

SUMMARY
Bone injury induces an inflammatory response that involves neutrophils, macrophages and other inflammatory cells. The recruitment of inflammatory cells to sites of injury occurs in response to specific signaling pathways. The CC chemokine receptor type 2 (CCR2) is crucial for recruiting macrophages, as well as regulating osteoclast function. In this study, we examined fracture healing in Ccr2−/− mice. We first demonstrated that the expression of Ccr2 transcripts and the filtration of macrophages into fracture calluses were most robust during the early phases of fracture healing. We then determined that the number of macrophages at the fracture site was significantly lower in Ccr2−/− mice compared with wild-type controls at 3 days after injury. As a result, impaired vascularization, decreased formation of callus, and delayed maturation of cartilage were observed at 7 days after injury in mutant mice. At day 14, Ccr2−/− mice had less bone in their calluses. At day 21, Ccr2−/− mice had larger calluses and more bone compared with wild-type mice, suggesting a delayed remodeling. In addition, we examined the effect of Ccr2 mutation on osteoclasts. We found that a lack of Ccr2 did not affect the number of osteoclasts within fracture calluses at 21 days after injury. However, Ccr2−/− osteoclasts exhibited a decreased ability to resorb bone compared with wild-type cells, which could contribute to the delayed remodeling of fracture calluses observed in Ccr2−/− mice. Collectively, these results indicate that a deficiency of Ccr2 reduces the infiltration of macrophages and impairs the function of osteoclasts, leading to delayed fracture healing.
INTRODUCTION
Unlike most vertebrate tissues that heal by forming scar tissue, bone heals through regeneration. Fracture repair occurs in three distinct but overlapping stages: the early inflammatory stage, the regenerative stage, and the later remodeling stage (Kalfas, 2001). Initially, inflammatory cells, including neutrophils and macrophages, infiltrate and debride the site of injury. Concomitantly, stem cells differentiate into chondrocytes and osteoblasts to form new cartilage and bone. The cartilage is subsequently replaced by bone through the process of endochondral ossification, and the callus is then remodeled until optimal biomechanical properties are achieved. Thus, fracture healing is a highly complex and coordinated process that involves interactions between many cell types. Our goal is to begin examining the interactions between various types of inflammatory cells that are involved in fracture repair, because a better understanding of this process may promote therapies that will improve bone healing.
The inflammatory stage may be crucial for successful fracture healing, because the chemical signals that are released during inflammation may initiate the cascade of events that culminates in skeletal repair. Several studies have shown that the use of anti-inflammatory or cytotoxic medications during the early stages of fracture healing may impair bone repair (Dimmen et al., 2008; Pountos et al., 2008; Simon and O’Connor, 2007). However, these studies have not established a direct relationship between inflammatory cells and bone fracture healing. In part, these agents may act on mesenchymal stem cells instead of inflammatory cells, to inhibit bone repair (Chang et al., 2007). Some anti-inflammatory agents, such as dexamethasaone and cyclooxygenase-2 (COX-2) inhibitors, affect the differentiation of mesenchymal stem cells into osteoblasts, and favor their differentiation into adipocytes (Ito et al., 2007; Kellinsalmi et al., 2007; Oshina et al., 2007). Thus, although the result is suppressed inflammation, the role that the inflammatory cells play remains unknown.
The recruitment of inflammatory cells to sites of injury is mediated by chemoattractive chemokines. There are four different subfamilies of chemokines (CC, CXC, CX3C and C) based on their biochemical, structural and functional properties. The CC chemokine CCL2 and its receptor, CCR2, are responsible for monocyte trafficking in the body. In mice that lack the Ccr2 gene, the recruitment of macrophages to sites of injury is impaired (Kuziel et al., 1997; Ma et al., 2002; Schober et al., 2004). CCR2/CCL2 signaling controls the movement of monocytes from the bone marrow into the bloodstream and from the circulatory system into sites of inflammation after injury (Tsou et al., 2007). CCR2 is also involved in osteoclast differentiation. Ccr2 mutant mice are osteopetrotic, and the reduced number and function of osteoclasts protects these mice from ovariectomy-induced osteoporosis (Binder et al., 2009). Therefore, we examined fracture healing in Ccr2−/− mutant mice to assess the role of macrophages and osteoclasts during bone fracture healing.
