

The secondary alcohol dehydrogenase mutant I86A from Thermoanaerobacter ethanolicus (TeSADH) was crystallized in novel crystallization conditions. Diffraction data to 3.2 Å were collected at the Canadian Light Source.
Keywords: secondary alcohol dehydrogenase, Thermoanaerobacter ethanolicus
Abstract
The Thermoanaerobacter ethanolicus secondary alcohol dehydrogenase I86A mutant is stereospecific for (R)-alcohols instead of (S)-alcohols. Pyramidal crystals grown in the presence of (R)-phenylethanol via the hanging-drop vapour-diffusion method diffracted to 3.2 Å resolution at the Canadian Light Source. The crystal belonged to the orthorhombic space group P212121, with unit-cell parameters a = 80.23, b = 124.90, c = 164.80 Å. The structure was solved by molecular replacement using the structure of T. brockii SADH (PDB entry 1ykf).
1. Introduction
Optically active alcohols are among the most important chiral building blocks. Alcohol dehydrogenases (ADHs) reversibly reduce carbonyl compounds to their corresponding alcohols. With their high stereoselectivity in the reduction direction (they produce one alcohol enantiomer from a prochiral ketone), ADHs are excellent candidate catalysts for chiral synthesis. The stereopreference of ADHs depends on the relative sizes of the two substituents at a prochiral ketone or those at the stereocenter of a secondary alcohol (Prelog, 1964 ▶). Only a few ADHs show an anti-Prelog stereopreference (Bradshaw et al., 1992 ▶; Kroutil et al., 2004 ▶) and only a few accept ketones with bulky side chains as substrates. Because of their applications as catalysts in asymmetric synthesis, it is of great interest to study and to expand the substrate specificity of ADHs.
The secondary ADHs from Thermoanaerobacter brockii and T. ethanolicus (TbSADH and TeSADH, respectively) are identical enzymes that show high thermostability, high resistance to solvents and broad substrate specificity. TbSADH was recognized early on as an enzyme that was useful for industrial synthesis (Lamed et al., 1981 ▶) and has been used to synthesize several useful chiral compounds (Wong et al., 1985 ▶; Keinan et al., 1986 ▶; Drueckhammer et al., 1988 ▶). The three-dimensional structure of TbSADH has been solved by X-ray crystallography in complex with NADP+ to 2.5 Å resolution in space group P65 (PDB entry 1ykf; Korkhin et al., 1998 ▶) and in complex with (S)-2-butanol to 2.99 Å resolution in space group P212121 (PDB entry 1bxz; Li et al., 1999 ▶). The substrate specificity and stereospecificity of the enzyme are determined by the structural and chemical properties of the substrate-binding site, which is formed by a large and a small pocket (Heiss et al., 2001 ▶). The current hypothesis explaining the stereospecificity of TeSADH is that if the larger of the two substituents of a ketone fits into the small pocket the enzyme will be likely to produce an (R)-alcohol. If the larger substituent is too large to fit into the small pocket, this substituent will bind in the large pocket, causing the enzyme to produce an (S)-alcohol (Keinan et al., 1986 ▶).
We are studying how the shape, size and physicochemical properties of the substrate-binding site of TeSADH affect its substrate specificity and stereospecificity. The TeSADH I86A mutant was constructed in order to determine whether a smaller side chain lining the small cavity would affect the stereoselectivity. TeSADH I86A was still very active towards 2-butanol, but it also became active towards acetophenone and (R)-1-phenylethanol, while it showed no activity towards (S)-1-phenylethanol. TeSADH I86A was also able to asymmetrically reduce benzylic and heteroaryl ketones to produce the corresponding (R)-alcohols (Musa et al., 2009 ▶). In this mutant enzyme the pro-(R) hydride is still delivered to the si face of the prochiral ketone (Fig. 1 ▶), indicating that the I86A mutation does not alter the orientation of NADPH in the enzyme (Musa et al., 2009 ▶).
Figure 1.

