

T. cruzi TcNDPK1 was overexpressed in Escherichia coli as an N-terminally poly-His-tagged fusion protein and crystallized.
Keywords: nucleoside dephosphate kinase 1, Trypanosoma cruzi
Abstract
The flagellated protozoan parasite Trypanosoma cruzi is the aetiological agent of Chagas disease. Nucleoside diphosphate kinases (NDPKs) are enzymes that are involved in energy management and nucleoside balance in the cell. T. cruzi TcNDPK1, a canonical isoform, was overexpressed in Escherichia coli as an N-terminally poly-His-tagged fusion protein and crystallized. Crystals grew after 72 h in 0.2 M MgCl2, 20% PEG 3350. Data were collected to 3.5 Å resolution using synchrotron X-ray radiation at the National Synchrotron Light Laboratory (Campinas, Brazil). The crystals belonged to the trigonal space group P3, with unit-cell parameters a = b = 127.84, c = 275.49 Å. Structure determination is under way and will provide relevant information that may lead to the first step in rational drug design for the treatment of Chagas disease.
1. Introduction
The flagellated protozoan parasite Trypanosoma cruzi is the aetiological agent of Chagas disease, which represents a serious health problem in the Americas, with 18 million people being infected and 100 million people being at risk (Barrett et al., 2003 ▶).
A common feature of parasitic protozoan organisms is their ability to adapt their metabolism in order to survive under a wide range of environmental conditions and selection pressures, including the availability and quality of carbon sources in the different mammalian and insect hosts (Tielens & Hellemond, 1998 ▶). One of the enzyme families that are involved in cell energy management is the nucleoside diphosphate kinases (NDPKs). These are enzymes that are involved not only in energy metabolism but also in the transfer of phosphates between nucleoside triphosphates and diphosphates, thus maintaining the nucleoside balance in the cell (Parks & Agarwal, 1973 ▶), according to the following reaction:
For a long time NDPKs were considered to be housekeeping enzymes; however, a wide variety of additional functions have recently been described. T. cruzi has four putative genes coding for NDPKs. Two of them have been characterized by our group and code for bona fide NDPK isoforms named TcNDPK1 and TcNDPK2 (GeneBank accession Nos. XP_820714 and XP_821632, respectively; Miranda et al., 2008a ▶). TcNDPK1 and TcNDPK2 present different kinetic parameters and regulation mechanisms. TcNDPK1 is a ‘short’ canonical isoform, whereas TcNDPK2 is a ‘long’ isoform containing an N-terminal extension called the DM10 motif. Interestingly, in addition to phosphotransferase activity, TcNDPK1 also has a high nuclease activity in vitro (Miranda et al., 2008b ▶). TcNDPK1 is a hexameric homoprotein (Ulloa et al., 1995 ▶) like all NDPKs from eukaryotes (Janin et al., 2000 ▶; Giraud et al., 2006 ▶) and archaea and from the bacteria Bacillus subtilis (Sedmak & Ramaley, 1971 ▶) and Mycobacterium tuberculosis (Chen et al., 2002 ▶), in contrast to the NDPKs from other bacteria [Escherichia coli (Ohtsuki et al., 1984 ▶; Almaula et al., 1995 ▶), Salmonella typhymurium (Ginther & Ingraham, 1974 ▶), Myxococcus xanthus (Munoz-Dorado et al., 1990 ▶; Williams et al., 1993 ▶) and Streptomyces coelicolor (Brodbeck et al., 1996 ▶)] which are tetrameric (Moynié et al., 2007 ▶). The TcNDPK1 monomer has a molecular weight of 16 878.3 Da.
Protozoan parasites, including T. cruzi, are not able to synthesize purines de novo and therefore rely upon the salvage pathway, in which free purines are converted to nucleotides by enzymes with NDPK activity (Hammond & Gutteridge, 1984 ▶; Marr, 1991 ▶). Biochemical and molecular-biology studies of the proteins involved in important metabolic pathways can help in understanding the biology of the parasite and thereby contribute to strategies for combatting Chagas disease.
