

Abstract
Purine nucleoside phosphorylase (PNP) catalyzes the synthesis and phosphorolysis of purine nucleosides, interconverting nucleosides with their corresponding purine base and ribose-1-phosphate. While PNP plays significant roles in human and pathogen physiology, we are interested in developing PNP as a catalyst for the formation of nucleoside analog drugs of clinical relevance. Towards this aim, we describe the engineering of human PNP to accept 2′,3′-dideoxyinosine (ddI, Videx®) as a substrate for phosphorolysis using a combination of site-directed mutagenesis and directed evolution. In human PNP, we identified a single amino acid, Tyr-88, as a likely modulator of ribose selectivity. RosettaLigand was employed to calculate binding energies for substrate and substrate analog transition state complexes for single mutants of PNP where Tyr-88 was replaced with another amino acid. In parallel, these mutants were generated by site-directed mutagenesis, expressed and purified. A tyrosine to phenylalanine mutant (Y88F) was predicted by Rosetta to improve PNP catalytic activity with respect to ddI. Kinetic characterization of this mutant determined a 9-fold improvement in kcat and greater than 2-fold reduction in KM. Subsequently, we used directed evolution to select for improved variants of PNP-Y88F in Escherichia coli cell extracts resulting in an additional 3-fold improvement over the progenitor strain. The engineered PNP may form the basis for catalysts and pathways for the biosynthesis of ddI.
Keywords: directed evolution, enzyme design, nucleoside analog, purine nucleoside phosphorylase, Rosetta
Introduction
Nucleoside analog drugs are primary therapeutics in the treatment of viral infections including HIV (Clerq, 2009) and hepatitis (Soriano et al., 2009). Indeed, it has recently been reported that of the 25 drugs currently approved in the USA for the treatment of HIV, 8 are nucleoside analogs (Clerq, 2009). Nucleoside analogs are also currently in advanced stage clinical trials for the treatment of various cancers including leukemia (Li et al., 2008). The broad efficacy of these compounds contrasts with their price. The cost of the manufacturing of the active ingredients of some of these drugs comprise up to 55–99% of the final therapeutic price (Pinheiro et al., 2006), a fact which has spurred the continuing development of new methods for the synthesis of nucleoside analogs.
We are interested in developing biocatalytic alternatives for the synthesis of nucleoside analogs. Correspondingly, we have targeted 2′,3′-dideoxyinosine (ddI, Didanosine, Videx®), a reverse transcriptase class nucleoside analog used in the treatment of HIV, as an attractive target for directed biosynthesis. Dideoxyinosine is representative of the broader class of dideoxynucleoside drugs and is a close analog of the primary metabolite inosine. Both de novo biosynthesis and purine salvage pathways for inosine have been extensively characterized in biochemical and structural studies (Pugmire and Ealick, 2002; Schramm, 2005). As a large fraction of nucleoside analogs are variants of 2′3′-dideoxynucleosides, methods developed for ddI may have broader application (Clerq, 2009).
Enzymes with new or improved functions are increasingly generated from existing enzymes or scaffolds by a 2-fold strategy consisting of (i) rational mutational active site remodeling, to modify binding specificity for a desired reaction or substrate, followed by (ii) optimization of global protein function by more stochastic methods such as directed evolution. Prerequisites to the first stage are the identification of a progenitor enzyme or scaffold with a suitable starting activity and acquisition of some knowledge of the active site geometry from structural data or homology models. If a suitable enzyme is identified, first or second shell interacting active site residues are selected for mutation and functional assessment (Voigt et al., 2001; Reetz et al., 2005; Jackel et al., 2008). As even the most prudently selected active site mutations may result in unexpected catalytic consequences, the complete complement of amino acids at targeted active site residues is often generated by saturation mutagenesis (Bernhardt et al., 2007).
To focus these efforts, it would be desirable to identify a subset of potentially beneficial mutations, particularly in cases in which permutations of several active site residues may be required. Many computational protein design strategies have been developed to engineer alternative ligand specificity into proteins and generate enzyme variants with improved activity on non-cognate substrates (Damborsky and Brezovsky, 2009). Recently, methods incorporated in the Rosetta program have been developed to estimate relative ligand–protein interaction energies with conformational flexibility. RosettaLigand employs a Monte Carlo-based search algorithm with protein side chains replaced by residues from a rotamer library (Meiler and Baker, 2006; Davis and Baker, 2009). It is easily combined with RosettaDesign (Kuhlman et al., 2003) allowing for redesign of binding pockets (Murphy et al., 2009). Indeed, the Rosetta framework has been successfully applied to the design of enzymes catalyzing both natural (retro-aldol (Jiang et al., 2008)) and non-natural (Kemp elimination (Rothlisberger et al., 2008)) reactions in the context of novel protein scaffolds (Zanghellini et al., 2006). The protocol identifies critical interactions between transition state models and catalytic residues. Subsequently, adjacent amino acids are placed in the binding site to optimize stability of the catalytic residues and specificity for the ligand. The initial computational designs displayed low catalytic activity and were further improved through directed evolution and screening.
Herein, we describe the identification of human purine nucleoside phosphorylase (hPNP) as an engineering candidate for nucleoside analog biocatalysis. hPNP catalyzes the reversible synthesis or phosphorolysis of 6-oxopurine (deoxy)nucleosides (Fig. 1). Previous biochemical and structural characterization of this enzyme and its transition state permitted the identification of a single first shell active site residue, Tyr-88 (Y88), as a potential modulator of ribose substrate selectivity (Stoeckler et al., 1980; Erion et al., 1997). Additionally, computational design of the hPNP binding site in the presence of inosine and ddI suggested Y88 as critical for differentiation of the two substrates. This result demonstrates that the computational method is generally applicable to focus experimental studies on specific sites and selected mutants thereby reducing experimental effort and accelerating research. RosettaLigand was used to predict changes in transition state binding free energy upon mutation of this position to all genetically encoded amino acids except glycine and proline. Mutation to phenylalanine was consistently predicted to increase catalytic activity with respect to ddI. Experimental testing finds Y88F is 23.6-fold more active for ddI and 2.9-fold less active for inosine. Systematic experimental analysis of all Y88X single mutants finds good agreement of Rosetta predictions with experimental data for all but negatively charged amino acids.
Fig. 1.

A depiction of interactions in the hPNP binding site. Mutations for rational design are limited to Y88 (bold) which hydrogen bonds to inosine at the 3′-OH.
The present study differs from previous enzyme designs in that Rosetta is tested in a limiting setup where scaffold, binding mode and even a single site of mutation are predetermined. Thereby success hinges on accurate prediction of binding free energy changes for mutations at the Y88 site. The results demonstrate Rosetta's general ability to identify favorable mutations in enzyme catalytic sites. They enabled a customization of the Rosetta energy function to improve correlation between predicted and experimentally determined transition state binding affinities to R = 0.65. However, these results point also to inaccuracies in handling electrostatics, in particular for charged amino acids, where further improvement of the Rosetta energy function is needed.
The Y88F variant then became the starting point for a directed evolution study. A high-throughput assay was developed and combined with error-prone PCR (epPCR) to generate and test libraries of PNP mutants. Three rounds of mutation and selection resulted in an enzyme with a modest 3-fold improvement in turnover compared with Y88F in in vitro assays.