A proposed model for the stereopreference of TeSADH I86A following Prelog’s rule for predicting the stereopreference of alcohol dehydrogenases. The mutation I86A allows large substituents to fit into the large pocket of I86A TeSADH, which corresponds to the small pocket in wild-type TeSADH. ADPR, adenosine diphosphoribose. This figure was adapted from Musa et al. (2009 ▶).
Not only is TeSADH I86A the first example of an anti-Prelog ADH engineered from a Prelog ADH with a single mutation, it also accepts more sterically demanding substrates than those accepted by wild-type TeSADH. To confirm that the inverse stereoselectivity of TeSADH I86A arises from the reverse fit of given substrates in the active site and to investigate how to further broaden the substrate specificity of TeSADH, we initiated structural analysis of this mutant enzyme. Here, we report new crystallization conditions and the preliminary crystallographic analysis of TeSADH I86A.
2. Materials and methods
2.1. Mutagenesis, protein expression and purification
Site-directed mutagenesis of the T. ethanolicus adhB gene carried out in plasmid pADHB1M1-kan (Burdette et al., 1997 ▶) is described by Musa et al. (2009 ▶). The mutated gene was then subcloned into pET24a(+). From this construct, I86A TeSADH was expressed with its two N-terminal methionines. The natural C-terminal residue of TeSADH is followed by residues Leu-Glu-His6, where Leu-Glu are encoded by the XhoI cloning site. TeSADH I86A was expressed in Escherichia coli HB101 (DE3) and purified as described by Musa et al. (2009 ▶). Briefly, after induction with isopropyl β-d-1-thiogalactopyranoside, cells were resuspended in Tris buffer pH 8.5 containing 1 mM phenylmethylsulfonyl fluoride. The cells were lysed in a French pressure cell and the soluble extract was heat-treated at 348 K for 15 min. After centrifugation, I86A TeSADH was purified from the cleared crude extract by Ni-affinity chromatography (Fig. 2 ▶).
Figure 2.
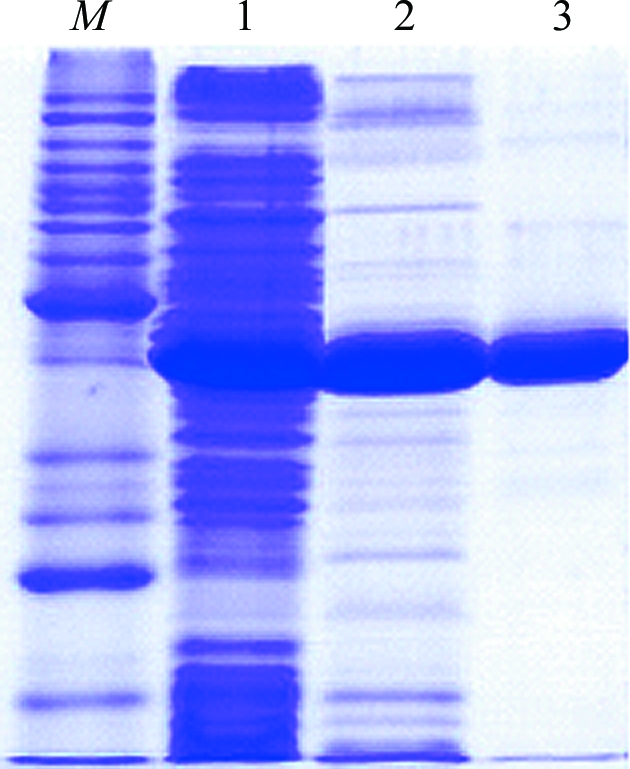
SDS–PAGE of TeSADH I86A purification. Lane M, molecular markers; lane 1, crude cell extract; lane 2, heat-treated soluble fraction; lane 3, TeSADH I86A after Ni-affinity purification.