This work describes how the overexpression and purification conditions of TcNDPK1 were optimized in order to obtain a higher yield of soluble protein. The crystallization conditions, results and preliminary X-ray analysis are also reported.
2. Materials and methods
2.1. Recombinant TcNDPK1 preparation
The recombinant plasmid was obtained as described previously (Miranda et al., 2008a ▶). We worked with the T. cruzi ndpk1 gene cloned into the pRSET-A plasmid. This vector system allows the expression of TcNDPK1 as a fusion protein with an N-terminal His tag. The plasmid was used to transform Escherichia coli BL21(DE3)pLys-S (Invitrogen, Carlsbad, California, USA) containing a lacUV5 promoter-driven T7 RNA polymerase. Competent bacteria were obtained by the CaCl2 method and the transformation was realised by thermal shock. The transformant clones were selected on solid LB–agar medium containing 100 µg ml−1 ampicillin and 70 µg ml−1 chloramphenicol. The selected colonies were grown in 5 ml LB medium containing 50 µg ml−1 ampicillin and 35 µg ml−1 chloramphenicol at 310 K overnight. A glycerol stock (80%) of each culture was stored at 193 K for future applications. Each overnight culture was separated into two 5 ml cultures and grown to an optical density of 0.6 at 600 nm. One culture corresponding to a single colony was induced with 0.4 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) and grown for an additional 4 h at the same temperature to express the fusion protein. The other culture from the same colony was used as a non-induced negative control. The cells were recovered by centrifugation and immediately lysed by three freeze–thaw cycles.
A starter culture of BL21(DE3)pLys-S harbouring pRSET-A-ndpk was grown in 5 ml LB medium containing 50 µg ml−1 ampicillin and 35 µg ml−1 chloramphenicol at 310 K overnight. This initial culture was increased to 3 l, grown to an optical density of 0.6 at 600 nm and induced with IPTG at a concentration of 0.2 mM. The bacteria were recovered by centrifugation at 4000g for 10 min at 277 K. The pellet was lysed by three freezing and defrosting cycles and was immediately resuspended in 150 ml native lysis buffer (20 mM Tris–HCl pH 7.3, 500 mM NaCl) and sonicated in ice by four cycles of 30 s with 25 s intervals between each pulse. The lysed sample was centrifuged at 17 700g for 45 min at 277 K. The pellet and supernatant obtained were analysed by 15% SDS–PAGE stained with Coomasie Brilliant Blue R-250 to check the efficiency of expression and lysis.
His-TcNDPK1 was purified using an Ni-Sepharose affinity column (Amersham). The crude extract was applied onto the affinity column and the flowthrough was collected. The column was washed with five volumes of 20 mM Tris–HCl pH 7.3, 500 mM NaCl buffer. The elution step was realised by application to the column of 10 ml of the same buffer containing an imidazole concentration in the range 5–500 mM. The obtained fractions were analysed by SDS–PAGE and stained with Coomassie Blue. The samples containing the recombinant protein were applied onto a Superdex 200 16/60 column. The purity of the eluates was evaluated by SDS–PAGE stained with Coomassie Blue. The samples corresponding to His-TcNDPK1 were concentrated using ultrafiltration cells (Amicon). The final protein sample was washed and quantified using the absorbance at 280 nm (Gill & von Hippel, 1989 ▶) and the Bradford assay (Bradford, 1976 ▶).
2.2. Crystallization assays
Initial attempts to crystallize His-TcNDPK1 were performed by sitting-drop vapour diffusion using the sparse-matrix screening method with Xtal Classic Suite, Xtal Classic Suite II and PEG II (Qiagen). The protein solution (10 mg ml−1 protein in 20 mM Tris–HCl pH 7.0, 20 mM NaCl, 10 mM ATP and 10 mM MgCl2) was mixed with an equal volume of reservoir solution and the drop was placed over 1 ml reservoir solution in a Linbro box. Protein crystals were observed in 72 h in a condition consisting of 0.2 M MgCl2, 0.1 M Tris–HCl pH 8.5, 30% PEG 4000. This condition was refined by the hanging-drop vapour-diffusion method at 293 and 277 K by using different PEG concentrations and molecular weights and varying the pH.