To the best of our knowledge, this study comprises the first engineering of a nucleoside phosphorylase for the biosynthesis of an unnatural dideoxynucleoside. This work demonstrates that a combination approach of targeted active site mutation and directed evolution may find future application in nucleoside analog biosynthetic pathway engineering.
Materials and methods
Transition state model
Transition state models for docking and calculation of binding energies incorporate critical characteristics of the mechanism and transition state.
The binding modes of substrates (PDB code: 1m73, 1a9s, 1rct, 1v2h, 1pwy, 1rfg), substrate analogs (PDB code: 1a9t, 1v41, 1v3q, 1v45, 1rt9) and transition state analog inhibitors (PDB code: 1pf7, 1rsz, 1rr6, 1b8o) crystallized with human and bovine PNP were studied for their resemblance to the experimentally derived transition state (Lewandowicz and Schramm, 2004). Coordinates of ligands with characteristics matching the known transition state were used as internal coordinates of the transition state model. Phosphate coordinates are derived from ribose-1-phosphate crystallized in bovine PNP (PDB code: 1a9t) as OP is oriented appropriately for nucleophilic attack. The P–OP bond in this structure is ∼0.22 Å longer than the other P–O bonds recapitulating the known bond lengthening of ∼0.23 Å as the reaction coordinate progresses (Deng et al., 2004). The purine of DADME-Immucilin-H (PDB code: 1rsz) is the aromatic base for both inosine and ddI transition state models (Ringia et al., 2006). Separate coordinates for the sugar residues of inosine and ddI transition states were taken from Immucilin-H (PDB code: 1pf7) and ddI (PDB code: 1v3q), respectively. The ring shape in ddI is flatter than that of nucleosides with hydroxyls at the 2′ or 3′ positions. Internal coordinates of the Immucilin-H sugar moiety were used for the inosine transition state model to best replicate the position of the 5′-OH over the 4′-O. The O5′–C5′–C4′–C3′ dihedral of Immucilin-H in 1pf7 is 62.72°. For ddI, the 5′-OH of the dideoxyribose moiety was rotated manually to match that of Immucilin-H. The ddI transition state model has an O5′–C5′–C4′–C3′ dihedral angle of 65.41°. The sugar moiety was aligned with the average C3′, C4′ and C5′ position from the aligned structures and a point directly between N9 of the purine and OP of the phosphate which put the anomeric carbon 2.56 Å from N9 and 2.66 Å from OP, both of which are shorter than the known value of 3 Å calculated from kinetic isotope effect (Lewandowicz and Schramm, 2004).
Computational mutation and docking
Trimeric hPNP coordinates were obtained from the Protein Data Bank (PDB code: 1rct, 1pf7, 1rr6, 1v3q) and relaxed using a combination of Monte Carlo rotamer replacement and gradient-based minimization (Qian et al., 2007) to generate an ensemble of 10 energy minimized models for each parent structure. Inosine and ddI transition state models were placed in the binding site of the minimized backbone ensemble for the ensemble derived from 1pf7 for docking and design.
Residues in the hPNP binding site involved in substrate selectivity of inosine over ddI were identified by redesigning the binding pocket around each transition state model. Correspondingly, 23 residues with C-α or C-β atoms within eight angstroms of the ligand and C-α/C-β vectors oriented toward the binding site were allowed to mutate (Fig. 2 and Supplementary data, Fig. S1A). To facilitate an unbiased design process, the amino acid identity for all 23 residues was converted to alanine. The position of inosine and ddI transition state models was optimized during design of these 23 residues along with concurrent repacking of additional side chains in the vicinity of the binding site. Three iterative rounds of design were performed to pare down the list of mutable residues. Residues that Rosetta filled with the wild-type amino acid were converted back to the wild-type identity after each round along with residues that lack interaction with substrate transition state models. After completion of this procedure, the Rosetta-suggested mutations for the remaining six sites were compared for inosine and ddI in order to identify sites for differentiation.
Fig. 2.

To identify positions which modulate substrate selectivity, amino acids oriented into the hPNP binding site were optimized with RosettaDesign for inosine and ddI transition state models. (A) C-α and C-β atoms of designed residues are shown as balls and sticks. The transition state analog Immucilin-H and sulfate are depicted (PDB code 1pf7). The position and identity of residues undergoing design in the final round are labeled. (B) Predicted ddI binding energy (ΔΔG) and differential binding energies (ΔΔΔG) for mutants suggested by Rosetta during the final round of design. Y88 mutations are expected to increase affinity to ddI while maintaining tight binding to the transition state.
Upon biochemical and computational confirmation of Y88 as a likely modulator of substrate selectivity, the identity of Y88 was altered to each possible amino acid (excluding Pro and Gly). All side chains in the active site were optimized using a backbone-dependent rotamer library (Dunbrack and Karplus, 1993; Bower et al., 1997), while all other amino acids were held in their minimized conformation. High-resolution docking of transition state models and optimization of side chain interactions was achieved through Monte Carlo minimization of side chain rotamers and intense sampling of the ligand orientation.
Energies for the unbound and bound forms of the enzyme were calculated using the Rosetta energy function. The function is a linear combination of weighted scores including a Lennard-Jones attractive and repulsive potential, an orientation-dependent hydrogen-bonding potential, Coulomb electrostatics and an implicit solvation model. Binding energy is calculated as ΔΔGbinding = ΔGTS_bound− ΔGunbound (Kortemme and Baker, 2002; Morozov et al., 2005; Kaufmann et al., 2009). Weights for individual parameters of the energy function were established using multiple linear regression. The correlation to the experimental activation energy (ΔG‡TS = −RT ln(kcat/KM)) (Fersht, 1974) of each mutant with each substrate was optimized in a leave-one-out (LOO) cross-validation scheme. For each substrate/mutant/backbone combination, the scores of the top 10 models were averaged to minimize noise in the predicted binding free energy (Popov et al., 2007).
Cloning, production and purification of hPNP and site-directed mutants
Expression vector pCRT7/NT-TOPO-PNP, containing wild-type hPNP, was generously provided by Prof. Vern Schramm (Lewandowicz and Schramm, 2004) and NdeI/HindIII restriction sites were added to facilitate cloning into pET28a (forward primer: 5′-CATATGGAGAACGG-ATACACCTATGAAGATTATAAG-3′, reverse primer: 5′-AAGCTTCAAGTGGCTTTGTCAG-GGAGTG-3′). Site-directed mutations were generated using the Quikchange-II system (Stratagene, La Jolla, CA, USA) (forward primer: 5′-GCAGGTTCCACATGXXXGAAGGGTACCCACTCTGG-3′, reverse primer: 5′-CCAGAGTGGGTACCCTTCXXXCATGTGGAACCTGC-3′, where XXX was the variable codon). Mutations were verified via DNA sequencing.
Individual PNP mutants were over-expressed as N-terminal hexahistidine tagged constructs in Escherichia coli BL21(DE3). Strains harboring mutant PNP constructs were grown with shaking in 500 ml of LB broth with 50 µg/ml kanamycin at 37°C. At OD600 ≈ 0.6 cultures were induced with 1 mM isopropyl-β-d-1-thiogalactopyranoside and allowed to incubate for an additional 6–10 h. Cells were harvested by centrifugation and cell pellets were frozen at −80°C until immediately before purification. Cells were resuspended in Binding Buffer (50 mM Na2HPO4, 300 mM NaCl, 10 mM Imidazole, pH 8), disrupted by passage through a French Pressure cell and centrifuged to remove cellular debris. The soluble proteins were purified in a single step via Ni-affinity chromatography using a HisTrap FF column on an AKTA FPLC (GE Healthcare Life Sciences). Proteins were eluted using a linear gradient from 100% Binding Buffer to 100% Elution Buffer (50 mM Na2HPO4, 300 mM NaCl, 500 mM Imidazole, pH 8). The sample was desalted and stored in exchange buffer (100 mM Tris–HCl, 0.1 mM EDTA, 0.1 mM DTT, pH 7.5) at −80°C (Deng et al., 2004). All enzyme concentrations were determined via λ280 measurements and extinction coefficients were estimated using Accelrys DSGene 1.5.