2.2. Protein crystallization
Initial crystallization conditions for the TeSADH I86A mutant protein were identified by high-throughput crystallization screening at the Hauptman–Woodword Medical Research Institute using the microbatch-under-oil method. The crystallization conditions were subsequently optimized using hanging-drop vapour diffusion in VDX Plates (Hampton Research, Aliso Viejo, California, USA). Crystals were obtained by combining 2 µl protein solution [8 mg ml−1 protein, 50 mM 2-(N-morpholino)ethanesulfonic acid pH 6.6, 20 mM NaCl, 0.1 mM dithiothreitol, 0.1 mM ethylenediaminetetraacetic acid, 2 mM (R)-1-phenylethanol], 2 µl Silver Bullets F2 [0.2% d-fructose 1,6-disphosphate trisodium salt octahydrate, 0.2% glycerol phosphate disodium salt hydrate, 0.2% l-O-phosphoserine, 0.2% O-phospho-l-tyrosine, 0.2% phytic acid sodium salt hydrate, 0.02 M 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) sodium pH 6.8; Hampton Research, Aliso Viejo, California, USA] and 1 µl reservoir solution [20%(v/v) polyethylene glycol 400, 0.1 M Na HEPES pH 7.0] in a drop over a reservoir-solution volume of 500 µl at room temperature (298 K). Pyramidal crystals appeared within 6–12 h, with the largest crystal being approximately 0.15 × 0.075 × 0.05 mm in size (Fig. 3 ▶). The crystals were harvested and directly placed into LV CryoOil (MiTeGen, Ithaca, New York, USA); the remaining mother liquor surrounding the crystal was removed prior to flash-cooling in liquid nitrogen.
Figure 3.

Crystals of TeSADH I86A mutant. The dimensions of the largest crystals are 0.15 × 0.075 × 0.05 mm. The picture was taken using a polarizing filter.
2.3. Data collection and processing
Diffraction data were collected on the Canadian Macromolecular Crystallography Facility (CMCF-1) beamline (08ID-1) at the Canadian Light Source (Saskatoon, Saskatchewan, Canada). The data were collected at a wavelength of 0.97949 Å as 180 images, each of which was collected with 3 s exposure over a 1° oscillation range at a crystal-to-detector distance of 260 mm. The data-collection statistics for the TeSADH I86A crystals are summarized in Table 1 ▶. Intensity data were indexed, integrated and scaled using XDS (Kabsch, 2010 ▶).
Table 1. Data-collection statistics.
| Beamline | 08ID-1, Canadian Light Source |
| Detector | MAR 300 CCD |
| Temperature (K) | 100 |
| Space group | P212121 |
| Unit-cell parameters (Å) | a = 80.23, b = 124.90, c = 164.80 |
| Resolution range (Å) | 50–3.20 (3.28–3.20) |
| Matthews coefficient (Å3 Da−1) | 2.58 |
| Solvent content (%) | 52 |
| No. of molecules in asymmetric unit | 4 |
| No. of measured reflections | 193080 (12759) |
| Total No. of unique reflections | 26008 (1708) |
| Completeness (%) | 92.9 (84.6) |
| Multiplicity | 7.42 (7.47) |
| Rmerge | 0.22 (0.81) |
| 〈I/σ(I)〉 | 10.06 (2.68) |
3. Results and discussion
The crystals of TeSADH I86A belonged to the orthorhombic space group P212121, with unit-cell parameters a = 80.23, b = 124.90, c = 164.80 Å. The Matthews coefficient (Matthews, 1968 ▶) was 2.58 Å3 Da−1, with a solvent content of 52%, assuming the presence of a tetramer in the asymmetric unit. A molecular-replacement solution was found using the structure of TbSADH (identical to TeSADH in sequence) in complex with NADP+. The structure was solved by molecular replacement using Phaser (Storoni et al., 2004 ▶). The solution had a log-likelihood gain (LLG) of 5996.63, with the next solution having an LLG of 3161. Preliminary structural refinement was completed using REFMAC5 (Murshudov et al., 1997 ▶). After three cycles of refinement, the R factor was 0.22 and R free was 0.28 at 3.2 Å resolution.
