

Abstract
Irreversible inhibition of cytochrome P450 enzymes can cause significant drug-drug interactions (DDIs). Formation of metabolites is fundamental for the inactivation of P450 enzymes. Of the 19 inactivators with a known mechanism of inactivation, 10 have circulating metabolites that are known to be on path to inactive P450. The fact that inactivation usually requires multiple metabolic steps implies that predicting in vivo interactions may require complex models, and in vitro data generated from each metabolite. The data reviewed here suggest that circulating metabolites are much more important in in vivo P450 inhibition than is currently acknowledged.
Keywords: mechanism-based inactivation, cytochrome P450 enzymes, circulating metabolites, drug-drug interactions, irreversible inhibition, in vitro-in vivo predictions
1. Introduction
Inhibitory drug-drug interactions (DDIs) are a considerable concern in drug development and in patients undergoing polytherapy. Inhibitory drug-drug interactions can be life-threatening by increasing the exposure to narrow therapeutic index drugs. Of the current drugs on the US market, 129 are in vivo inhibitors of P450 enzymes, and of these inhibitors, 80% have circulating metabolites[1]. However, the quantitative role of inhibitory metabolites in in vivo DDIs is rarely known. Several examples of reversible P450 inhibitors, which have circulating metabolites that are predicted to contribute to in vivo interactions do, however, exist. For example, the CYP2C9 inhibitor sulfinpyrazone and CYP3A4 inhibitor itraconazole have circulating metabolites that also inhibit CYP2C9 and CYP3A4, respectively. Based on plasma concentrations and in vitro Ki values, CYP2C9 inhibition after sulfinpyrazone administration can be attributed to the circulating sulfide metabolite that has a 15-fold lower Ki in HLMs than the parent drug and circulates at comparable concentrations with the parent[2, 3]. Three circulating metabolites of itraconazole are predicted to contribute approximately 50% of the total CYP3A4 inhibition in vivo[4].
In addition to reversible interactions, many clinically important pharmacokinetic drug-drug interactions result from inhibition of metabolic clearance via mechanism-based inactivation (MBI) of cytochrome P450 (CYPs) enzymes. By definition, MBI involves metabolism of the inhibitor to a reactive metabolite, which modifies the P450 enzyme and results in irreversible loss of enzyme activity[5]. Enzymatic activity can only be restored by de novo protein synthesis. Even though metabolites play a fundamental role in the MBI of P450's, and MBI of P450 enzymes has been studied for multiple decades, in many instances the reactive metabolite responsible for inhibition of the P450 enzyme is unknown. General understanding of the biological fate of metabolites, especially reactive metabolites, has surfaced as an important part of the drug discovery process, and increasing attention has been paid to predicting and identifying circulating human metabolites[6-9]. Despite this increasing interest, known circulating metabolites are often not characterized for MBI of P450 enzymes and are usually only tested for pharmacological activity. As a result, the mechanism and extent of P450 inhibition by inhibitory metabolites is not well established. This review presents the current knowledge of the role of metabolites in MBI of P450 enzymes, focusing on the circulating metabolites that contribute to P450 inactivation and the complex metabolic pathways that lead to inactive enzyme. For detailed review on the chemical and biochemical mechanisms of MBI by the various compounds presented the reader is referred to Kalgutkar et al 2007[10]
2. Classification of mechanism-based inhibitors
From the literature, 31 in vitro mechanism-based inhibitors were indentified. A drug was considered an MBI if any reports existed of irreversible or time-dependent P450 inhibition by the given drug, regardless of other reports, which may have determined reversible inhibition parameters for the same drug. Generally, MBI of P450s can be classified into two groups: protein and/or heme alkylation and metabolic-intermediate (MI) complex formation.
The general pathways to formation of inactive P450 via metabolites are shown in Figure 1 and a typical pathway for MI complex formation is indicated by (a). An MI complex refers to a tight, coordinate bond between the metabolite and the reduced state of the P450 heme iron. Although the interaction between the final ligand and the heme iron is strong, no covalent bond is formed and the complex can be reversed under experimental conditions by the addition of potassium ferricyanide. The MI complex is catalytically inactive. MI complexes have distinct spectral properties and are measured by difference spectroscopy using the UV/Vis detectable Soret peak centered on λmax of 452-457 nm[11]. MI complexes are believed to be reasonably stable, and have been observed in microsomes prepared from animals and liver biopsy specimens from humans treated with troleandomycin[12, 13].
Figure 1.
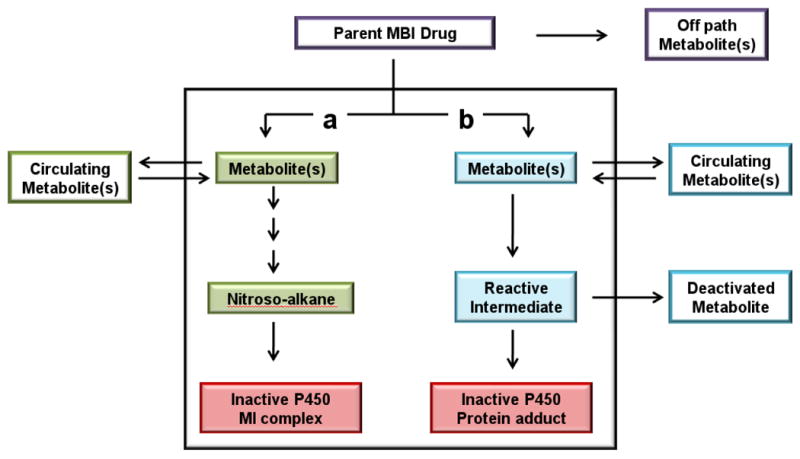
Pathways to irreversible inhibition of P450 enzymes. Pathways a and b indicate the two metabolic routes to inactivate P450 (red boxes) by MI complex formation (green boxes) or by protein alkylation (blue boxes). The off path metabolites (purple box) are not directly involved in the inactivation of the P450 enzymes. Circulating metabolites (pathway a and b) are able to rebind to the P450 enzymes and are further metabolized to inactivate the P450 enzymes. The deactivated metabolites (pathway b) are dead end products and often detected in vitro.
Protein and heme adducts to P450 enzymes are generally characterized by a covalent bond forming between a bioactivated, electrophilic drug and a nucleophilic amino acid in the P450's active site or the heme prosthetic group. As shown in Figure 1, pathway b, the reactive intermediate may bind to the P450 enzymes, forming an irreversible protein adduct, or the reactive intermediate may be released into the media and inactivated, in many instances by reacting with glutathione. The resulting glutathione and other conjugates can often be detected in plasma or urine. P450 protein adducts are usually measured by detection of radioactive drug binding to the P450 or by identification of adducted peptides via mass spectroscopy. In addition, detection of the deactivated metabolites is used as support for determining the mechanism of inactivation.
Nine of the total 31 inhibitors (29%) were classified as irreversible inhibitors by protein alkylation (Table 1). Protein alkylation was defined based on reports of a covalent bond forming to an amino acid of the P450 enzymes, trapping of an electrophilic, reactive intermediate with glutathione, or evidence of a covalent adduct to the P450 heme pyrrole nitrogen. Fourteen inhibitors (45%) were classified as MI complex forming inhibitors (Table 2) based on reports of time- and NADPH-dependent formation of a Soret peak between 452-456 nm. The remaining eight drugs (26%) were classified as MBIs with an unknown mechanism of inactivation (Table 3), based on concentration- and time-dependent inactivation assays, but lack of reports of a protein adduct, heme destruction, trapping of a reactive intermediate or detection of a spectral MI complex.
Table 1.
Inhibitors that inactivate CYP enzymes by protein adduct formation. The inactivating metabolites were classified by determining the proximal reactive species reported on the pathway to the irreversible inhibition. Π circulating metabolite, * indicated DDI in vivo (w= weak, m=moderate, p=potent as indicated in text), ‡ inhibition by metabolite only, N negative control study of inhibition reported (< 20% inhibition of an FDA approved probe drug).
| Parent | Inactivating metabolites | 1A2 | 2B6 | 2C8 | 2C9 | 2C19 | 2D6 | 3A4 | Off path circulating metabolites | Ref |
|---|---|---|---|---|---|---|---|---|---|---|
| clopidogrel | 2-oxo-clopidogrel (thiolactone)Π | X*w | SR26334 (clopidogrel acid) | [79,80] | ||||||
| dasatinib | para-quinone-imine intermediate | X,N | M4, M5, M6, M20, and M24 | [15,81] | ||||||
| diclofenac | benzoquinone imine intermediate | X | 3-OH,4methoxy-diclofenac, 4-OH-diclofenac | [46,82] | ||||||
| efavirenz | 8-hydroxyefavirenzΠ | X*m ‡ | [33.35] | |||||||
| gemfibrozil | gemfibrozil 1-O-β-glucuronideΠ | X*m | [41,42] | |||||||
| nefazodone | para-quinone-imine intermediate | X*p | mCPP,hydroxynefazodone,desethylhydroxy | [17,51] | ||||||
| raloxifene | diquinone methide intermediate | X | [83] | |||||||
| ticlopidine | S-oxide ticlopidineΠ (2C19), thiolactone (2B6) | X*w | X*p | metabolites 85-95% of plasma radioactivity | [79,84] | |||||
| zafirlukast | α,β-unsaturated iminium ion intermediate | X | hydroxylated metabolites | [19] |
Table 2.