Biochemical assays of PNP
PNP assays were performed in the phosphorolysis direction by continuously monitoring the formation of hypoxanthine (Degroot et al., 1985). A catalytic excess of xanthine oxidase was used in a tandem assay converting hypoxanthine to uric acid with concomitant reduction of iodonitrotetrazolium (INT) chloride to form a formazan chromophore (λmax = 546 nm). Assay Mix buffer contained 50 mM potassium phosphate saturated with O2, 50 mM HEPES, 0.075% Triton X-100, 1 mM INT and xanthine oxidase from buttermilk (Sigma). Substrates were dissolved in Assay Mix at concentration ranges of 20–200 µM or 100–1000 µM inosine and 250–2500 µM or 750–7500 µM ddI (Fluka and 3B Medical Systems, Inc.) depending on preliminary substrate concentration response curves. For each mutant, the concentration of enzymes diluted in Assay Mix were adjusted to produce rates within the dynamic range of the assay and <15% consumption of substrate was observed over time course measurements. Correspondingly, PNP concentration ranges of 0.006–2 µM for measuring inosine rates and 0.02–10 µM for measuring ddI rates were used with substrate ranges spanning the KM region, wherever possible. The assays were performed as follows: 100 µl of substrate dissolved in Assay Mix was transferred into wells of a flat-bottomed 96-well plate, equilibrated at 25°C, followed by addition of 100 µl of assay mix containing PNP. The rate of hypoxanthine formation was observed by monitoring tandem formazan formation by its unique absorbance at 546 nm over 5 min. A hypoxanthine standard curve was performed in parallel with each concentration series to convert absorbance numbers into molar turnover values.
hPNP library generation
Mutant libraries were generated via epPCR using MutazymeII (Stratagene, Inc.) with forward primer, 5′-GCAGCAGCCATCATCATCATC-3′ and reverse primer, 5′-GGATCTCAGTGGTGGTGGTGG-3′ flanking the PNP coding region of pET28a-hPNP template constructs. To ensure efficient restriction digestion prior to ligation into pET28a, primers were designed to generate PCR product with overhangs 45 bp upstream and downstream of NdeI and HindIII restriction sites. pET28a-hPNP plasmid preparations were used as the template for directed evolution and subsequent rounds of directed evolution used plasmid preparations from the previous round as template. The rate of mutation was adjusted by (i) varying template concentration in PCR reactions and (ii) the number of rounds of PCR. The mutation rate was titrated so that ca. 30% of PNP mutant subcloned into pET28a demonstrated <5% activity (assay described below). Correspondingly, template concentrations were varied from 0.03 to 20 ng/µl with 20–30 cycles. The desired mutation rate was obtained with 20 ng/µl and 20 cycles. DNA sequencing of 10 random mutants at the 30% dead rate indicated a mutation rate of ∼1.5–2 base pairs per kb.
PCR products were gel purified to remove template, digested with NdeI/HindIII and gel purified again prior to ligation into correspondingly restricted pET28a. Transformation of ligation reactions into E.coli BL21(DE3) cells was performed using Promega T4 DNA ligase with 17 ng insert and 100 ng vector per 10 µl for 180 min. A random sampling of clones indicated all contained PNP insert.
Screening and selection of improved hPNP mutants
Individual transformants were picked into 300 µl round bottomed 96-well plates containing 75 µl LB medium with 50 µg/ml kanamycin and grown to confluence (24 h at 37°C with shaking at 220 rpm). Glycerol stocks of the library were generated by plate replication in LB medium prior to cells being collected by centrifugation at 3000 rpm. Supernatant was removed by inversion and the resulting pellets were frozen and stored at −80°C until ready for assay. Directly prior to assay, frozen cell pellets were thawed and resuspended in 200 µl of a lysis mixture containing 50 mM phosphate buffer, 0.5 mg/ml egg white lysozyme (Sigma), 20 µg/ml deoxyribonuclease I (Sigma) and 0.1–0.125 mg/ml xanthine oxidase (to consume endogenous hypoxanthine) followed by a single freeze/thaw cycle from −80 to 37°C (Hsu et al., 2005). Following centrifugation, 25 µl of cell-free extract from each well was transferred into 384-well flat-bottom plates and assayed by the addition of 50 µl of buffer containing 50 mM phosphate, pH 7, 50 mM HEPES, pH 7, 125 µM ddI, 0.0375% Triton X-100, 2 mM INT and 0.4–0.6 mg/ml xanthine oxidase. The reactions were followed continuously at 546 nm for 60 s. Hits from the primary screen were replicated from glycerol stocks (1–5 per plate) and re-assayed with wild-type PNP containing cells as a benchmark to eliminate false positives.
Plasmid preps from the best candidates were retransformed by electroporation and 1 ml of an overnight culture from the fresh transformants was used to inoculate 50 ml of LB broth and grown to OD600 ≈ 0.6–0.8 before the addition of 1 mM isopropyl-β-d-1-thiogalactopyranoside. After 3 h of incubation at 37°C, 2 ml aliquots were pelleted by centrifugation at 13 000 rpm and frozen at −80°C. Pellets were lysed using BugBuster protein extraction reagent (Novagen Inc.) and the resulting cell-free extract was diluted in Assay Mix to titrate the specific activity within the dynamic range of the kinetic assay as described above. Upon addition of substrate (125 µM ddI or 20 µM inosine), hypoxanthine release was measured continuously over 60 s. Turnover rates were calculated and normalized to the cell density (OD600) at the time of harvest to yield a per-cell turnover rate for each PNP mutant. The kinetic parameters for selected mutant PNPs with high per-cell turnover rates (2–3 per round) were established as described above. Of the mutants characterized in each round, those with the most favorable kinetic parameters for ddI were selected as the source of template DNA for the next round of epPCR and screening.
Results
Design rationale
Based on the analysis of ribose binding interactions in bacterial (Bennett et al., 2003) and mammalian (Fedorov et al., 2001; de Azevedo et al., 2003; Canduri et al., 2004) PNP structures (Pugmire and Ealick, 2002), human PNP (hPNP) was selected as a starting point for the development of dideoxynucleoside phosphorylase catalytic activity. Throughout the reaction coordinate of hPNP, the purine ring is positioned via hydrogen-bonding interactions with Asn-243 and Glu-201. The nucleophilic phosphate is positioned by a complex network of hydrogen-bonding interactions (Fig. 1) and activated for addition to C1′ by a His-86/Glu-89 diad. Notably, there appear to be relatively fewer significant ribose binding interactions, namely His-257 contacting O5′ and Y88, which forms a hydrogen bond with O3′. The O3′-hydroxyl is also implicated in coordinating to the phosphate ligand and has been suggested to play a role in optimizing the geometry of the transition state (Erion et al., 1997). This analysis proposes Y88 as a primary contributor to substrate selectivity in the ribose binding region as side chain contacts between the ligand and substituted sugar atoms are limited to this residue.