2.3. Diffraction and data collection
The crystals obtained after 72 h were flash-cooled in liquid nitrogen using a cryogenic solution consisting of 20% glycerol mixed with 80% reservoir buffer. Each crystal was harvested using a nylon loop and transferred from the crystallization drop to 5 µl cryogenic solution for a few seconds. The crystals were immediately flash-cooled to 100 K in a nitrogen stream. These crystals were used for data collection. Diffraction data were collected using synchrotron X-ray radiation at the National Synchrotron Light Laboratory (LNLS, Campinas, Brazil). The data set was reduced and merged using the MOSFLM (Leslie, 1992 ▶) and SCALA (Collaborative Computational Project, Number 4, 1994 ▶) programs.
3. Results and discussion
The expression clones were selected on LB plates containing 100 µg ml−1 ampicillin and 70 µg ml−1 chloramphenicol. The colonies isolated from the agar plates were used for overexpression of TcNDPK1 with satisfactory results. SDS–PAGE analysis showed that high-level expression of a 20 kDa polypeptide which corresponded to the recombinant TcNDPK1 was obtained in the soluble fraction. A high yield of soluble recombinant protein was obtained after 6 h of IPTG induction using E. coli strain BL21(DE3)pLysS. The crude extract was applied onto an Ni-Sepharose affinity column and fractions were eluted using imidazole as described in §2. The recombinant protein was eluted in fractions corresponding to high imidazole concentrations of approximately 500 mM. The obtained fractions were concentrated and applied onto a Superdex-200 16/60 column. A single peak was observed in the first fractions, indicating that the protein might be in a polymeric form. This result was confirmed using dynamic light-scattering and native PAGE assays, which suggested the presence of a hexameric complex in solution. The purity of the protein as analyzed by SDS–PAGE showed that the sample was suitable for crystallographic assays (Fig. 1 ▶). Protein crystals were obtained by hanging-drop vapour-diffusion assays at 277 K using 10 mg ml−1 recombinant protein in 20 mM Tris–HCl pH 7.0, 20 mM NaCl, 10 mM ATP and 10 mM MgCl2 as the protein solution and 18–20% PEG 3350 and 200 mM MgCl2 solution as the reservoir; 1.5 µl protein solution was mixed with an equal volume of reservoir solution (Fig. 2 ▶). A data set was collected to 3.5 Å resolution using synchrotron X-ray radiation at LNLS (Table 1 ▶). The crystals belonged to the trigonal space group P3, with unit-cell parameters a = b = 127.84, c = 275.49 Å. Initial analysis of the crystal solvent content using the Matthews coefficient (Matthews, 1968 ▶) suggested that the asymmetric unit contains 24 molecules with 61.72% solvent content, 30 molecules with 52.15% solvent content or 36 molecules with 42.58% solvent content. Structure determination is under way.
Figure 1.

The final protein sample obtained after the purification protocol (right) was very pure compared with the soluble fraction (left) obtained after the lysis step. The protein efficiently purified by affinity and size-exclusion chromatographic techniques is shown on 15% SDS–PAGE stained with Coomassie Blue.
Figure 2.

Crystals of His-TcNDPK1 used for diffraction analysis.
Table 1. X-ray data-collection statistics.
| Wavelength (Å) | 1.45 |
| Resolution range (Å) | 86.38–3.00 |
| Space group | P3 |
| Unit-cell parameters (Å, °) | a = b = 127.84, c = 275.49, α = β = 90, γ = 120 |
| Completeness (%) | 92.3 |
| Redundancy | 1.8 |
| Rmerge† | 0.34 |
| Average I/σ(I) | 2.7 |
| Total reflections | 103460 |
| Unique reflections | 58612 |
R
merge = 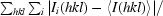
 , where I
i(hkl) is the intensity of the ith observation and I(hkl) is the mean intensity of the reflections.
, where I
i(hkl) is the intensity of the ith observation and I(hkl) is the mean intensity of the reflections.