Inhibitors that inactivate CYP enzymes by MI complex formation. Π circulating metabolite, * indicated DDI in vivo (w= weak, m=moderate, p=potent as indicated in the text), ‡ inhibition by metabolite only, # inhibition by both parent and metabolites, N negative control study of inhibition reported (< 20% inhibition of an FDA approved probe drug), º parent did not form MI complex but was an MBI of unknown mechanism, 1 metabolite may be on path to inactivation
| Parent | Inactivating metabolites | 1A2 | 2B6 | 2C8 | 2C9 | 2C19 | 2D6 | 3A4 | Off path circulating metabolites | Ref |
|---|---|---|---|---|---|---|---|---|---|---|
| Alkyl amines | ||||||||||
| amiodarone | N-desethylamiodaroneΠ | X‡ | X‡ | Xº | Xº | X*w ‡ | X*w | [70,75] | ||
| amitriptyline | nortriptyllineΠ | X# | X‡ | X# | X‡ | X‡ | [76,85] | |||
| clarithromycin | N-desmethylclarithromycin | X*p | 14-hydroxyclarithromycin | [86] | ||||||
| diltiazem | N-demethyldiltiazemΠ | X*m # | N-deacetyldiltiazem | [65,87] | ||||||
| erythromycin | N-desmethylerythromycin | X*m | [28,88] | |||||||
| fluoxetine | norfluoxetineΠ | X | X*m # | X*w,N | [26,66,76] | |||||
| tamoxifen | N-desmethyltamoxifenΠ | X | X# | 4OH-tamoxifen, N-didesmethyltamoxifen1 | [89,90] | |||||
| troleandomycin | N-desmethyltroleandomycin | X*p | [12,26] | |||||||
| verapamil | N-desalkylverapamil, norverapamilΠ | X | X*m | D-617, D-620 | [54,76] | |||||
| Protease inhibitors | ||||||||||
| amprenavir | unknown | X*m | [30] | |||||||
| indinavir | unknown | X*p | [30] | |||||||
| nelfinavir | unknown | X*p | Hydroxy-t-butylamide nelfinavir | [30] | ||||||
| ritonavir | unknown | X*p | M-2 (Hydroxy-isopropyl ritonavir) | [30] | ||||||
| Other | ||||||||||
| paroxetine | methylenedioxy carbene intermediate | X*p | [32] |
Table 3.
Inhibitors that inactivate CYP enzymes by unknown mechanism. * indicated DDI in vivo (w= weak, m=moderate, p=potent as indicated in the text), 1 P450 heme-destruction implied based on mechanism of action.
| Parent | Inactivating metabolites | 1A2 | 2B6 | 2C8 | 2C9 | 2C19 | 2D6 | 3A4 | circulating metabolites | Ref |
|---|---|---|---|---|---|---|---|---|---|---|
| cimetidine | unknown | X*w | sulfoxide, hydroxymethyl cimetidine | [91,92] | ||||||
| delavirdine | unknown | X*w | N-desisopropyl, N-desakyl delavirdine | [93] | ||||||
| lopinavir | unknown | X*p | [30] | |||||||
| phenelzine | unknown1 | X | X | X | X | [76,94] | ||||
| saquinavir | unknown | X*p | mono- and di-hydroxylated saquinavir | [30] | ||||||
| thiotepa | unknown | X | tepa | [95-97] | ||||||
| zileuton | unknown | X*m | N-dehydroxylated | [98] | ||||||
| zolpidem | unknown | X*w | [99] |
Overall, 54% of the inhibitors that displayed in vitro P450 inactivation kinetics also inhibited the same P450 enzyme in vivo, defined as at least a 1.25-fold increase in the AUC of the probe drug. Of these in vivo inhibitors, ten were potent (>5-fold increase in the substrate AUC), eight were moderate (2-5-fold AUC increase) and eight were weak (1.25-2-fold AUC increase) as classified based on the FDA recommendations[14]. The mechanism of P450 inactivation did not correlate with magnitude of clinical interactions. Six of the ten potent inhibitors, five of the seven moderate inhibitors, and three of the eight weak inhibitors were MI complex forming drugs. Interestingly, CYP3A4 appeared to be most susceptible to interactions via MBI. Eight (89%) out of the nine potent inhibitors displayed potent interactions with CYP3A4 probes in vivo. In fact, 22 of 31 (71%) MBIs were reported to inactivate CYP3A4, whereas CYP2C9 had only two reports.
All but five of the 31 irreversible inhibitors indentified have confirmed circulating metabolites (Tables 1-3). However, the role of the circulating metabolites in P450 inactivation is equivocal for many of these metabolites. Based on our current classification, inactivating metabolites were defined as such, if the metabolite was shown to inactivate the enzyme, and was on the metabolic route to the reactive intermediate (Figure 1, pathway a, b). Based on these criteria, nine (29%) of the inhibitors have circulating metabolites that have a confirmed role in the inactivation of the P450 enzymes. Of these circulating metabolites, six result in MI complex formation and three contribute to protein adduct formation. It is important to note that the reported inactivating metabolites (Tables 1 and 2) often undergo further metabolism before irreversible inhibition is sustained. As depicted in Figure 1, the parent drug has three possible routes of metabolism. The first is an off path metabolite, which results from metabolism removed from the inactivation site, however this metabolite may still undergo convergent secondary metabolism to P450 inactivation. The two other metabolic pathways (a, b) represent formation of metabolites that are precursors to the reactive species that leads to inactivation. These precursor metabolites can stay on path to the inactivation of the enzyme, or can be released into the media and circulate, and rebind to the P450 enzyme to cause inactivation
3. Role of metabolites in irreversible inhibition by protein adduct formation
The inhibitors that formed protein adducts (Table 1) were commonly oxidized to an electrophilic species that either binds to the P450 protein or is released from the active site and binds to other macromolecules. Hence, circulating metabolites associated with the reactive species were rarely observed. The reactive intermediates, usually quinone imine-derivatives, are commonly identified as glutathione adducts. For example, dasatinib, diclofenac, nefazodone, raloxifene, and zafirlukast have identified glutathione adducts but only one of these five drugs, nefazodone, displays in vivo inhibition of the inactivated enzyme[15-19]. Clopidogrel and ticlopidine, that both result in protein adduct formation, have similar pharmacological actions as well as similar inactivation mechanisms. The primary reactive metabolites identified for both clopidogrel and ticlopidine are further oxidized by CYP enzymes before forming a protein adduct, and in each case, there is controversy on the exact species responsible for the inactivation[20]. The remaining inhibitors in this group, efavirenz and gemfibrozil, have unique mechanism of inactivation and are discussed in detail as case studies. It is interesting to note that all of the protein adduct forming inhibitors that cause in vivo interactions, also have circulating metabolites that can contribute to the P450 inactivation.
4. Role of metabolites in irreversible inhibition by MI complex formation
Of the drugs characterized as MI complex forming inhibitors, 64% contained an alkyl amine moiety at the site of P450 metabolism. Alkyl amines are not only common moieties known to cause P450 inactivation, but are also important entities to the pharmacological activity of the drugs. The formation of MI complexes from alkyl amine containing drugs is believed to occur via a multistep process, which results in the eventual inactivation of the P450 (Figure 2). As shown in Figure 2, there are multiple oxidative steps required to form an MI complex with the P450 enzyme. In addition, the intermediate metabolites, such as the primary amines often have high affinity for binding to the P450 enzymes. It is commonly believed that a primary amine is required for MI complex formation via hydroxylation of the amine nitrogen to form the primary hydroxylamine and further oxidation to a nitroso group that coordinates to the ferrous state of the heme iron[21]. It is important to note that the metabolite responsible for coordination to the heme iron has not been fully characterized and several different chemical pathways to inactive enzyme have been proposed[22, 23]. For example, Lindeke et al. examined MI complex formation from the secondary amine, N-methylamphetamine, and proposed that the secondary amine is N-oxygenated to the secondary hydroxylamine, and then proceeds through further oxidative steps to coordination of the heme iron (Figure 2)[24]. Hence, the pathway to the formation of an MI complex from alkyl amine drugs is a complex process involving a variable number of metabolic steps.
Figure 2.