To demonstrate the applicability of the method for the identification of specificity encoding sites, the RosettaDesign algorithm was used to identify amino acids in the binding site that maintain tight binding upon mutation but display a differential mutation profile in the presence of inosine and ddI. Using this approach, 23 amino acids were identified possessing C-α or C-β atoms within eight angstroms of the transition state analog Immucilin-H (PDB code: 1pf7) and pointing toward the binding site (Fig. 2A). Each of these amino acids was allowed to mutate to any other amino acid in the presence of ddI or inosine transition state analogs. The frequency of each amino acid in each position was evaluated using the publicly available WebLogo server (Crooks et al., 2004). Residues populated by Rosetta with the wild-type identity are not expected to affect a change in substrate binding characteristics. Similarly, residues designed by Rosetta to interact with neighboring amino acids, or residues turned toward solvent would not be expected to alter the substrate activity profile. In this analysis, 17 residues were reverted to their wild-type identity after two rounds of iterative refinement (Supplementary data, Fig. S1B–D). Five of the six remaining residues (F159, Y88, A116, F200, M219 and V245) interacted with the sugar moiety of the transition state models. The obtained mutation profiles were similar suggesting that minimal differentiation between the two ligands may be achieved. Suggested mutations for Y88 indicated that tyrosine is ideal for this location; secondary predictions, however, differ significantly for inosine and ddI (H and N, respectively) indicating the potential for modulating the substrate selectivity and specificity by mutating this position.
Next, step-wise analysis of all proposed single mutants was performed. All six positions were mutated sequentially to all amino acids proposed by Rosetta. Affinity for the ddI transition state model as well as preference for the ddI transition state model above the inosine transition state model were subsequently computed (Fig. 2B). Y88 remained as the only position that fulfills criteria for both tight binding to transition state models and displays a differential mutation profile.
Binding energy calculations
An initial gradient-based energy minimization (Qian et al., 2007) of the experimental protein structure was carried out in the absence of substrate to obtain an unbiased starting conformation of the protein that occupies a minimum in the energy landscape. The lowest energy structure had a C-alpha rmsd of 0.64 Å from the experimental coordinates (PDB code: 1pf7). The average C-alpha rmsd of the 10 lowest energy minimized structures was ∼0.86 Å.
Transition state models for inosine and ddI were constructed based on experimental and crystallographic data (see Materials and Methods section). Docking of the transition state models allows for the calculation of transition state binding energies. For this purpose, the transition state models were placed into the minimized protein structures and Y88 was mutated in silico through replacement of the wild-type side chain. The position and orientation of the transition state model was optimized together with the conformations of residues near the binding site using the RosettaLigand energy function (Meiler and Baker, 2006; Davis and Baker, 2009).
Using this protocol, a first round of seven single mutants was initially characterized: Y88F/A/C/I/L/W/K. These mutants were selected for predicted high ddI activity (Y88F/L), a range in predicted activity and variety in the side chain character. In a second round of experiments, all Y88X mutants were generated, purified and characterized to comprehensively analyze RosettaLigand's ability to rank individual mutations (Table I). Mutants predicted to form tight complexes with the ddI transition state model include Y88F, Y88L and Y88M as well as the negatively charged amino acid mutations Y88D and Y88E (Fig. 3A).
Table I.
Kinetic characteristics for hPNP-Y88Xa
| Variant |
kcat (s−1) |
KM (µM) |
Kcat/KM (s−1 M−1) |
|||
|---|---|---|---|---|---|---|
| Inosine | ddI | Inosine | ddI | Inosine (×102) | ddI | |
| Wild type | 43.9 ± 0.6 | 0.9 ± 0.01 | 48 ± 2 | 1030 ± 20 | 9210 ± 140 | 875 ± 8 |
| Y88F | 28.3 ± 0.7 | 10.5 ± 0.1 | 73 ± 4 | 450 ± 10 | 3890 ± 110 | 23 140 ± 230 |
| Y88H | 8.6 ± 0.2 | 2.5 ± 0.1 | 74 ± 3 | 675 ± 20 | 1160 ± 23 | 3670 ± 60 |
| Y88W | 5.6 ± 0.1 | 0.09 ± 0.01 | 500 ± 25 | 980 ± 40 | 113 ± 3 | 91 ± 2 |
| Y88A | 17.6 ± 0.4 | 2.8 ± 0.05 | 80 ± 3 | 1670 ± 50 | 2190 ± 51 | 1670 ± 22 |
| Y88V | 2.4 ± 0.1 | 0.6 ± 0.01 | 1200 ± 60 | 4410 ± 160 | 19.6 ± 0.7 | 144 ± 3 |
| Y88L | 15.9 ± 0.3 | 6.1 ± 0.05 | 94 ± 4 | 391 ± 8 | 1690 ± 34 | 15 670 ± 130 |
| Y88I | 1.3 ± 0.1 | 0.5 ± 0.02 | 1420 ± 55 | 4670 ± 270 | 9.4 ± 0.3 | 115 ± 4 |
| Y88M | 12.8 ± 0.1 | 5.2 ± 0.1 | 345 ± 7 | 645 ± 20 | 371 ± 3 | 8090 ± 90 |
| Y88C | 5.5 ± 0.2 | 2.5 ± 0.1 | 170 ± 10 | 810 ± 30 | 321 ± 13 | 3070 ± 60 |
| Y88S | 8.8 ± 0.1 | 2.7 ± 0.1 | 430 ± 10 | 4060 ± 110 | 203 ± 3 | 652 ± 10 |
| Y88T | 2.3 ± 0.1 | 0.4 ± 0.01 | 1200 ± 70 | 8160 ± 280 | 19 ± 1 | 44 ± 1 |
| Y88N | 9.7 ± 0.2 | 1.5 ± 0.1 | 845 ± 25 | 9530 ± 540 | 114 ± 2 | 153 ± 6 |
| Y88Q | 1.4 ± 0.1 | 0.15 ± 0.01 | 855 ± 50 | 860 ± 25 | 17 ± 0.6 | 177 ± 2 |
| Y88D | 0.8 ± 0.1 | 0.02 ± 0.001 | 2600 ± 120 | 12 700 ± 500 | 2.9 ± 0.1 | 1.8 ± 0.5 |
| Y88E | 0.7 ± 0.1 | 0.01 ± 0.001 | 4860 ± 410 | 14 900 ± 1200 | 1.4 ± 0.1 | 0.9 ± 0.1 |
| Y88K | ND | ND | ND | ND | ND | ND |
| Y88R | 0.01 ± 0.001 | ND | 370 ± 20 | ND | 0.0184 ± 0.0005 | ND |
aAssay conditions: 50 mM phosphate buffer, pH 7, 50 mM HEPES, pH 7, 0.075% Triton X-100, 1 mM INT, xanthine oxidase. Inosine: 20–200 or 100–1000 µM. ddI: 250–2500 or 750–7500 µM. Enzymes were assayed at concentrations which ensure steady state conditions.
Fig. 3.