RESULTS
Recruitment of macrophages to the fracture site during early bone healing
To begin our analysis, we examined the expression of CCR2, its ligand CCL2, and other important chemoattractants during early fracture healing. These chemoattractants include CCL7 [chemokine (CC motif) ligand 7, a chemoattractant for monocytes and macrophages], CCL8 [chemokine (CC motif) ligand 8, a chemoattractant for many different inflammatory cells] and MSR1 (macrophage scavenger receptor 1, which is expressed on macrophages and mediates endocytosis). The expression of CCL2, CCR2, CCL7 and MSR1 are closely related to the recruitment and function of macrophages. By contrast, CCL8 is chemotactic for different types of inflammatory cells. Real-time (RT)-PCR demonstrated that all of these chemoattractants were expressed at low levels in uninjured mouse tibiae. Their expression was significantly increased at 2 days after bone injury. At 7 days after injury, the expression of all of these chemoattractants except CCL8 returned to levels close to normal (Fig. 1A).
Fig. 1.

Mobilization of macrophages during normal fracture healing. (A) RT-PCR assay of the expression levels of molecules related to macrophage recruitment or endocytosis. CCL2, CCL7 and CCL8 are chemokine (CC motif) ligand 2, 7 and 8, respectively. CCR2 is CC chemokine receptor type 2. MSR1 is macrophage scavenger receptor 1. (B,C) In normal unfractured tibiae, macrophages are detected in subcutaneous tissues. Macrophages are stained brown after immunohistochemistry using anti-F4/80 antibody. (D) At 3 days after fracture, abundant macrophages are present in granulation tissues (E), between injured muscles (F), and in the region of periosteal reaction (G). (H) At 7 days after fracture, macrophages are observed in fibrous callus (I) and between newly formed trabecular bone (J), but not in cartilage (K). (L) At day 14, macrophages are present at the periphery of fracture callus (M). A small number of macrophages were also detected at the front of endochondral ossification (N, arrow) and in the new bone (O, arrow). (P) At day 21, macrophages are observed at the periphery of callus (Q). (R)Macrophages were also detected lining trabecular bone in the callus (arrows). The dashed lines in B, D, H, L and P show fracture ends. The dashed line in G shows cortical bone. Bars, 1 mm (B,D,H,L,P); 50 μm (C,E–G,I–K,M–O,Q,R).
We used immunohistochemistry to visualize macrophages in the fracture site. Immunostaining with anti-F4/80 antibody, a panmacrophage marker, illustrated that some macrophages were present in the uninjured hindlimbs (Fig. 1B,C). At 3 days after fracture, granulation tissue formed at the fracture site and there were a large number of F4/80-positive macrophages within the callus (Fig. 1D,E), in the injured muscles (Fig. 1F), and in the region of periosteal reaction (Fig. 1G). At 7 days, newly formed cartilage and bone were present in the fracture callus. Macrophages were observed in fibrous callus tissues (Fig. 1H,I) and in a portion of the newly formed bone (Fig. 1J). However, these cells were not detected within newly formed cartilage (Fig. 1K). At 14 days, endochondral ossification was robust. Macrophages were mainly located at the periphery of the fracture callus (Fig. 1L,M). A small number of F4/80-positive cells were observed at the front of endochondral ossification (Fig. 1N) and lining trabecular bone (Fig. 1O). At 21 days, all fractures healed by bony bridging. Macrophages were present in the periphery of the fracture callus (Fig. 1P,Q). Some cells lining the trabecular bone also appeared to stain positively for F4/80 (Fig. 1R).
Recruitment of macrophages to the fracture site is altered in Ccr2−/− mice
Since the recruitment of macrophages is most robust during the early stages of fracture healing, we quantified the number of F4/80-positive cells at the fracture site in wild-type and Ccr2−/− mice at 3 days after injury. A large number of F4/80-positive cells were present in the callus of wild-type mice at this time (Fig. 2A,B,I). By contrast, only a few F4/80-positive cells were detected at the fracture site in Ccr2−/− mice (P<0.01) (Fig. 2E,F,I). These observations were confirmed using another macrophage marker, Mac-3. Much less Mac-3 immunostaining was detected in Ccr2-null mice compared with wild-type animals (supplementary material Fig. S1). In addition, the number of neutrophils was also quantified. A similar number of neutrophils were observed at the fracture site in both wild-type and mutant mice (P=0.27) (Fig. 2C,D,G–I). Thus, a deficiency of Ccr2 impairs the recruitment of macrophages but not neutrophils after bone injury.