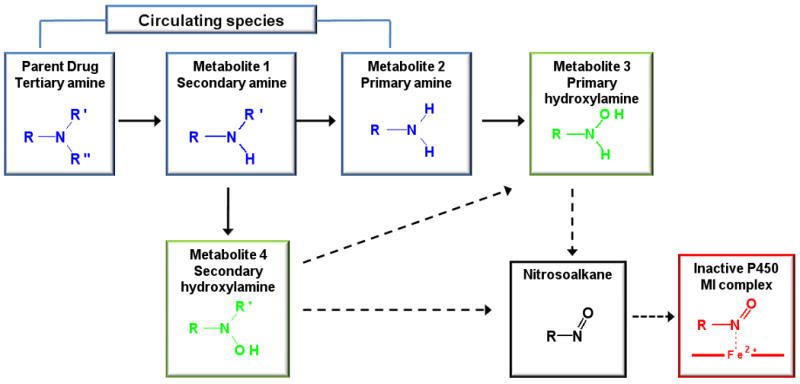
Proposed route of alkyl amine metabolism to MI complex. Solid arrows indicate P450 metabolic reactions indentified in HLMs, dashed arrows are proposed metabolic reactions on route to MI complex formation but have not been verified. Blue compounds are known circulating metabolites, green compounds have been shown to form MI complexes but are not known circulating metabolites, the black colored compound is the proposed proximal species to MI complex formation, the red colored compound is the inactivated P450 enzyme. The pathways were adapted from references [21, 22, 24]
The most remarkable feature of the alkylamine drugs is that with the exception of troleandomycin and erythromycin, they all have characterized circulating metabolites. Most commonly, the N-dealkylated metabolites circulate (Table 2) and their plasma concentrations are higher than the parent drug. Based on current mechanistic understanding, it is assumed that these N-dealkylated metabolites also contribute to P450 inactivation, although very little evidence is available of inactivation of P450 enzymes by the primary amine metabolites of the drugs listed in Table 2. It is interesting that despite the fact that the primary amines are believed to be the direct precursors of MI complexes, there is no direct correlation between formation of the primary amine by a given P450 enzyme and inactivation of that same P450 enzyme by MI complex formation. For example, CYP2D6 has the lowest reported Km (2.1μM) [Clint = 2.9 μM-1min-1] with regards to the N-demethylation of fluoxetine to norfluoxetine, but this CYP does not form an MI complex with fluoxetine; whereas, CYP2C19 which has a 82-fold higher Km (172μM) [Clint= 0.23 μM-1min-1] for norfluoxetine formation undergoes rapid inactivation by fluoxetine[25, 26]. At present, it is not known why some P450 enzymes are capable of oxidizing the parent alkyl amine drug to inhibitory metabolites but are incapable of forming an MI complex. A plausible explanation may be the structure of the P450 active site, given the prevalence of CYP3A4 to form MI complexes (100%), and the absence of an MI complex formation with CYP2C9 (0%). Given CYP3A4 promiscuity to metabolize a large number of drugs and its large active site, it may be more prone to forming and binding reactive metabolites. However, the prevalence of reports of MI complexes with CYP3A4 may be a result of more studies conducted with this enzyme. There may also be a concentration dependence to the inactivation of P450 enzymes as with CYP3A4 and troleandomyocin[27]. At concentrations higher than 10μM, troleandomycin no longer displays inactivation kinetics due to the amount of competitive primary and secondary metabolites formed that are able to compete for the active site of CYP3A4.
Due to the complex, and potentially multiple, parallel pathways to MI complex formation with P450's, it is unlikely that the inactivation kinetics of the P450 by a tertiary amine or secondary amine (parent drug) will correctly represent the cellular or in vivo system, in which multiple inactivating metabolites are present and may dominate the overall in vivo inactivation. In addition, the metabolites may be potent reversible inhibitors of the relevant P450 adding further complexity into the characterization of inhibition. Hall et al. recently proposed the need to add both the competitive and irreversible inhibition, when determining the extent of inhibition in vivo with the alkyl amine drugs, erythromycin and diltiazem[28]. A semiphysiological model was presented, which suggested that both diltiazem and its metabolite N-desmethyldiltiazem contributed to the overall inhibitory effect after diltiazem administration in vivo[29]. Unfortunately, sufficient data in vivo and in vitro for the metabolites is lacking to develop a more detailed model of in vivo MI complex formation kinetics.
Four of the MI complex forming drugs are anti-HIV protease inhibitors (amprenavir, indinavir, nelfinavir and ritonavir). Figure 3 shows the structures of these four protease inhibitors and their primary metabolic sites. No apparent reactive metabolite(s) or intermediates that would lead to MI complex formation have been identified in vitro for any of these drugs. As is evident from the structures, no obvious mechanism for the MI complex formation that would agree with the pathway in Figure 2 can be readily proposed. However, a spectroscopic detection of a peak at ∼455 nm after incubation of HLM's and recombinant CYP3A4 with these compounds suggested MI complex formation, and warrants further mechanistic studies with these compounds with focus on metabolites[30].
Figure 3.
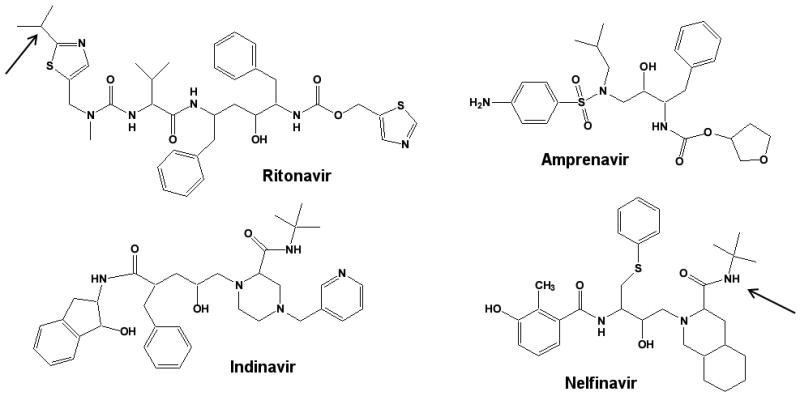
Structures of anti-HIV protease inhibitors that irreversibly inhibit CYP3A4 via an MI complex formation. The arrows indicate sites of metabolism for known circulating metabolites (Table 2).
Finally, paroxetine forms a distinctive type of MI complex with CYP2D6. Paroxetine contains a methylenedioxyphenyl moiety, a structural alert known to exhibit MBI of P450 enzyme[31]. P450-catalyzed metabolism of the methylenedioxyphenyl substituent results in initial hydroxylation at the methylene carbon forming an unstable intermediate. This unstable intermediate can partition between demethylenation yielding a cathecol metabolite or dehydration to a carbene, which forms an MI complex with the P450 enzyme[32]. Based on the mechanism it is unlikely that circulating metabolites play a role in CYP2D6 inactivation by paroxetine.
5. MBIs of unknown mechanisms of inhibition
Eight (26%) of the inhibitors identified have an unknown mechanism of inactivation based on lack of any described mechanism in the literature. These inhibitors were characterized as MBIs based on reports of concentration- and time-dependent inactivation assays, which are not necessarily conclusive in characterizing a drug as an MBI. Six of the eight (75%) drugs indentified have in vivo interactions with probe drug substrates. Based on the structures of these eight drugs, it could be hypothesized that MI complex is unlikely for all of the drugs, with the exception of delavirdine, which contains an alkyl amine moiety.
6. Case Studies
6.1 Irreversible Inhibition by P450 protein adduct
6.1A
Efavirenz (Figure 4) is metabolized mainly by CYP2B6 to two metabolites: 8-hydroxyefavirenz (major) and 8,14-dihydroxyefavirenz (minor)[33], and is an example of a drug that has a circulating metabolite that appears to be responsible for protein adduct formation. Efavirenz and both metabolites contain an ethynyl group, which is a known alert for P450 mechanism-based inactivation, particularly with the CYP2B family of enzymes[34]. Both efavirenz and its major metabolite 8-hydroxyefavirenz inhibit CYP2B6 but the inactivation of CYP2B6 by 8- hydroxyefavirenz was markedly different from the inactivation by efavirenz[35]. A comparison of the kinact/KI-ratio (efavirenz =0.0013; 8-hydroxyefavirenz =0.0094) shows that the metabolite is a more efficient inactivator than the parent compound. Interestingly, the metabolism of efavirenz and 8-hydroxylefavirenz by CYP2B6 leads to inhibition by two distinct mechanism although the reactive species responsible for the inactivation are not yet known. Efavirenz was a potent apparent reversible inhibitor (time- and concentration-dependent inactivation yet reversible by dialysis), whereas the 8-hydroxylefavirenz was an irreversible inhibitor (not reversible by dialysis). Based on this in vitro data, it appears that for in vitro to in vivo extrapolation of CYP2B6 inactivation after efavirenz administration, 8-hydroxyefavirenz rather than efavirenz should be modeled.
Figure 4.