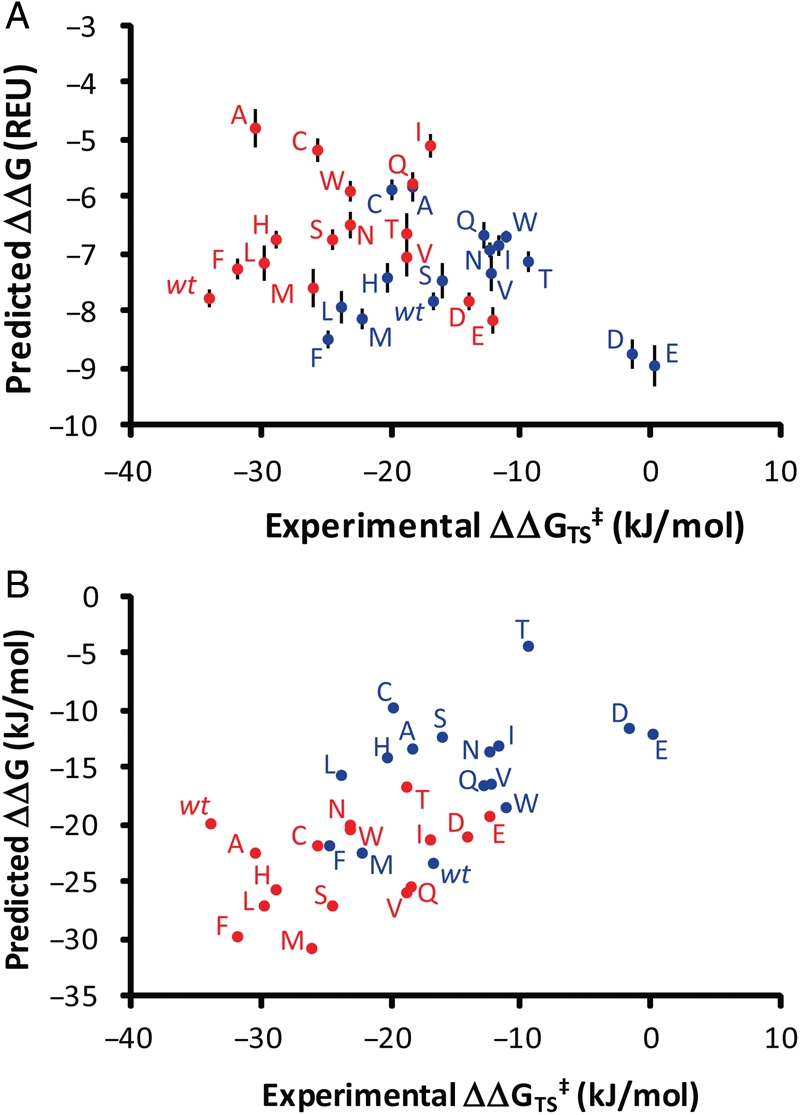
Plots of experimental binding energies versus binding energies using (A) the Protein:Ligand weight set and (B) a customized weight set.  , inosine;
, inosine;  , ddI. REU are Rosetta Energy Units. Error bars indicate standard deviation of top 10 transition state bound models. Numerical values of predicted binding energies are available in Supplementary data, Table S1.
, ddI. REU are Rosetta Energy Units. Error bars indicate standard deviation of top 10 transition state bound models. Numerical values of predicted binding energies are available in Supplementary data, Table S1.
In wild-type hPNP, the phosphorolysis of inosine is three orders of magnitude more efficient than that of ddI. The values of KM and kcat reported here are similar to those previously reported for inosine (Stoeckler et al., 1980; Erion et al., 1997). Each single mutant had decreased kcat and increased KM values for inosine. Positively charged Y88K and Y88R mutants were not sufficiently active for kinetic characterization at enzyme concentrations more than 500 times those used for the wild-type enzyme; accordingly, they were not included in the in silico analyses. In line with the in silico prediction, the Y88F mutant displayed the second highest catalytic activity.
Six mutants demonstrated a higher catalytic efficiency for ddI than the wild-type enzyme. A similar profile for toleration of mutations was seen with ddI as with inosine; however, it appears that almost any mutation allows for some improvement in the catalytic efficiency ratio relative to wild-type hPNP. As predicted, in silico Y88F, Y88L and Y88M displayed the highest catalytic activities. Of these, hPNP-Y88F displayed the highest overall kcat/KM and the highest turnover rate. Y88D/E mutants did not show the predicted high activity. We expect that the addition of a negative charge to the binding site disrupts the sensitive arrangements of partial charges in the catalytic site, an effect that is not considered by RosettaLigand as it solely optimizes binding affinity and has no means to directly assess catalytic competence of an active site from a mechanistic perspective.
With a comprehensive data set in hand, we set out to test which fraction of the differences in experimental and predicted transition state binding free energies can be attributed to inaccurate weighting of the energy terms in RosettaLigand and which part must be attributed to inaccuracies in the RosettaLigand energy function and structural models used. Weights were developed for individual components of the Rosetta energy function using multi-linear regression to optimize the correlation of predicted binding energies to experimental binding energies. Transition state binding energies were calculated from experimental data as ΔΔG‡TS = −RT ln(kcat/KM)(Fersht, 1974) while predicted binding energies were calculated as ΔΔG = Σwi · [si(bound) − si(unbound)], where wi is the weight applied to a particular score si (Kortemme and Baker, 2002; Morozov et al., 2005; Kaufmann et al., 2009). Using a LOO cross-validation analysis, weights are generated without the use of one data point and then binding energy predictions made for the data point left out.
The greatest correlation between experimental and predicted ΔΔG's was found with a weight set involving a combination of the Lennard-Jones attractive and repulsive terms, a solvation term and a Generalized Borne solvation term which aids in differentiation of electrostatic properties (Table II). The correlation between independently predicted binding energies and those found experimentally has an R-value of 0.65 and the average unsigned error of the predicted binding energies is 5.3 kJ/mol (Fig. 3B).
Table II.
Comparison of established weights
| Score | PNP:nucleoside | Protein:ligand | Protein:protein | Standard |
|---|---|---|---|---|
| Attractive | 1 ± 0.05 | 1 | 1 | 1 |
| Repulsive | 0.33 ± 0.02 | 0.75 | 0.16 | 0.91 |
| Solvation | 0.56 ± 0.04 | 0.63 | 0.73 | 0.65 |
| GBSol | 0.37 ± 0.04 | – | – | – |
| Hydrogen bonding | – | 1.5 | 1.11 | 1.74 |
| Pair Elec. | – | 0.63 | – | 0.34 |
| Rotamer probability | – | 0.4 | 0.64 | 0.4 |
| Phi-psi probability | – | 0.4 | – | 0.51 |
Directed evolution
To further optimize the hPNP-Y88F mutant for turnover of ddI, we developed and implemented a 96-well plate assay method for screening epPCR-generated mutant libraries. Diverse libraries were generated using an error-prone polymerase (Mutazyme, Stratagene, Inc.) to amplify hPNP with Y88F hPNP cloned into pET28a serving as the template DNA. Primers were designed to be >45 bp upstream and downstream of restriction sites (NdeI and HindIII) in order to ensure efficient restriction of PCR products prior to ligation reactions. The rate of mutation was adjusted by varying the amount of template and number of PCR cycles such that the dead rate (colonies with <5% activity) was approximately 30–40%. Library sizes of 1000–2000 colonies had an average mutation rate of 1.5–2 mutations per kb, as established by sequencing 10 randomly selected clones.