Fig. 2.

Quantification of macrophages and neutrophils at the fracture sites at 3 days after injury. Macrophages were detected by using the anti-F4/80 antibody. Neutrophils were detected with an MCA771G antibody. Macrophages (A,B) and neutrophils (C,D) are abundant at the fracture site in wild-type mice (WT). (E,F) In Ccr2−/− mice, very few macrophages are present at the fracture site. (G,H) Abundant neutrophils are detected in mutant mice. (I) Further quantitative analysis confirms that the number of macrophages, but not neutrophils, is significantly lower in mutant mice than wild-type mice. Dashed lines in A, C, E and G show fracture ends. *P<0.01. Bars, 1 mm (A,C,E,G); 50 μm (B,D,F,H).
Loss of Ccr2−/− delays fracture healing
To assess the effect that the absence of Ccr2 would have on bone repair, we examined fracture healing in Ccr2−/− mice. We compared the size and composition of the callus in control mice with Ccr2−/− mice at various times after injury, and determined that the healing process in the Ccr2−/− mice was delayed compared with wild-type mice (Fig. 3). At day 7, the callus in the Ccr2−/− mice was significantly smaller than that in wild-type mice (P<0.05), but we did not detect any difference in the total volume of new bone or cartilage in the callus. At this time, the majority of bone is forming by intramembranous ossification in the periosteum or marrow cavity adjacent to the fracture. Since we observed no difference in the amount of bone at this time point, intramembranous ossification does not appear to be affected by the absence of Ccr2. In situ hybridization revealed that cartilage maturation was delayed in Ccr2−/− mice. Compared with control mice, less of the cartilage in the callus of the mutant mice appeared to be comprised of chondrocytes that had matured and that were expressing collagen type 10 (Fig. 4, and supplementary material Fig. S2). At day 14, there was no difference in callus size, but there was significantly less new bone in Ccr2−/− mice than in the wild-type mice (P=0.01), which may have resulted from delayed maturation of cartilage and endochondral ossification. At day 21, both callus size and volume of new bone remained large in the Ccr2−/− mice, but were significantly decreased in the wild-type mice (P=0.016 and P=0.03, respectively). There was no cartilage left in mutant and control animals at this time point. These results indicate that callus formation and callus remodeling were delayed in Ccr2−/− mice compared with wild-type mice.
Fig. 3.

Histomorphometric analysis of fracture callus in Ccr2−/− mice. (A) The callus size is smaller at day 7, but larger at day 21, in Ccr2−/− mice compared with wild-type mice. (B) The volume of new bone is smaller at day 14, but larger at day 21, in Ccr2−/− mice compared with wild-type mice. (C) There is no difference in the volume of new cartilage between these two groups at all time points. Data in graphs are mean ± standard deviation (S.D.). *P<0.05.
Fig. 4.

Delayed chondrocyte maturation in Ccr2−/− mice. (A,B) In situ hybridization shows that Col2 (A) and Col10 (B) transcripts are present in the fracture callus of wild-type mice at day 7. (C,D) In Ccr2−/− mice, abundant Col2 expression is observed at this time (C), but Col10 expression is greatly reduced compared with controls (D). Bar, 1 mm.
Vascular response to fracture is altered in Ccr2−/− mice
Since angiogenesis plays an essential role during fracture healing, we examined functional parameters of the vascular system in the callus at day 7 after fracture. The length density of blood vessels in the callus was not significantly different between Ccr2−/− mice and wild-type mice. However, the surface density of blood vessels in the callus was significantly lower in the Ccr2−/− mice (Fig. 5). Thus, the lower surface density suggests that there is an impaired angiogenic response in the mutant animals.
Fig. 5.