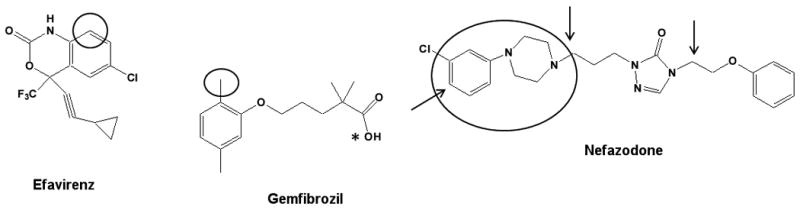
Structures of three drugs that form protein adducts with P450 enzymes. Circles indicate the site of metabolism that leads to inactivation, the arrows indicate site of metabolism for off path circulating metabolites (Table 1). *indicates the site of glucuronidation to form the inactivating glucoronide metabolite.
6.1B
Gemfibrozil (Figure 4) is known to be a more potent in vitro inhibitor of CYP2C9 than CYP2C8[36-38]. However, in vivo, gemfibrozil is an inhibitor of CYP2C8 but not of CYP2C9[39]. An important step in providing a potential explanation of this in vitro to in vivo discrepancy was provided by demonstrating that gemfibrozil 1-O-β-glucuronide, the circulating metabolite of gemfibrozil, is a more potent inhibitor than gemfibrozil of CYP2C8[40]. The mechanism of inactivation was recently characterized to occur via formation of a heme adduct[41]. It is important to note that the MBI of CYP2C8 was not observed with the parent drug in microsomes and phase II metabolites are not routinely tested for MBI, suggesting that testing for MBI in hepatocytes may be advantageous[42]. Oxidation of phase II metabolites by P450 enzymes appears rare; however, some examples include the oxidation of sulfate conjugates of testosterone, dehydroepiandrosterone, and estrogens[43-45] and oxidation of the acyl glucuronide of diclofenac by CYP2C8[46]. Gemfibrozil 1-O-β-glucuronide is the first report of a phase II metabolite that irreversibly inhibits CYP2C8 but this type of inhibition may be more widespread than currently acknowledged and true incidence may not be appreciated, especially given that phase II metabolites are rarely evaluated.
6.1C
Nefazodone (Figure 4) is known to have incidences of idiosyncratic hepatotoxicity, and the reactive intermediates may be responsible for liver injury[47-49]. Nefazodone has both on path and off path metabolites that circulate and two circulating metabolites are known to inactivate CYP3A4[50, 51]. Nefazodone is oxidized by P450 enzymes to the major circulating metabolite, hydroxynefazodone, which is then further oxidized by CYP3A4 to an electrophilic quinonoid intermediate. The structure of this reactive intermediate was inferred through the characterization of the corresponding glutathione conjugate. A second circulating metabolite, para-hydroxyl-m-CPP (mCPP), which is formed mainly by CYP2D6, can also be activated to quinone-imine by CYP3A4 and may play a role in the inactivation of CYP3A4[17]. It is interesting that P450 inactivation by nefazodone may involve different pathways in single enzyme systems and in more complex matrices due to the involvement of different P450's in the formation of the intermediates. Due to the multi-enzyme involvement in inactivation and the presence of two distinct inactivating metabolites, in vitro to in vivo extrapolation of CYP3A4 inactivation by nefazodone is expected to require complex models.
6.2 Irreversible Inhibition of P450 by MI complex formation
6.2A
Verapamil (Figure 5) has two major metabolites formed by CYP3A4 via N-dealkylation: N-desalkylverapamil (D-617) and norverapamil[52, 53]. Verapamil and these two major metabolites form MI complexes with CYP3A4[54]. Based on the ratio of kinact to KI, the inactivation potency was norverapamil > verapamil > D-617. Although the plasma concentration of D-617 is comparable to verapamil and norverapamil, and D-617 is a secondary amine, the potency of inactivation is weak and D-617 probably does not contribute to the in vivo inactivation. However, the steady-state levels of norverapamil reach that of verapamil and incorporating the inactivation kinetics of the secondary amine metabolite improved the in vivo predictions[55].
Figure 5.
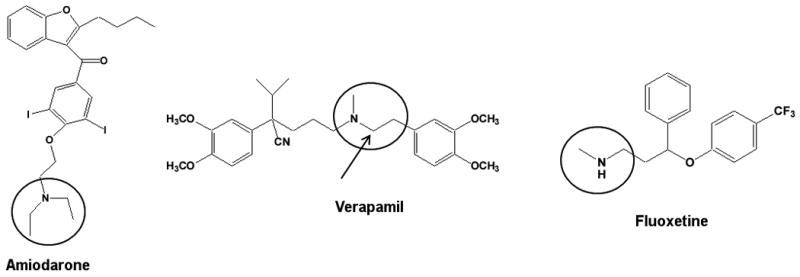
Structures of three drugs that result in MI complex formation with P450 enzymes. Circles indicate the site of metabolism that leads to inactivation, the arrows indicate site of metabolism for off path circulating metabolites (Table 2).
6.2B
Fluoxetine (Figure 5), is a substrate and inhibitor of multiple CYP enzymes. The major route of fluoxetine metabolism is by N-demethylation to norfluoxetine by CYP2D6 and other P450 isoforms, including CYP3A4 and CYP2C9[56, 57]. Fluoxetine also undergoes CYP2C19-mediated O-dealkylation to a p-trifluoromethylphenol metabolite[58]. Fluoxetine and norfluoxetine have been shown to be reversible inhibitors of CYP2D6[59, 60], CYP2C19[61, 62], CYP3A4[63] and CYP2C9[64]. In all cases of CYP inhibition, norfluoxetine was a more potent competitive inhibitor than the parent compound fluoxetine. However, fewer studies have been conducted on the MBI potential of fluoxetine and norfluoxetine. Mayhew et al.[65] showed fluoxetine to be an MBI of CYP3A4 and McGinnity et al.[26] demonstrated time- and concentration-dependent inhibition of CYP3A4 and CYP2C19 in multiple in vitro systems, including hepatocytes. Recently, Stresser et al.[66] reported that norfluoxetine exhibited an 11-fold shift in IC50 value when tested in human liver microsomes with the CYP2C19 probe (S)-mephenytoin, suggesting that conversion of fluoxetine to norfluoxetine represents a metabolic pathway leading to time-dependent inhibition. As mentioned previously, it is important to point out that not all the enzymes that form norfluoxetine are inactivated by fluoxetine. CYP2D6 and CYP2C9 both form norfluoxetine efficiently, but do not undergo MBI by fluoxetine. More so, fluoxetine is an MBI of CYP2C19 but CYP2C19 has a relatively high Km (172 ± 25 μM) for the formation of norfluoxetine[25]. It is possible that norfluoxetine formed by CYP2D6 and CYP2C9 is released and then inactivates other enzymes such as CYP3A4 and CYP2C19, again emphasizing the importance of testing for MBI in multiple CYP systems. Given that the route to inactivation is a multi-step process and metabolites are released into circulation, it is not clear which alkyl amine metabolites are important in making accurate predictions of in vivo inhibition. For example, CYP3A4 is clearly inactivated in vitro by fluoxetine but there is no in vivo interaction with CYP3A4 probes[67, 68]. In contrast, in vitro inactivation of CYP2C19 is associated with moderate inhibition of CYP2C19 in vivo[69].
6.2C
Amiodarone (Figure 5) is a tertiary amine that interacts in vivo with a number of drugs metabolized by CYP1A2, CYP2C9, CYP2D6 and CYP3A4 but the in vitro inactivation profile for amiodarone is not fully established[70-72]. Amiodarone is known to form an MI complex in rodents, which is consistent with tertiary amine metabolism to the nitrosoalkane reactive intermediate (Figure 2, [73]). The identity CYP isozymes inactivated by MI complex formation is unclear as the major circulating metabolite of amiodarone, N-desethylamiodarone, inactivates different isozymes than the parent drug. Ohyama, K et al.[74] report a kinact value of 0.06 min-1 and a KI value of 13.4μM for CYP3A4 and amiodarone but no inactivation of CYP3A4 by N-desethylamiodarone. In contrast, both N-desethylamiodarone and amiodarone were recently shown to be time- and concentration- dependent inactivators of CYP3A4[75]. N-desethylamiodarone, but not amiodarone, is consistently reported as an MBI of CYP2D6 highlighting the importance of studying metabolites separately for MBI[74, 75]. Interestingly, CYP2D6 is capable of N-dealkylation reaction with an in vitro intrinsic clearance to the secondary amine of 106.8 μM/min/nmol CYP[74]. Although, CYP2D6 is very efficient in forming N-desethylamiodarone, the inhibitory metabolite, MBI of CYP2D6 by the parent drug is not observed.
Amiodarone also inactivates CYP2C8 and CYP2C9 (Polasek, PM et al. 2004, Mori, K. et al.) and CYP2C8 plays a significant role in amiodarone deethylation[70, 75, 76]. However, spectral studies did not detect MI complex formation or heme loss with CYP2C8 and the exact mechanism by which amiodarone inactivates CYP2C8 remains unclear[76]. N-desethylamiodarone may play a role in CYP2C8 inactivation, as the inhibition by N-desethylamiodarone increased by 42% between co- and pre-incubated samples[76]. CYP2C9 has similar time- and concentration-dependent inactivation by both amiodarone and N-desethylamiodarone. It is likely that N-desethlyamiodarone is essential for CYP2C9 inactivation as a correlation was found between the ΔINR/ warfarin dose and plasma concentration of N-desethylamiodarone but not with amiodarone concentration[77]. This again emphasizes the critical role of the metabolites in CYP inactivation and suggests that accounting of the metabolites is important for quantitative understanding and predictions of in vivo CYP inactivation.