Subsequent to transformation into electrocompetent E.coli BL21(DE3), single colonies were transferred into 96-well round bottom plates. The plates were incubated with shaking for 24 h and grown to confluence. Cells were harvested by centrifugation and lysed by addition of lysozyme and a single freeze/thaw cycle (Hsu et al., 2005). Upon removal of insoluble debris by centrifugation, cell-free extracts were transferred to 384-well flat-bottom plates to assay phosphorylase activity via a modified hypoxanthine formation assay. The assay is based on a continuous colorimetric assay for the conversion of hypoxanthine to uric acid via xanthine oxidase which is coupled to the reduction of INT chloride to a purple formazan dye (λmax = 546 nm) (Degroot et al., 1985). Reactions were initiated by the addition of 125 µM ddI (∼1/4 of KM) and the amount of enzyme was titrated to assure linearity of the reaction during the measurement window (≥60 s). To test the reproducibility of the screening methodology, a single sequence library containing hPNP-Y88F was assayed. Under final assay conditions, the single sequence library had a coefficient of variance of 11.6% within each plate. Top hits from each plate were collected and re-screened from glycerol stocks of the error-prone library to eliminate false positives.
Over three rounds of directed evolution the turnover rate of enzyme in E.coli extracts steadily increased, albeit modestly (Fig. 4). The first round mutant, hPNP-19E2, has an increased normalized turnover rate for ddI with a concomitant decrease in turnover of inosine. Subsequent rounds provided a further increase in turnover of ddI but also an increase in inosine turnover. Catalytic constants, however, do not consistently improve for ddI throughout the directed evolution process (Table III). The first round mutant hit (hPNP-19E2) has a kcat and KM of 5.8 s−1 and 230 µM which is a decrease in turnover rate but a significant improvement in KM over the template. Kinetic parameters of hits from ensuing rounds of selection (hPNP-30F2 and hPNP-46D6) do not improve despite a regular increase in turnover in cell-free extracts.
Fig. 4.

Turnover of ddI increases throughout the directed evolution process. Turnover rates are normalized to cell density. Inosine, grey; ddI, white.
Table III.
Table of characteristics for directed evolution
| Round | Variant | Acquired mutation | Codon change |
kcat (s−1) |
KM (µM) |
kcat/KM (M−1 s−1) |
|||
|---|---|---|---|---|---|---|---|---|---|
| Inosine | ddI | Inosine | ddI | Inosine (×102) | ddI | ||||
| Wild type | – | – | 43.9 ± 0.6 | 0.90 ± 0.01 | 48 ± 2 | 1030 ± 20 | 9210 ± 140 | 875 ± 8 | |
| Rational | Y88F | Y88F | TAT→TTT | 28.3 ± 0.8 | 10.5 ± 0.1 | 73 ± 4 | 450 ± 10 | 3890 ± 110 | 23 140 ± 230 |
| DE-1 | 19E2 | M170T | ATG→ACG | 9.6 ± 0.2 | 5.8 ± 0.1 | 26 ± 1 | 230 ± 15 | 3710 ± 70 | 25 160 ± 620 |
| DE-2 | 30F2 | G4E | GGA→GAA | 14.5 ± 0.3 | 6.1 ± 0.1 | 37 ± 2 | 247 ± 10 | 3970 ± 90 | 24 650 ± 330 |
| Q172L | CAG→CTG | ||||||||
| C206C | TGT→TGC | ||||||||
| DE-3 | 46D6 | T177A | ACC→GCC | 14.7 ± 0.7 | 4.5 ± 0.1 | 33 ± 4 | 235 ± 11 | 4390 ± 280 | 19 240 ± 320 |
In addition to improving the turnover and kinetic characteristics of the mutant enzymes, we observed a marked improvement in substrate selectivity for ddI. Each mutant has an improved specificity ratio (wild-type efficiency/mutant efficiency) though none of the variants are selective for ddI over inosine. Wild-type hPNP possesses a specificity ratio for inosine of >1000:1, whereas selected hPNP mutants attained a specificity ratio of 15:1 subsequent to the directed evolution process. The final clone, hPNP-46D6, contained five amino acid mutations (G4E, Y88F, M170T, Q172L, T177A) with the gene containing a sixth silent mutation (Table III).
Discussion
We describe a two-step process of structure-based rational design coupled with directed evolution to engineer the function of hPNP for an unnatural nucleoside substrate. Based on the analysis of previously reported structures and activity profiles in addition to computational design studies, we selected a single active site residue, Y88, as a likely hotspot for improvements in sugar analog binding and created a comprehensive library of substitutions at this position. RosettaLigand was used to estimate the binding energies of inosine and ddI transition state models to in silico models of these mutations.
The present experiment evaluates Rosetta in a scenario likely to occur in enzyme design—assessing the change of enzyme specificity for a different but related ligand. Simultaneously, this study evaluates a scenario in which options for computational design were limited to a single active site residue, Y88. The accuracy of the Rosetta algorithm needs to be sufficiently high to discriminate improved substrate/transition state binding properties for a single active site ensemble (hPNP). By comparing Rosetta calculated binding energies to energies derived from experimentally measured kinetic parameters for every possible mutation, we were able to assess the ability of Rosetta to rank substitutions at Y88. RosettaLigand ranked the Y88F mutant in the top three for transition state stabilization. However, RosettaLigand generally underestimated energies of mutants with improved catalytic function and overestimated energies of mutants with decreased function; this trend was particularly evident in the case of wild-type hPNP which was predicted to process ddI with greater efficiency than inosine. Phenomena outside the scope of the method may contribute to errors in the prediction of protein variant binding energies. For example, wt-PNP crystallized with ddI has an ordered water molecule in the binding pocket which forms a hydrogen bond with Y88 and is situated proximal to the position of the 3′-hydroxyl in inosine-bound structures. The presence of this water molecule was not evaluated in the Rosetta calculations and might be one source of error. We speculate that the removal of the hydrogen bond acceptor by mutation to phenylalanine may be a significant source of improvement. Additionally, mutation to β-branch amino acids Y88V/I/T reduced the function of the enzyme relative to structurally similar counterparts, such as Y88L/S. Rosetta models predicted these mutants to have a destabilizing effect in the enzyme, evident by an increase in the total energy score caused by clashing between the C-β-methyl substituent and Pro-198, which resides on an adjacent loop involved in purine binding. This destabilization is not revealed by binding energy calculations as the residue–residue clash is present in both bound and unbound structures. This has been previously noted in the design of protein:protein interfaces (Sammond et al., 2007) and is confirmed experimentally in this work. The empirical and computational data sets resulting from this experiment permitted the optimization of Rosetta parameters resulting in appreciable correlation with R = 0.65 (Fig. 3B). Reweighting of the scoring function corrects the gross mis-identification of charged amino acids as catalytically advantageous and aligns inosine and ddI with respect to one another. This reweighting, while advantageous in terms of correlating global substrate preference, decreases the quality of ranking mutants with a particular substrate. Conversely, the unchanged RosettaLigand score function properly identifies the wt and Y88F varieties as most catalytically active for Inosine and ddI (Fig. 3A).