Acknowledgments
This work was supported by grants from the international agreement between Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Brazil and Secretaría de Ciencia y Tecnología (Secyt), Argentina, from Agencia Nacional de Promoción Científica y Tecnológica (FONCyT) PICTR 2003-300 and PICT-2005 33431, from the National Research Council of Argentina (CONICET) PIP 5492 and from Secretaría de Ciencia y Técnica de la Universidad Nacional de San Luis, Argentina. CAP is a member of the career of scientific investigator of CONICET (Argentina) and MRM is a research fellow of Fundación YPF.
References
- Almaula, N., Lu, Q., Delgado, J., Belkin, S. & Inouye, M. (1995). J. Bacteriol.177, 2524–2529. [DOI] [PMC free article] [PubMed]
- Barrett, M. P., Burchmore, R. J., Stich, A., Lazzari, J. O., Frasch, A. C., Cazzulo, J. J. & Krishna, S. (2003). Lancet, 362, 1469–1480. [DOI] [PubMed]
- Bradford, M. M. (1976). Anal. Biochem.72, 248–254. [DOI] [PubMed]
- Brodbeck, M., Rohling, A., Wohlleben, W., Thompson, C. J. & Süsstrunk, U. (1996). Eur. J. Biochem.239, 208–213. [DOI] [PubMed]
- Chen, Y., Moréra, S., Mocan, J., Lascu, I. & Janin, J. (2002). Proteins, 47, 556–557. [DOI] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Gill, S. C. & von Hippel, P. H. (1989). Anal. Biochem.182, 319–326. [DOI] [PubMed]
- Ginther, C. L. & Ingraham, J. L. (1974). J. Biol. Chem.249, 3406–3411. [PubMed]
- Giraud, M. F., Georgescauld, F., Lascu, I. & Dautant, A. (2006). J. Bioenerg. Biomembr.38, 261–264. [DOI] [PubMed]
- Hammond, D. J. & Gutteridge, W. E. (1984). Mol. Biochem. Parasitol.13, 243–261. [DOI] [PubMed]
- Janin, J., Dumas, C., Moréra, S., Xu, Y., Meyer, P., Chiadmi, M. & Cherfils, J. (2000). J. Bioenerg. Biomembr.32, 215–225. [DOI] [PubMed]
- Leslie, A. G. W. (1992). Jnt CCP4/ESF–EACBM Newsl. Protein Crystallogr.26
- Marr, J. J. (1991). J. Lab. Clin. Med.118, 111–119. [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed]
- Miranda, M. R., Canepa, G. E., Bouvier, L. A. & Pereira, C. A. (2008a). Exp. Parasitol.120, 103–107. [DOI] [PubMed]
- Miranda, M. R., Canepa, G. E., Bouvier, L. A. & Pereira, C. A. (2008b). Parasitology, 135, 1661–1666. [DOI] [PubMed]
- Moynié, L., Giraud, M. F., Georgescauld, F., Lascu, I. & Dautant, A. (2007). Proteins, 67, 755–765. [DOI] [PubMed]
- Munoz-Dorado, J., Inouye, S. & Inouye, M. (1990). J. Biol. Chem.265, 2707–2712. [PubMed]
- Ohtsuki, K., Yokoyama, M., Koike, T. & Ishida, N. (1984). Biochem. Int.8, 715–723. [PubMed]
- Parks, R. E. Jr & Agarwal, R. P. (1973). The Enzymes, 3rd ed., edited by P. D. Boyer, Vol. 8, pp. 307–334. New York: Academic Press.
- Sedmak, J. & Ramaley, R. (1971). J. Biol. Chem.246, 5365–5372. [PubMed]
- Tielens, A. G. & Van Hellemond, J. J. (1998). Parasitol. Today, 14, 265–272. [DOI] [PubMed]
- Ulloa, R. M., Muschietti, J. P., Veron, M., Torres, H. N. & Tellez-Iñón, M. T. (1995). Mol. Biochem. Parasitol.70, 119–29. [DOI] [PubMed]
- Williams, R. L., Oren, D. A., Munoz-Dorado, J., Inouye, S., Inouye, M. & Arnold, E. (1993). J. Mol. Biol.234, 1230–1247. [DOI] [PubMed]