7. Conclusions
Given the limited number of predictive models for complex DDIs involving parent drugs and their metabolites, it is difficult to fully evaluate the importance of inhibitory metabolites. Many drugs that display in vitro MBI kinetics, do not display significant in vivo DDIs. In fact only 9 of the 31 inhibitors (29%) are potent inhibitors in vivo. On the other hand, drugs such as mibefradil, a potent MBI of CYP3A4, was withdrawn from the market as a result of unpredicted CYP inhibition, most likely due to interactions caused by a metabolite[78]. From the available data for the compounds reviewed here, it appears that circulating metabolites are much more important in in vivo CYP inactivation than is currently acknowledged. The data suggests that CYP inactivation by circulating primary and secondary metabolites needs to be characterized for accurate predictions as well as for better mechanistic understanding of in vivo MBI. Due to the multi-enzyme involvement of CYP inactivation, testing for MBI in complex enzyme systems such as hepatocytes, may help in the overall understanding of the inhibition and significantly improve in vitro to in vivo predictions.
Acknowledgments
This work was partially supported by an NIH grant P01 GM32165. The authors wish to thank Dr. Kent Kunze for helpful discussion during the preparation of this manuscript.
Abbreviations
- CYP
cytochrome P450 enzyme
- DDI
drug-drug interaction
- HLMs
human liver microsomes
- kinact
maximum inactivation rate constant
- KI
inactivation binding constant
- Ki
reversible binding constant
- MBI
mechanism-based inactivation (inhibitor)
- MI complex
metabolic-intermediate complex
References
- 1.Isoherranen N, Hachad H, Yeung CK, Levy RH. Qualitative analysis of the role of metabolites in inhibitory drug-drug interactions: literature evaluation based on the metabolism and transport drug interaction database. Chem Res Toxicol. 2009;22:294–298. doi: 10.1021/tx800491e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.He M, Kunze KL, Trager WF. Inhibition of (S)-warfarin metabolism by sulfinpyrazone and its metabolites. Drug Metab Dispos. 1995;23:659–663. [PubMed] [Google Scholar]
- 3.He M, Rettie AE, Neal J, Trager WF. Metabolism of sulfinpyrazone sulfide and sulfinpyrazone by human liver microsomes and cDNA-expressed cytochrome P450s. Drug Metab Dispos. 2001;29:701–711. [PubMed] [Google Scholar]
- 4.Templeton IE, Thummel KE, Kharasch ED, Kunze KL, Hoffer C, Nelson WL, Isoherranen N. Contribution of Itraconazole Metabolites to Inhibition of CYP3A4 In Vivo. Clin Pharmacol Ther. 2007;83:77–85. doi: 10.1038/sj.clpt.6100230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Silverman RB. Mechanism-based Enzyme Inactivation: Chemistry and Enzymology. CRC Press; 1988. [Google Scholar]
- 6.Atrakchi AH. Interpretation and Considerations on the Safety Evaluation of Human Drug Metabolites. Chemical Research in Toxicology. 2009 doi: 10.1021/tx900124j. [DOI] [PubMed] [Google Scholar]
- 7.Baillie TA. Metabolism and toxicity of drugs. Two decades of progress in industrial drug metabolism. Chem Res Toxicol. 2008;21:129–137. doi: 10.1021/tx7002273. [DOI] [PubMed] [Google Scholar]
- 8.Smith DA, Obach RS. Metabolites in Safety Testing (MIST): Considerations of Mechanisms of Toxicity with Dose, Abundance, and Duration of Treatment. Chemical Research in Toxicology. 2009;22:267–279. doi: 10.1021/tx800415j. [DOI] [PubMed] [Google Scholar]
- 9.Smith DA, Obach RS. Metabolites and safety: What are the concerns, and how should we address them? Chem Res Toxicol. 2006;19:1570–1579. doi: 10.1021/tx0602012. [DOI] [PubMed] [Google Scholar]
- 10.Kalgutkar AS, Obach RS, Maurer TS. Mechanism-based inactivation of cytochrome P450 enzymes: chemical mechanisms, structure-activity relationships and relationship to clinical drug-drug interactions and idiosyncratic adverse drug reactions. Curr Drug Metab. 2007;8:407–447. doi: 10.2174/138920007780866807. [DOI] [PubMed] [Google Scholar]; * A thorough review of chemical and biochemical reactions
- 11.Waley SG. Kinetics of suicide substrates: Practical procedures for determining parameters. Biochemical Journal. 1985;227:843–849. doi: 10.1042/bj2270843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pessayre Dominique, L D, Vitauxa Jean, Breila Philippe, Belghitia Jacques, Benhamoua Jean-Pierre. Formation of an inactive cytochrome P-450 Fe(II)-metabolite complex after administration of troleandomycin in humans. Biochemical Pharmacology. 1981;31:1699–1704. doi: 10.1016/0006-2952(82)90671-2. [DOI] [PubMed] [Google Scholar]
- 13.Pessayre D, Tinel M, Larrey D, Cobert B, Funck-Brentano C, Babany G. Inactivation of cytochrome P-450 by a troleandomycin metabolite. Protective role of glutathione. J Pharmacol Exp Ther. 1983;224:685–691. [PubMed] [Google Scholar]
- 14.Administration, F.a.D. Guidance for Industry Drug Interaction Studies — Study Design, Data Analysis, and Implications for Dosing and Labeling. 2006. Draft Guidance. [DOI] [PubMed] [Google Scholar]
- 15.Li X, He Y, Ruiz CH, Koenig M, Cameron MD. Characterization of dasatinib and its structural analogs as CYP3A4 mechanism-based inactivators and the proposed bioactivation pathways. Drug Metab Dispos. 2009;37:1242–1250. doi: 10.1124/dmd.108.025932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu LJ, Chen Y, Deninno MP, O'Connell TN, Hop CE. Identification of a novel glutathione adduct of diclofenac, 4′-hydroxy-2′-glutathion-deschloro-diclofenac, upon incubation with human liver microsomes. Drug Metab Dispos. 2005;33:484–488. doi: 10.1124/dmd.104.002840. [DOI] [PubMed] [Google Scholar]
- 17.Kalgutkar AS, Vaz AD, Lame ME, Henne KR, Soglia J, Zhao SX, Abramov YA, Lombardo F, Collin C, Hendsch ZS, Hop CE. Bioactivation of the nontricyclic antidepressant nefazodone to a reactive quinone-imine species in human liver microsomes and recombinant cytochrome P450 3A4. Drug Metab Dispos. 2005;33:243–253. doi: 10.1124/dmd.104.001735. [DOI] [PubMed] [Google Scholar]
- 18.Chen Q, Ngui JS, Doss GA, Wang RW, Cai X, DiNinno FP, Blizzard TA, Hammond ML, Stearns RA, Evans DC, Baillie TA, Tang W. Cytochrome P450 3A4-mediated bioactivation of raloxifene: irreversible enzyme inhibition and thiol adduct formation. Chem Res Toxicol. 2002;15:907–914. doi: 10.1021/tx0200109. [DOI] [PubMed] [Google Scholar]
- 19.Kassahun K, Skordos K, McIntosh I, Slaughter D, Doss GA, Baillie TA, Yost GS. Zafirlukast metabolism by cytochrome P450 3A4 produces an electrophilic alpha,beta-unsaturated iminium species that results in the selective mechanism-based inactivation of the enzyme. Chem Res Toxicol. 2005;18:1427–1437. doi: 10.1021/tx050092b. [DOI] [PubMed] [Google Scholar]
- 20.Nishiya Y, Hagihara K, Kurihara A, Okudaira N, Farid NA, Okazaki O, Ikeda T. Comparison of mechanism-based inhibition of human cytochrome P450 2C19 by ticlopidine, clopidogrel, and prasugrel. Xenobiotica. 2009 doi: 10.3109/00498250903191427. [DOI] [PubMed] [Google Scholar]
- 21.Ortiz de Montellano PR, editor. Cytochrome P450: Structure, Mechanism, and Biochemistry. Third edition. New York: Kluwer Academic/Plenum Publishers; 2005. [Google Scholar]