The arrangement of the molecules in the asymmetric unit was found to be the same as in TbSADH: a tetramer formed by a dimer of dimers. Each dimer consisted of two monomers, each with a molecular weight of 37 590 Da (352 amino-acid residues). The (R)-1-phenylethanol molecule is speculated to exist near Trp110 and density was observed in the difference map. However, at the resolution of these data we were unable to model the substrate properly. Further refinement and modelling of (R)-1-phenylethanol are pending owing to the lack of high-resolution data.
Comparison with the previously solved stucture of TbSADH in complex with (S)-2-butanol (PDB code 1bxz) showed that both studies resulted in basically the same crystal form, despite different crystallization conditions that were independently obtained. The crystallization conditions used to crystallize 1bxz differed in pH, salt, molecular weight of polyethylene glycol, additives and substrates compared with the conditions for our TeSADH I86A crystals (the conditions for 1bxz were 2 mM NADP, 50 mM Tris–HCl, 50 mM NaCl, 0.1 mM DTT, 50 µM ZnCl2, 14% PEG 4000 pH 8.3; Korkhin et al., 1996 ▶). In the previous study magnesium and chloride ions were found to be involved in crystal contacts. To investigate such a role in our crystals, the structure of TbSADH in complex with (S)-2-butanol (PDB code 1bxz) was superimposed on the structure of TeSADH (r.m.s.d. 0.46 Å). There were no Mg2+ or Cl− ions present to form contacts at the interface of the two subunits in the TeSADH I86A structure as previously observed in the 1bxz structure.
Acknowledgments
LTJD was a Tier 1 Canada Research Chair in Structural Biochemistry (chair 206885). We thank CRC and the US National Science Foundation (grant No. 0445511 to CV) for support of this research. The research described in this paper was performed at the Canadian Light Source, which is supported by NSERC, NRC, CIHR and the University of Saskatchewan.
References
- Bradshaw, C. W., Hummel, W. & Wong, C. (1992). J. Org. Chem.57, 1532–1536.
- Burdette, D. S., Secundo, F., Phillips, R. S., Dong, J., Scott, R. A. & Zeikus, J. G. (1997). Biochem. J.326, 717–724. [DOI] [PMC free article] [PubMed]
- Drueckhammer, D. G., Barbas, C. F. III, Nozaki, K., Wong, C.-H., Wood, C. Y. & Ciufolini, M. A. (1988). J. Org. Chem.53, 1607–1611.
- Heiss, C., Laivenieks, M., Zeikus, J. G. & Phillips, R. S. (2001). Bioorg. Med. Chem.9, 1659–1666. [DOI] [PubMed]
- Kabsch, W. (2010). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Keinan, E., Hafeli, E. K., Seth, K. K. & Lamed, R. (1986). J. Am. Chem. Soc.108, 162–169.
- Korkhin, Y., Frolow, F., Bogin, O., Peretz, M., Kalb (Gilboa), A. J. & Burstein, Y. (1996). Acta Cryst. D52, 882–886. [DOI] [PubMed]
- Korkhin, Y., Kalb, A. J., Peretz, M., Bogin, O., Burstein, Y. & Frolow, F. (1998). J. Mol. Biol.278, 967–981. [DOI] [PubMed]
- Kroutil, W., Mang, H., Edegger, K. & Faber, K. (2004). Curr. Opin. Chem. Biol.8, 120–126. [DOI] [PubMed]
- Lamed, R., Keinan, E. & Zeikus, J. G. (1981). Enzyme Microb. Technol.3, 144–148.
- Li, C., Heatwole, J., Soelaiman, S. & Shoham, M. (1999). Proteins, 37, 619–627. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed]
- Musa, M. M., Lott, N., Laivenieks, M., Watanabe, L., Vieille, C. & Phillips, R. S. (2009). ChemCatChem, 1, 89–93.
- Prelog, V. (1964). Pure Appl. Chem.9, 119–130.
- Storoni, L. C., McCoy, A. J. & Read, R. J. (2004). Acta Cryst. D60, 432–438. [DOI] [PubMed]
- Wong, C.-H., Drueckhammer, D. G. & Sweers, H. M. (1985). J. Am. Chem. Soc.107, 4028–4031.