Predicted binding energies were correlated to experimental data by the transition state binding energy (ΔG‡TS = −RT ln(kcat/KM)). By performing the design and docking studies with the transition state models, mutants with greater predicted binding energy should possess a higher turnover rate. However, RosettaLigand does not directly evaluate the mechanistic aspects of turnover, a situation that is also evidenced when predicting binding for mutants with charged residues. Therefore, one might argue that predicted transition state binding energies correlate more accurately with KM, where ΔG‡TS = RT ln(KM). However, at least in the present studies, this analysis yields somewhat reduced correlation coefficients for inosine and ddI, where R = 0.236 and 0.284, respectively, using binding energies established with the RosettaLigand weight set and excluding charged residues. These values are 0.362 and 0.438 when correlating predicted binding energies to kcat/KM. All terms in the RosettaLigand energy function were applied in the generation of enzyme:substrate ensembles but the attractive and repulsive, solvation and generalized Borne solvation terms were most critical for the final evaluation of binding energy. There are several notable differences in the weights developed here with those reported for use in protein:protein binding, the RosettaLigand weights used during docking and the standard weight set. Relative to the Lennard-Jones attractive weight, the weight for the Lennard-Jones repulsive score is twice as large as that used in protein docking but much lower than the original ligand docking and standard weights. The weight for solvation is also slightly decreased compared with typical Rosetta weights. Of note is the absence of a hydrogen-bonding term from the evaluation; while this term was used during the docking portion of the experiment, its inclusion in the final weight set does not improve the correlation to experiment. It is not surprising that the weights of electrostatic terms were increased considering the number and role of charged and polar residues in binding and catalysis. The best point mutant, hPNP-Y88F, was subjected to directed evolution by epPCR and in vitro selection for improved phosphorolysis in cell-free extracts. Three rounds of directed evolution resulted in a mutant with six mutations (Fig. 5) and a ddI turnover rate 36 times that of wild-type. The acquired M170T mutation in hPNP-19E2 provides significant improvement in binding affinity with a subsequent decrease in turnover rate despite its location ∼25 Å from the reaction center. Successive rounds of directed evolution identified Q172L and T177A mutations which occur on the same helix as M170T and may counteract any destabilizing effects of the first mutation. Based on its location, the G4E mutant may re-orient and stabilize the recombinant hexahistidine tag which is not removed prior to screening or characterization. Acquired mutations may improve expression efficiency or RNA stability as evidenced by the considerable increase in turnover of enzyme in E.coli extracts despite nominal improvements in catalytic efficiency.
Fig. 5.

Mutations in the final variant, hPNP-46D6, are mapped onto the wild-type structure (PDB code: 1rct). Mutations are colored by lineage: cyan, rational; blue, DE-1; yellow, DE-2; red, DE-3.
Directed evolution strategies that target residues in the first and second shell of the binding site (i.e. CASTing (Fazelinia et al., 2007)) have found success in modifying or broadening the substrate specificity of enzymes. It is possible that additional mutations in the vicinity of Y88F may further improve the turnover of ddI as mutations closer to the active site are frequently observed to have greater effect on substrate selectivity (Morley and Kazlauskas, 2005). It is not surprising, however, that mutations in the first or second shell of the binding site were not identified in this work considering the moderate library sizes employed.
Conclusions
The process outlined herein was applied to improve the catalytic properties of hPNP for an alternate substrate, dideoxyinosine. The Rosetta method proved capable of identifying mutations which are likely to improve catalysis in a test case made particularly difficult by constraint to a single amino acid substitution and high substrate similarity. In this environment, charged residues had a major impact on computational and experimental results. Addition of an electrostatic term allowed for moderate improvement in the results. The makeup of the scoring function highlights the characteristics of this particular test and points to the broad scope of applications available for the Rosetta algorithm. Many other nucleoside analogs contain functionality on the sugar moiety providing a handle for successful design.
Directed evolution using whole-gene epPCR and an in vitro selection scheme allowed for the identification of residues distant to the binding site which improve the binding affinity of the phosphorylase for ddI. Individual rounds resulted in modest improvements in kinetic parameters and/or turnover. The robustness of the in vitro tandem enzymatic assay is demonstrated by its ability to discriminate incremental improvements in activities. The final mutant (hPNP-46D6) has a catalytic efficiency 22 times greater than the wild type and the specificity ratio was shifted to 15:1 from a starting point of 1000:1. On a per-cell basis turnover was 36-fold improved, the additional improvement likely originating from increased protein production. As these improvements were products of small libraries, we feel it is likely that further rounds of directed evolution can improve the catalytic properties of the enzyme and identify residues in other regions of the protein that may enhance catalysis of ddI or further shift the selectivity.
In principle, catalysts for the phosphorolysis and formation of a variety of nucleoside analogs can be accessed using the methods described herein. Indeed, we have tested hPNP-46D6 for the biocatalytic generation of ddI as a component of tandem reactions and found it to be kinetically competent in the synthesis direction (to be presented in a forthcoming publication). Several nucleoside analogs with ribose substitution at 2′ and 3′ positions are of significant clinical importance as are nucleoside analogs with nucleobase substitutions (Li et al., 2008; Clerq, 2009; Soriano et al., 2009). Of note, relatively few biotransformation-based approaches have been described for biocatalysis of dideoxynucleosides and other analogs (Shirae et al., 1989; Rogert et al., 2002; Komatsu et al., 2003; Kaminski et al., 2008; Medici et al., 2008). Ongoing experiments in our laboratories endeavor to generate optimized enzymes and pathways capable of generating ribose precursors for PNP variants. Engineered pathways for nucleoside analogs have potential to provide economical alternatives to chemical synthesis of these valuable pharmaceuticals.
Supplementary data
Funding
B.O.B. acknowledges support from the Vanderbilt Institute of Chemical Biology and NIH R01 GM077189. D.P.N. was also supported by National Institutes of Health 5 T90 DA022873. J.M. recieved support from the Defense Advanced Research Projects Agency Protein design processes and K.W.K. was supported by National Institutes of Health 1F31DA024528.
Supplementary Material
Acknowledgements
We thank the Vanderbilt University Advanced Computing Center for Research and Education for use of the cluster for computations and the Defense Advanced Research Projects Agency Protein Design Project for partial funding.