- 22.Mansuy D, Gans P, Chottard JC, Bartoli JF. Nitrosoalkanes as Fe(II) Ligands in the 455-nm- Absorbing Cytochrome P-450 Complexes Formed from Nitroalkanes in Reducing Conditions. European Journal of Biochemistry. 1977;76:607–615. doi: 10.1111/j.1432-1033.1977.tb11631.x. [DOI] [PubMed] [Google Scholar]
- 23.Bensoussan C, Delaforge M, Mansuy D. Particular ability of cytochrome P450 3A to form inhibitory P450-iron-metabolite complexes upon metabolic oxidation of aminodrugs. Biochem Pharmacol. 1995;49:591–602. doi: 10.1016/0006-2952(94)00477-4. [DOI] [PubMed] [Google Scholar]
- 24.Jonsson KH, Lindeke B. Cytochrome P-455 nm complex formation in the metabolism of phenylalkylamines. XII. Enantioselectivity and temperature dependence in microsomes and reconstituted cytochrome P-450 systems from rat liver. Chirality. 1992;4:469–477. doi: 10.1002/chir.530040803. [DOI] [PubMed] [Google Scholar]
- 25.Margolis JM, O'Donnell JP, Mankowski D, Ekins S, Obach RS. (R)-, (S)-, and Racemic Fluoxetine N-Demethylation by Human Cytochrome P450 Enzymes. Drug Metabolism and Disposition. 2000;28:1187–1191. [PubMed] [Google Scholar]
- 26.McGinnity DF, Berry AJ, Kenny JR, Grime K, Riley RJ. Evaluation of Time-Dependent Cytochrome P450 Inhibition Using Cultured Human Hepatocytes. Drug Metabolism and Disposition. 2006;34:1291–1300. doi: 10.1124/dmd.106.009969. [DOI] [PubMed] [Google Scholar]
- 27.Xue-Jun Zhao TI. Metabolic interactions of selected antimalarial and non-antimararial drugs with the major pathway (3-hydroxylation) of quinine in human liver microsomes. Br J Clin Phamacol. 1997;44:505–511. doi: 10.1046/j.1365-2125.1997.t01-1-00619.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang X, Jones DR, Hall SD. Prediction of the Effect of Erythromycin, Diltiazem, and their Metabolites, Alone and in Combination, on CYP3A4 Inhibition. Drug Metab Dispos. 2008 doi: 10.1124/dmd.108.022178. dmd.108.022178. [DOI] [PubMed] [Google Scholar]
- 29.Zhang X, Quinney SK, Gorski JC, Jones DR, Hall SD. Semiphysiologically based pharmacokinetic models for the inhibition of midazolam clearance by diltiazem and its major metabolite. Drug Metab Dispos. 2009;37:1587–1597. doi: 10.1124/dmd.109.026658. [DOI] [PubMed] [Google Scholar]
- 30.Ernest CS, 2nd, Hall SD, Jones DR. Mechanism-based inactivation of CYP3A by HIV protease inhibitors. J Pharmacol Exp Ther. 2005;312:583–591. doi: 10.1124/jpet.104.075416. [DOI] [PubMed] [Google Scholar]
- 31.Fontana E, D PM, Poli SM. Cytochrome P450 Enzyme Mechanism Based Inhibitors:Commmon Sub-Structures and Reactivity. Current Drug Metabolism. 2005;6:413–454. doi: 10.2174/138920005774330639. [DOI] [PubMed] [Google Scholar]
- 32.Bertelsen KM, Venkatakrishnan K, von Moltke LL, Obach RS, Greenblatt DJ. Apparent Mechanism-based Inhibition of Human CYP2D6 in Vitro by Paroxetine: Comparison with Fluoxetine and Quinidine. Drug Metab Dispos. 2003;31:289–293. doi: 10.1124/dmd.31.3.289. [DOI] [PubMed] [Google Scholar]
- 33.Ward BA, Gorski JC, Jones DR, Hall SD, Flockhart DA, Desta Z. The cytochrome P450 2B6 (CYP2B6) is the main catalyst of efavirenz primary and secondary metabolism: implication for HIV/AIDS therapy and utility of efavirenz as a substrate marker of CYP2B6 catalytic activity. J Pharmacol Exp Ther. 2003;306:287–300. doi: 10.1124/jpet.103.049601. [DOI] [PubMed] [Google Scholar]
- 34.Foroozesh M, Primrose G, Guo Z, Bell LC, Alworth WL, Guengerich FP. Aryl acetylenes as mechanism-based inhibitors of cytochrome P450-dependent monooxygenase enzymes. Chem Res Toxicol. 1997;10:91–102. doi: 10.1021/tx960064g. [DOI] [PubMed] [Google Scholar]
- 35.Bumpus NN, Kent UM, Hollenberg PF. Metabolism of efavirenz and 8-hydroxyefavirenz by P450 2B6 leads to inactivation by two distinct mechanisms. J Pharmacol Exp Ther. 2006;318:345–351. doi: 10.1124/jpet.106.102525. [DOI] [PubMed] [Google Scholar]
- 36.Fujino H, Yamada I, Shimada S, Hirano M, Tsunenari Y, Kojima J. Interaction between fibrates and statins--metabolic interactions with gemfibrozil. Drug Metabol Drug Interact. 2003;19:161–176. doi: 10.1515/dmdi.2003.19.3.161. [DOI] [PubMed] [Google Scholar]
- 37.Wang JS, Wen X, Backman JT, Neuvonen PJ. Effect of albumin and cytosol on enzyme kinetics of tolbutamide hydroxylation and on inhibition of CYP2C9 by gemfibrozil in human liver microsomes. J Pharmacol Exp Ther. 2002;302:43–49. doi: 10.1124/jpet.302.1.43. [DOI] [PubMed] [Google Scholar]
- 38.Wen X, Wang JS, Backman JT, Kivisto KT, Neuvonen PJ. Gemfibrozil is a potent inhibitor of human cytochrome P450 2C9. Drug Metab Dispos. 2001;29:1359–1361. [PubMed] [Google Scholar]
- 39.Lilja JJ, Backman JT, Neuvonen PJ. Effect of gemfibrozil on the pharmacokinetics and pharmacodynamics of racemic warfarin in healthy subjects. Br J Clin Pharmacol. 2005;59:433–439. doi: 10.1111/j.1365-2125.2004.02323.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shitara Y, Hirano M, Sato H, Sugiyama Y. Gemfibrozil and its glucuronide inhibit the organic anion transporting polypeptide 2 (OATP2/OATP1B1:SLC21A6)-mediated hepatic uptake and CYP2C8-mediated metabolism of cerivastatin: analysis of the mechanism of the clinically relevant drug-drug interaction between cerivastatin and gemfibrozil. J Pharmacol Exp Ther. 2004;311:228–236. doi: 10.1124/jpet.104.068536. [DOI] [PubMed] [Google Scholar]
- 41.Baer BR, DeLisle RK, Allen A. Benzylic oxidation of gemfibrozil-1-O-beta-glucuronide by P450 2C8 leads to heme alkylation and irreversible inhibition. Chem Res Toxicol. 2009;22:1298–1309. doi: 10.1021/tx900105n. [DOI] [PubMed] [Google Scholar]
- 42.Ogilvie BW, Zhang D, Li W, Rodrigues AD, Gipson AE, Holsapple J, Toren P, Parkinson A. Glucuronidation converts gemfibrozil to a potent, metabolism-dependent inhibitor of CYP2C8: implications for drug-drug interactions. Drug Metab Dispos. 2006;34:191–197. doi: 10.1124/dmd.105.007633. [DOI] [PubMed] [Google Scholar]
- 43.Berg A, Carlstrom K, Gustafsson JA, Ingelman-Sundberg M. Demonstration of a cytochrome P-450-dependent steroid 15beta-hydroxylase in Bacillus megaterium. Biochem Biophys Res Commun. 1975;66:1414–1423. doi: 10.1016/0006-291x(75)90517-3. [DOI] [PubMed] [Google Scholar]
- 44.Kitada M, Kamataki T, Itahashi K, Rikihisa T, Kanakubo Y. Significance of cytochrome P-450 (P-450 HFLa) of human fetal livers in the steroid and drug oxidations. Biochem Pharmacol. 1987;36:453–456. doi: 10.1016/0006-2952(87)90350-9. [DOI] [PubMed] [Google Scholar]
- 45.Ohmori S, Fujiki N, Nakasa H, Nakamura H, Ishii I, Itahashi K, Kitada M. Steroid hydroxylation by human fetal CYP3A7 and human NADPH-cytochrome P450 reductase coexpressed in insect cells using baculovirus. Res Commun Mol Pathol Pharmacol. 1998;100:15–28. [PubMed] [Google Scholar]
- 46.Kumar S, Samuel K, Subramanian R, Braun MP, Stearns RA, Chiu SH, Evans DC, Baillie TA. Extrapolation of diclofenac clearance from in vitro microsomal metabolism data: role of acyl glucuronidation and sequential oxidative metabolism of the acyl glucuronide. J Pharmacol Exp Ther. 2002;303:969–978. doi: 10.1124/jpet.102.038992. [DOI] [PubMed] [Google Scholar]
