa suggest that in this crystal form one monomer of PPM is present in each asymmetric unit, with a Matthews coefficient (V M; Matthews, 1968 ▶) of 2.8 Å3 Da−1 and a solvent content of 56%.
Figure 2.

Crystals and diffraction image from PPM crystallized without Mn2+. (a) Crystals of PPM without additional manganese. Crystals of PPM that grew without manganese in reservoir solution containing 33–36% PEG 6000, 100 mM MES pH 6.5, 100–300 mM ammonium sulfate at 291 K had a needle morphology. The black bar represents 100 µm. (b) Diffraction from crystals of PPM without additional manganese. Diffraction from these crystals identified that they belonged to space group P212121. The crystals diffracted to ∼2 Å but the spots were oblate and split in the outer resolution shells (inset). A data set consisting of 120 frames using 1° oscillations per frame that is 97.1% complete to 2.5 Å was collected on beamline ID-21-G at APS.
Table 1. Data-collection statistics.
Values in parentheses are for the highest resolution shell.
| Crystal form | P212121 | P21 |
|---|---|---|
| Wavelength (Å) | 0.979 | 0.979 |
| Beamline | APS 21-ID-G | APS 21-ID-G |
| Resolution (Å) | 50–2.50 (2.59–2.50) | 50–1.85 (1.92–1.85) |
| Unit-cell parameters (Å, °) | a = 68.2, b = 75.7, c = 96.1 | a = 91.2, b = 76.6, c = 106.6, β = 108.9 |
| Total reflections | 67394 | 361062 |
| Unique reflections | 17382 | 116902 |
| Multiplicity | 3.9 (3.6) | 3.1 (2.9) |
| Mean I/σ(I) | 23.2 (5.0) | 13.3 (3.0) |
| Completeness (%) | 97.1 (97.8) | 98.6 (99.1) |
| Rmerge† | 0.112 (0.391) | 0.080 (0.369) |
| No. of molecules in asymmetric unit | 1 | 3 |
| Matthews coefficient (Å3 Da−1) | 2.8 | 2.7 |
| Solvent content (%) | 56 | 54 |
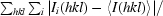
 , where Ii(hkl) is the ith instance of the intensity at position hkl and 〈I(hkl)〉 is the average of all instances of the reflection at position hkl.
, where Ii(hkl) is the ith instance of the intensity at position hkl and 〈I(hkl)〉 is the average of all instances of the reflection at position hkl.
3.3. Improved crystals of PPM identified by supplementing sparse-matrix screens with Mn2+
Repeating sparse-matrix screening with 50 mM MnCl2 included in the protein buffer conditions identified a second crystal form of PPM (Fig. 3 ▶ a). These crystals of PPM grown in the presence of Mn2+ were significantly easier to manipulate than crystals grown in the absence of Mn2+. The diffraction spots exhibited near-ideal profiles and were observed to better than 2 Å resolution (Fig. 3 ▶ b). Data merging to 1.85 Å resolution (Table 1 ▶) was 98.6% complete overall and 99.1% complete in the outer shell. These crystals formed in space group P21, with unit-cell parameters a = 91.2, b = 76.6, c = 106.6 Å, α = γ = 90, β = 108.9°. Specific volume calculations based on the unit-cell parameters and the molecular weight suggest that in this P21 crystal form there are three monomers of PPM in each asymmetric unit, with a Matthews coefficient (V M) of 2.7 Å3 Da−1 and a solvent content of 54% (Matthews, 1968 ▶).
Figure 3.

Crystals and a sample diffraction image from PPM cystallized with Mn2+. (a) Crystals of PPM grown in the presence of 50 mM MnCl2. Optimized crystals of PPM that were identifed after supplementing the protein with 50 mM MnCl2. Crystals grew from 100 mM bis-tris pH 5.5, 50 mM MnCl2, 13–16% PEG 3350 and 25–100 mM ammonium acetate and appeared in 3–5 d at 291 K. The black bar represents 100 µm. (b) Diffraction from crystals of PPM grown in the presence of 50 mM MnCl2. Diffraction images collected at APS on beamline ID-21-G using 1° oscillations per frame revealed that these crystals belonged to space group P21. A data set consisting of 180 frames and is 98.6% complete to 1.85 Å was collected.
3.4. Potential application of the supplementation method
The idea of including a catalytic metal during purification and crystallization is common practice when working with metalloproteins with known biochemical properties. However, advances in bioinformatics and robotics have made the crystallization of a protein before or in parallel with biochemical characterization increasingly common. While the inclusion of potentially catalytically relevant metals in the protein buffer during sparse-matrix screening may not result in completely uniform metal incorporation, it can be easily adapted to modern high-throughput screening techniques. Since robotics easily allow parallel crystallizations of a protein in the presence of several different metals, this may be particularly useful for cases where the metal-binding properties of the protein have not yet been established.
Acknowledgments
Use of the Advanced Photon Source was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor for the support of this research program (Grant 085P1000817). Use of the Stanford Synchrotron Radiation Lightsource was supported by Stanford University and the US Department of Energy. TMI acknowledges support from R01-GM079419. BOB acknowledges support from The Vanderbilt Institute of Chemical Biology and R01-GM077189. TDP acknowledges support from T32 NS07491 and T32 GM008320. DPN acknowledges support from T90 DA022873.
References
- Galperin, M. Y., Bairoch, A. & Koonin, E. V. (1998). Protein Sci.7, 1829–1835. [DOI] [PMC free article] [PubMed]
- Hammer-Jespersen, K. & Munch-Petersen, A. (1970). Eur. J. Biochem.17, 397–407. [DOI] [PubMed]
- Horzempa, J., Carlson, P. E., O’Dee, D. M., Shanks, R. M. Q. & Nau, G. J. (2008). BMC Microbiol.8, 17. [DOI] [PMC free article] [PubMed]
- Kanehisa, M., Goto, S., Kawashima, S. & Nakaya, A. (2002). Nucleic Acids Res.30, 42–46. [DOI] [PMC free article] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Reiter, T. A., Reiter, N. J. & Rusnak, F. (2002). Biochemistry, 41, 15404–15409. [DOI] [PubMed]
- Tainer, J. A., Roberts, V. A. & Getzoff, E. D. (1991). Curr. Opin. Biotechnol.2, 582–591. [DOI] [PubMed]
- Tozzi, M. G., Camici, M., Mascia, L., Sgarrella, F. & Ipata, P. L. (2006). FEBS J.273, 1089–1101. [DOI] [PubMed]