Footnotes
Edited by Alan Fersht
References
- Bennett E.M., Li C.L., Allan P.W., Parker W.B., Ealick S.E. J. Biol. Chem. 2003;278:47110–47118. doi: 10.1074/jbc.M304622200. [DOI] [PubMed] [Google Scholar]
- Bernhardt P., Mccoy E., O'Connor S.E. Chem. Biol. 2007;14:888–897. doi: 10.1016/j.chembiol.2007.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bower M.J., Cohen F.E., Dunbrack R.L. J. Mol. Biol. 1997;267:1268–1282. doi: 10.1006/jmbi.1997.0926. [DOI] [PubMed] [Google Scholar]
- Canduri F., dos Santos D.M., Silva R.G., Mendes M.A., Basso L.A., Palma M.S., de Azevedo W.F., Santos D.S. Biochem. Biophys. Res. Commun. 2004;313:907–914. doi: 10.1016/j.bbrc.2003.11.179. [DOI] [PubMed] [Google Scholar]
- Clerq E.D. Int. J. Antimicrob. Agents. 2009;33:307–320. doi: 10.1016/j.ijantimicag.2008.10.010. [DOI] [PubMed] [Google Scholar]
- Crooks G.E., Hon G., Chandonia J.M., Brenner S.E. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damborsky J., Brezovsky J. Curr. Opin. Chem. Biol. 2009;13:26–34. doi: 10.1016/j.cbpa.2009.02.021. [DOI] [PubMed] [Google Scholar]
- Davis I.W., Baker D. J. Mol. Biol. 2009;385:381–392. doi: 10.1016/j.jmb.2008.11.010. [DOI] [PubMed] [Google Scholar]
- de Azevedo W.F., Canduri F., dos Santos D.M., Pereira J.H., Dias M.V.B., Silva R.G., Mendes M.A., Basso L.A., Palma M.S., Santosce D.S. Biochem. Biophys. Res. Commun. 2003;309:917–922. doi: 10.1016/j.bbrc.2003.08.094. [DOI] [PubMed] [Google Scholar]
- Degroot H., Degroot H., Noll T. Biochem. J. 1985;230:255–260. doi: 10.1042/bj2300255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng H., Lewandowicz A., Schramm V.L., Callender R. J. Am. Chem. Soc. 2004;126:9516–9517. doi: 10.1021/ja049296p. [DOI] [PubMed] [Google Scholar]
- Dunbrack R.L., Karplus M. J. Mol. Biol. 1993;230:543–574. doi: 10.1006/jmbi.1993.1170. [DOI] [PubMed] [Google Scholar]
- Erion M.D., Takabayashi K., Smith H.B., Kessi J., Wagner S., Honger S., Shames S.L., Ealick S.E. Biochemistry. 1997;36:11725–11734. doi: 10.1021/bi961969w. [DOI] [PubMed] [Google Scholar]
- Fazelinia H., Cirino P.C., Maranas C.D. Biophys. J. 2007;92:2120–2130. doi: 10.1529/biophysj.106.096016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedorov A., et al. Biochemistry. 2001;40:853–860. doi: 10.1021/bi002499f. [DOI] [PubMed] [Google Scholar]
- Fersht A.R. Proc. R. Soc. London Ser. B. 1974;187:397–407. doi: 10.1098/rspb.1974.0084. [DOI] [PubMed] [Google Scholar]
- Hsu C.C., Hong Z., Wada M., Franke D., Wong C.H. Proc. Natl Acad. Sci. USA. 2005;102:9122–9126. doi: 10.1073/pnas.0504033102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackel C., Kast P., Hilvert D. Annu. Rev. Biophys. 2008;37:153–173. doi: 10.1146/annurev.biophys.37.032807.125832. [DOI] [PubMed] [Google Scholar]
- Jiang L., et al. Science. 2008;319:1387–1391. doi: 10.1126/science.1152692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaminski P.A., Dacher P., Dugue L., Pochet S. J. Biol. Chem. 2008;283:20053–20059. doi: 10.1074/jbc.M802706200. [DOI] [PubMed] [Google Scholar]
- Kaufmann K.W., Dawson E.S., Henry L.K., Field J.R., Blakely R.D., Meiler J. Proteins. 2009;74:630–642. doi: 10.1002/prot.22178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu H., Awano H., Ishibashi H., Oikawa T., Ikeda I., Araki T. Nucleic Acids Res. Suppl. 2003;3:101–102. doi: 10.1093/nass/3.1.101. [DOI] [PubMed] [Google Scholar]
- Kortemme T., Baker D. Proc. Natl Acad. Sci. USA. 2002;99:14116–14121. doi: 10.1073/pnas.202485799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlman B., Dantas G., Ireton G.C., Varani G., Stoddard B.L., Baker D. Science. 2003;302:1364–1368. doi: 10.1126/science.1089427. [DOI] [PubMed] [Google Scholar]
- Lewandowicz A., Schramm V.L. Biochemistry. 2004;43:1458–1468. doi: 10.1021/bi0359123. [DOI] [PubMed] [Google Scholar]
- Li F., Maag H., Alfredson T. J. Pharm. Sci. 2008;97:1109–1134. doi: 10.1002/jps.21047. [DOI] [PubMed] [Google Scholar]
- Medici R., Lewkowicz E.S., Iribarren A.M. FEMS Microbiol. Lett. 2008;289:20–26. doi: 10.1111/j.1574-6968.2008.01349.x. [DOI] [PubMed] [Google Scholar]
- Meiler J., Baker D. Proteins. 2006;65:538–548. doi: 10.1002/prot.21086. [DOI] [PubMed] [Google Scholar]
- Morley K.L., Kazlauskas R.J. Trends Biotechnol. 2005;23:231–237. doi: 10.1016/j.tibtech.2005.03.005. [DOI] [PubMed] [Google Scholar]
- Morozov A.V., Havranek J.J., Baker D., Siggia E.D. Nucleic Acids Res. 2005;33:5781–5798. doi: 10.1093/nar/gki875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy P.M., Bolduc J.M., Gallaher J.L., Stoddard B.L., Baker D. Proc. Natl Acad. Sci. USA. 2009;106:9215–9220. doi: 10.1073/pnas.0811070106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinheiro E., Vasan A., Kim J.Y., Lee E., Guimier J.M., Perriens J. AIDS. 2006;20:1745–1752. doi: 10.1097/01.aids.0000242821.67001.65. [DOI] [PubMed] [Google Scholar]
- Popov V.M., Yee W.A., Anderson A.C. Proteins. 2007;66:375–387. doi: 10.1002/prot.21201. [DOI] [PubMed] [Google Scholar]
- Pugmire M.J., Ealick S.E. Biochem. J. 2002;361:1–25. doi: 10.1042/0264-6021:3610001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian B., Raman S., Das R., Bradley P., McCoy A.J., Read R.J., Baker D. Nature. 2007;450:259–264. doi: 10.1038/nature06249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reetz M.T., Bocola M., Carballeira J.D., Zha D., Vogel A. Angew. Chem. Int. Ed. Engl. 2005;44:4192–4196. doi: 10.1002/anie.200500767. [DOI] [PubMed] [Google Scholar]
- Ringia E.A.T., Tyler P.C., Evans G.B., Furneaux R.H., Murkin A.S., Schramm V.L. J. Am. Chem. Soc. 2006;128:7126–7127. doi: 10.1021/ja061403n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogert M.C., Trelles J.A., Porro S., Lewkowicz E.S., Iribarren A.M. Biocatal. Biotransform. 2002;20:347–351. [Google Scholar]
- Rothlisberger D., et al. Nature. 2008;453:190–194. doi: 10.1038/nature06879. [DOI] [PubMed] [Google Scholar]
- Sammond D.W., Eletr Z.M., Purbeck C., Kimple R.J., Siderovski D.P., Kuhlman B. J. Mol. Biol. 2007;371:1392–1404. doi: 10.1016/j.jmb.2007.05.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schramm V.L. Arch. Biochem. Biophys. 2005;433:13–26. doi: 10.1016/j.abb.2004.08.035. [DOI] [PubMed] [Google Scholar]
- Shirae H., Kobayashi K., Shiragami H., Irie Y., Yasuda N., Yokozeki K. Appl. Environ. Microbiol. 1989;55:419–424. doi: 10.1128/aem.55.2.419-424.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano V., Peters M.G., Zeuzem S. Clin. Infect. Dis. 2009;48:313–320. doi: 10.1086/595848. [DOI] [PubMed] [Google Scholar]
- Stoeckler J.D., Cambor C., Parks R.E., Jr Biochemistry. 1980;19:102–107. doi: 10.1021/bi00542a016. [DOI] [PubMed] [Google Scholar]
- Voigt C.A., Mayo S.L., Arnold F.H., Wang Z.G. Proc. Natl Acad. Sci. USA. 2001;98:3778–3783. doi: 10.1073/pnas.051614498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanghellini A., Jiang L., Wollacott A.M., Cheng G., Meiler J., Althoff E.A., Rothlisberger D., Baker D. Protein Sci. 2006;15:2785–2794. doi: 10.1110/ps.062353106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.