- 47.Carvajal Garcia-Pando A, Garcia del Pozo J, Sanchez AS, Velasco MA, Rueda de Castro AM, Lucena MI. Hepatotoxicity associated with the new antidepressants. J Clin Psychiatry. 2002;63:135–137. doi: 10.4088/jcp.v63n0208. [DOI] [PubMed] [Google Scholar]
- 48.Stewart DE. Hepatic adverse reactions associated with nefazodone. Can J Psychiatry. 2002;47:375–377. doi: 10.1177/070674370204700409. [DOI] [PubMed] [Google Scholar]
- 49.Dykens JA, Jamieson JD, Marroquin LD, Nadanaciva S, Xu JJ, Dunn MC, Smith AR, Will Y. In vitro assessment of mitochondrial dysfunction and cytotoxicity of nefazodone, trazodone, and buspirone. Toxicol Sci. 2008;103:335–345. doi: 10.1093/toxsci/kfn056. [DOI] [PubMed] [Google Scholar]
- 50.DeVane CL, Donovan JL, Liston HL, Markowitz JS, Cheng KT, Risch SC, Willard L. Comparative CYP3A4 inhibitory effects of venlafaxine, fluoxetine, sertraline, and nefazodone in healthy volunteers. J Clin Psychopharmacol. 2004;24:4–10. doi: 10.1097/01.jcp.0000104908.75206.26. [DOI] [PubMed] [Google Scholar]
- 51.von Moltke LL, Greenblatt DJ, Granda BW, Grassi JM, Schmider J, Harmatz JS, Shader RI. Nefazodone, meta-chlorophenylpiperazine, and their metabolites in vitro: cytochromes mediating transformation, and P450-3A4 inhibitory actions. Psychopharmacology (Berl) 1999;145:113–122. doi: 10.1007/s002130051039. [DOI] [PubMed] [Google Scholar]
- 52.Kroemer HK, Echizen H, Heidemann H, Eichelbaum M. Predictability of the in vivo metabolism of verapamil from in vitro data: contribution of individual metabolic pathways and stereoselective aspects. J Pharmacol Exp Ther. 1992;260:1052–1057. [PubMed] [Google Scholar]
- 53.Shen L, Fitzloff JF, Cook CS. Differential enantioselectivity and product-dependent activation and inhibition in metabolism of verapamil by human CYP3As. Drug Metab Dispos. 2004;32:186–196. doi: 10.1124/dmd.32.2.186. [DOI] [PubMed] [Google Scholar]
- 54.Wang YH, Jones DR, Hall SD. Prediction of cytochrome P450 3A inhibition by verapamil enantiomers and their metabolites. Drug Metab Dispos. 2004;32:259–266. doi: 10.1124/dmd.32.2.259. [DOI] [PubMed] [Google Scholar]
- 55.Wang YH, Jones DR, Hall SD. Differential mechanism-based inhibition of CYP3A4 and CYP3A5 by verapamil. Drug Metab Dispos. 2005;33:664–671. doi: 10.1124/dmd.104.001834. [DOI] [PubMed] [Google Scholar]
- 56.Margolis JM, O'Donnell JP, Mankowski DC, Ekins S, Obach RS. (R)-, (S)-, and racemic fluoxetine N-demethylation by human cytochrome P450 enzymes. Drug Metab Dispos. 2000;28:1187–1191. [PubMed] [Google Scholar]
- 57.Mandrioli R, Forti GC, Raggi MA. Fluoxetine metabolism and pharmacological interactions: the role of cytochrome p450. Curr Drug Metab. 2006;7:127–133. doi: 10.2174/138920006775541561. [DOI] [PubMed] [Google Scholar]
- 58.Liu ZQ, Tan ZR, Wang D, Huang SL, Wang LS, Zhou HH. Simultaneous determination of fluoxetine and its metabolite p-trifluoromethylphenol in human liver microsomes using a gas chromatographic-electron-capture detection procedure. J Chromatogr B Analyt Technol Biomed Life Sci. 2002;769:305–311. doi: 10.1016/s1570-0232(02)00016-8. [DOI] [PubMed] [Google Scholar]
- 59.Brosen K, Skjelbo E. Fluoxetine and norfluoxetine are potent inhibitors of P450IID6--the source of the sparteine/debrisoquine oxidation polymorphism. Br J Clin Pharmacol. 1991;32:136–137. doi: 10.1111/j.1365-2125.1991.tb05630.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stevens JC, Wrighton SA. Interaction of the enantiomers of fluoxetine and norfluoxetine with human liver cytochromes P450. J Pharmacol Exp Ther. 1993;266:964–971. [PubMed] [Google Scholar]
- 61.Kobayashi K, Yamamoto T, Chiba K, Tani M, Ishizaki T, Kuroiwa Y. The effects of selective serotonin reuptake inhibitors and their metabolites on S-mephenytoin 4′-hydroxylase activity in human liver microsomes. Br J Clin Pharmacol. 1995;40:481–485. doi: 10.1111/j.1365-2125.1995.tb05793.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Foti RS, Wahlstrom JL. CYP2C19 inhibition: the impact of substrate probe selection on in vitro inhibition profiles. Drug Metab Dispos. 2008;36:523–528. doi: 10.1124/dmd.107.019265. [DOI] [PubMed] [Google Scholar]
- 63.von Moltke LL, Greenblatt DJ, Schmider J, Duan SX, Wright CE, Harmatz JS, Shader RI. Midazolam hydroxylation by human liver microsomes in vitro: inhibition by fluoxetine, norfluoxetine, and by azole antifungal agents. J Clin Pharmacol. 1996;36:783–791. doi: 10.1002/j.1552-4604.1996.tb04251.x. [DOI] [PubMed] [Google Scholar]
- 64.Hemeryck A, De Vriendt C, Belpaire FM. Inhibition of CYP2C9 by selective serotonin reuptake inhibitors: in vitro studies with tolbutamide and (S)-warfarin using human liver microsomes. Eur J Clin Pharmacol. 1999;54:947–951. doi: 10.1007/s002280050580. [DOI] [PubMed] [Google Scholar]
- 65.Mayhew BS, Jones DR, Hall SD. An In Vitro Model for Predicting In Vivo Inhibition of Cytochrome P450 3A4 by Metabolic Intermediate Complex Formation. Drug Metabolism and Disposition. 2000;28:1031–1037. [PubMed] [Google Scholar]
- 66.Stresser DM, Mason AK, Perloff ES, Ho T, Crespi CL, Dandeneau AA, Morgan L, Dehal SS. Differential time- and NADPH-dependent inhibition of CYP2C19 by enantiomers of fluoxetine. Drug Metab Dispos. 2009;37:695–698. doi: 10.1124/dmd.108.025726. [DOI] [PubMed] [Google Scholar]
- 67.Wright CE, Lasher-Sisson TA, Steenwyk RC, Swanson CN. A pharmacokinetic evaluation of the combined administration of triazolam and fluoxetine. Pharmacotherapy. 1992;12:103–106. [PubMed] [Google Scholar]
- 68.Lam YW, Alfaro CL, Ereshefsky L, Miller M. Pharmacokinetic and pharmacodynamic interactions of oral midazolam with ketoconazole, fluoxetine, fluvoxamine, and nefazodone. J Clin Pharmacol. 2003;43:1274–1282. doi: 10.1177/0091270003259216. [DOI] [PubMed] [Google Scholar]
- 69.Dingemanse J, Wallnofer A, Gieschke R, Guentert T, Amrein R. Pharmacokinetic and pharmacodynamic interactions between fluoxetine and moclobemide in the investigation of development of the “serotonin syndrome”. Clin Pharmacol Ther. 1998;63:403–413. doi: 10.1016/S0009-9236(98)90035-2. [DOI] [PubMed] [Google Scholar]
- 70.Ohyama K, Nakajima M, Suzuki M, Shimada N, Yamazaki H, Yokoi T. Inhibitory effects of amiodarone and its N-deethylated metabolite on human cytochrome P450 activities: prediction of in vivo drug interactions. Br J Clin Pharmacol. 2000;49:244–253. doi: 10.1046/j.1365-2125.2000.00134.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kozlik P, Ha HR, Stieger B, Bigler L, Follath F. Metabolism of amiodarone (Part III): identification of rabbit cytochrome P450 isoforms involved in the hydroxylation of mono-N-desethylamiodarone. Xenobiotica. 2001;31:239–248. doi: 10.1080/00498250110046442. [DOI] [PubMed] [Google Scholar]
- 72.Heimark LD, Wienkers L, Kunze K, Gibaldi M, Eddy AC, Trager WF, O'Reilly RA, Goulart DA. The mechanism of the interaction between amiodarone and warfarin in humans. Clin Pharmacol Ther. 1992;51:398–407. doi: 10.1038/clpt.1992.39. [DOI] [PubMed] [Google Scholar]
- 73.Larrey D, Tinel M, Letteron P, Geneve J, Descatoire V, Pessayre D. Formation of an inactive cytochrome P-450Fe(II)-metabolite complex after administration of amiodarone in rats, mice and hamsters. Biochem Pharmacol. 1986;35:2213–2220. doi: 10.1016/0006-2952(86)90594-0. [DOI] [PubMed] [Google Scholar]
- 74.Ohyama K, Nakajima M, Nakamura S, Shimada N, Yamazaki H, Yokoi T. A significant role of human cytochrome P450 2C8 in amiodarone N-deethylation: an approach to predict the contribution with relative activity factor. Drug Metab Dispos. 2000;28:1303–1310. [PubMed] [Google Scholar]
- 75.Mori K, Hashimoto H, Takatsu H, Tsuda-Tsukimoto M, Kume T. Cocktail-substrate assay system for mechanism-based inhibition of CYP2C9, CYP2D6, and CYP3A using human liver microsomes at an early stage of drug development. Xenobiotica. 2009;39:415–422. doi: 10.1080/00498250902822204. [DOI] [PubMed] [Google Scholar]
- 76.Polasek TM, Elliot DJ, Lewis BC, Miners JO. Mechanism-based inactivation of human cytochrome P4502C8 by drugs in vitro. J Pharmacol Exp Ther. 2004;311:996–1007. doi: 10.1124/jpet.104.071803. [DOI] [PubMed] [Google Scholar]
- 77.Naganuma M, Shiga T, Nishikata K, Tsuchiya T, Kasanuki H, Fujii E. Role of desethylamiodarone in the anticoagulant effect of concurrent amiodarone and warfarin therapy. J Cardiovasc Pharmacol Ther. 2001;6:363–367. doi: 10.1177/107424840100600405. [DOI] [PubMed] [Google Scholar]
- 78.Wandel C, Kim RB, Guengerich FP, Wood AJ. Mibefradil is a P-glycoprotein substrate and a potent inhibitor of both P-glycoprotein and CYP3A in vitro. Drug Metab Dispos. 2000;28:895–898. [PubMed] [Google Scholar]
- 79.Richter T, Murdter TE, Heinkele G, Pleiss J, Tatzel S, Schwab M, Eichelbaum M, Zanger UM. Potent mechanism-based inhibition of human CYP2B6 by clopidogrel and ticlopidine. J Pharmacol Exp Ther. 2004;308:189–197. doi: 10.1124/jpet.103.056127. [DOI] [PubMed] [Google Scholar]
- 80.Hagihara K, Nishiya Y, Kurihara A, Kazui M, Farid NA, Ikeda T. Comparison of human cytochrome p450 inhibition by the thienopyridines prasugrel, clopidogrel, and ticlopidine. Drug Metab Pharmacokinet. 2008;23:412–420. doi: 10.2133/dmpk.23.412. [DOI] [PubMed] [Google Scholar]
- 81.Wang L, Christopher LJ, Cui D, Li W, Iyer R, Humphreys WG, Zhang D. Identification of the human enzymes involved in the oxidative metabolism of dasatinib: an effective approach for determining metabolite formation kinetics. Drug Metab Dispos. 2008;36:1828–1839. doi: 10.1124/dmd.107.020255. [DOI] [PubMed] [Google Scholar]
- 82.Masubuchi Y, Ose A, Horie T. Diclofenac-induced inactivation of CYP3A4 and its stimulation by quinidine. Drug Metab Dispos. 2002;30:1143–1148. doi: 10.1124/dmd.30.10.1143. [DOI] [PubMed] [Google Scholar]
- 83.Baer BR, Wienkers LC, Rock DA. Time-dependent inactivation of P450 3A4 by raloxifene: identification of Cys239 as the site of apoprotein alkylation. Chem Res Toxicol. 2007;20:954–964. doi: 10.1021/tx700037e. [DOI] [PubMed] [Google Scholar]
- 84.Ha-Duong NT, Dijols S, Macherey AC, Goldstein JA, Dansette PM, Mansuy D. Ticlopidine as a selective mechanism-based inhibitor of human cytochrome P450 2C19. Biochemistry. 2001;40:12112–12122. doi: 10.1021/bi010254c. [DOI] [PubMed] [Google Scholar]
- 85.Polasek TM, Miners JO. Time-dependent inhibition of human drug metabolizing cytochromes P450 by tricyclic antidepressants. Br J Clin Pharmacol. 2008;65:87–97. doi: 10.1111/j.1365-2125.2007.02964.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ohmori S, Ishii I, Kuriya S, Taniguchi T, Rikihisa T, Hirose S, Kanakubo Y, Kitada M. Effects of clarithromycin and its metabolites on the mixed function oxidase system in hepatic microsomes of rats. Drug Metab Dispos. 1993;21:358–363. [PubMed] [Google Scholar]
- 87.Jones DR, Gorski JC, Hamman MA, Mayhew BS, Rider S, Hall SD. Diltiazem inhibition of cytochrome P-450 3A activity is due to metabolite intermediate complex formation. J Pharmacol Exp Ther. 1999;290:1116–1125. [PubMed] [Google Scholar]
- 88.McConn DJ, 2nd, Lin YS, Allen K, Kunze KL, Thummel KE. Differences in the inhibition of cytochromes P450 3A4 and 3A5 by metabolite-inhibitor complex-forming drugs. Drug Metab Dispos. 2004;32:1083–1091. doi: 10.1124/dmd.32.10.. [DOI] [PubMed] [Google Scholar]
- 89.Zhao XJ, Jones DR, Wang YH, Grimm SW, Hall SD. Reversible and irreversible inhibition of CYP3A enzymes by tamoxifen and metabolites. Xenobiotica. 2002;32:863–878. doi: 10.1080/00498250210158230. [DOI] [PubMed] [Google Scholar]
- 90.Sridar C, Kent UM, Notley LM, Gillam EM, Hollenberg PF. Effect of tamoxifen on the enzymatic activity of human cytochrome CYP2B6. J Pharmacol Exp Ther. 2002;301:945–952. doi: 10.1124/jpet.301.3.945. [DOI] [PubMed] [Google Scholar]
- 91.Madeira M, Levine M, Chang TK, Mirfazaelian A, Bellward GD. The effect of cimetidine on dextromethorphan O-demethylase activity of human liver microsomes and recombinant CYP2D6. Drug Metab Dispos. 2004;32:460–467. doi: 10.1124/dmd.32.4.460. [DOI] [PubMed] [Google Scholar]
- 92.Steiner E, Spina E. Differences in the inhibitory effect of cimetidine on desipramine metabolism between rapid and slow debrisoquin hydroxylators. Clin Pharmacol Ther. 1987;42:278–282. doi: 10.1038/clpt.1987.147. [DOI] [PubMed] [Google Scholar]
- 93.Voorman RL, Maio SM, Payne NA, Zhao Z, Koeplinger KA, Wang X. Microsomal metabolism of delavirdine: evidence for mechanism-based inactivation of human cytochrome P450 3A. J Pharmacol Exp Ther. 1998;287:381–388. [PubMed] [Google Scholar]
- 94.Polasek TM, Elliot DJ, Somogyi AA, Gillam EM, Lewis BC, Miners JO. An evaluation of potential mechanism-based inactivation of human drug metabolizing cytochromes P450 by monoamine oxidase inhibitors, including isoniazid. Br J Clin Pharmacol. 2006;61:570–584. doi: 10.1111/j.1365-2125.2006.02627.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Harleton E, Webster M, Bumpus NN, Kent UM, Rae JM, Hollenberg PF. Metabolism of N,N′,N″-triethylenethiophosphoramide by CYP2B1 and CYP2B6 results in the inactivation of both isoforms by two distinct mechanisms. J Pharmacol Exp Ther. 2004;310:1011–1019. doi: 10.1124/jpet.104.069112. [DOI] [PubMed] [Google Scholar]
- 96.Richter T, Schwab M, Eichelbaum M, Zanger UM. Inhibition of human CYP2B6 by N,N′,N″-triethylenethiophosphoramide is irreversible and mechanism-based. Biochem Pharmacol. 2005;69:517–524. doi: 10.1016/j.bcp.2004.10.008. [DOI] [PubMed] [Google Scholar]
- 97.Walsky RL, Obach RS. A comparison of 2-phenyl-2-(1-piperidinyl)propane (ppp), 1,1′,1″-phosphinothioylidynetrisaziridine (thioTEPA), clopidogrel, and ticlopidine as selective inactivators of human cytochrome P450 2B6. Drug Metab Dispos. 2007;35:2053–2059. doi: 10.1124/dmd.107.015883. [DOI] [PubMed] [Google Scholar]
- 98.Lu P, Schrag ML, Slaughter DE, Raab CE, Shou M, Rodrigues AD. Mechanism-based inhibition of human liver microsomal cytochrome P450 1A2 by zileuton, a 5-lipoxygenase inhibitor. Drug Metab Dispos. 2003;31:1352–1360. doi: 10.1124/dmd.31.11.1352. [DOI] [PubMed] [Google Scholar]
- 99.Bomsien S, Skopp G. An in vitro approach to potential methadone metabolic-inhibition interactions. Eur J Clin Pharmacol. 2007;63:821–827. doi: 10.1007/s00228-007-0327-z. [DOI] [PubMed] [Google Scholar]