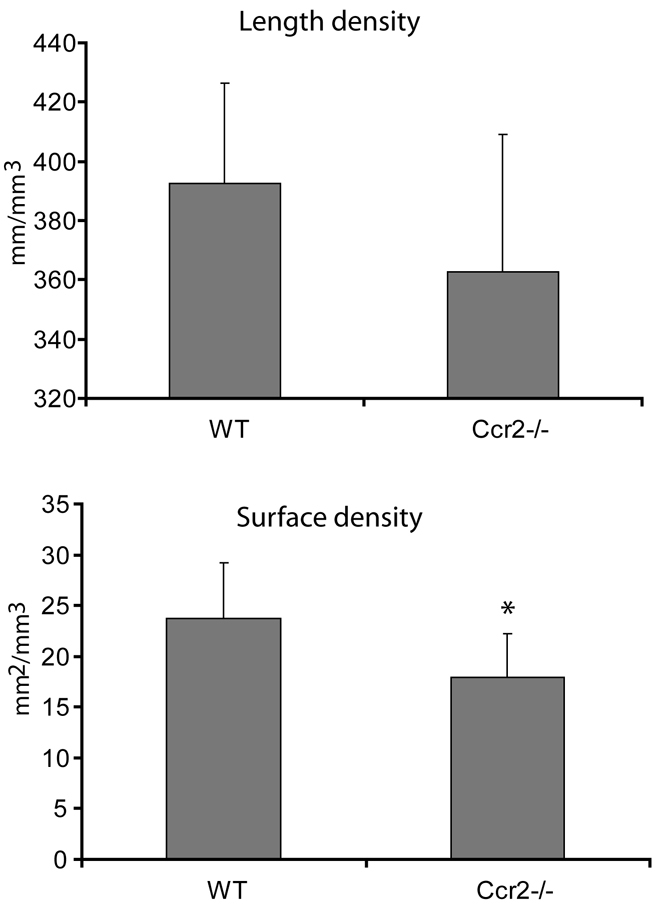
Quantification of blood vessels in the callus at day 7 after fracture. There is no difference in the length density of blood vessels between Ccr2−/− mice and wild-type mice. The surface density of blood vessels in the fracture callus is significantly lower in Ccr2−/− mice than that in the wild-type mice. Data in graphs are mean ± S.D. *P<0.05.
Function of osteoclasts is impaired in Ccr2−/− mice
We quantified osteoclasts at the fracture site at 21 days after injury and found that, based on tartrate-resistant acidic phosphatase (TRAP) staining, Ccr2−/− mice had similar numbers of osteoclasts when compared with wild-type mice (Fig. 6A). To test the possibility that Ccr2 deficiency may affect the function of osteoclasts, in vitro differentiation and bone resorption assays were performed on wild-type and mutant osteoclasts. We found that the differentiation of osteoclasts was not significantly affected by deficiency of Ccr2. As shown in Fig. 6B, bone marrow monocytes/macrophages were collected from wild-type or mutant mice and gave rise to similar numbers of osteoclasts when they were cultured in a medium with RANKL (receptor activator of nuclear factor-kappa B ligand) and macrophage-colony stimulating factor (M-CSF). Further functional analysis demonstrated that that the capability of bone resorption was significantly reduced in Ccr2−/− mutant osteoclasts compared with wild-type osteoclasts. Ccr2−/− mutant osteoclasts created smaller resorption pits and a decreased resorption area (Fig. 6C–F).
Fig. 6.

Osteoclast number and function. (A) There is no significant difference in the number of osteoclasts in fracture calluses of Ccr2−/− mice and wild-type mice at day 21, and (B) the differentiation of osteoclasts in vitro is not altered in the mutants. (C,D)Micrographs of the pits created in discs by wild-type (C) and mutant (D) osteoclasts. (E,F) Ccr2−/− osteoclasts create smaller numbers of larger pits (E) and resorb less surface area (F) in vitro than wild-type osteoclasts. MNC refers to multinucleated cells. *P<0.05, **P<0.01.
DISCUSSION
Injuries in adult animals elicit complex inflammatory responses. Inflammation is a defense mechanism by which the host eradicates pathogens and stimulates the healing process (Contreras-Shannon et al., 2007). Inflammatory cells are a rich source of signaling molecules that coordinate the early phases of healing (reviewed in Lloberas and Celada, 2002; Werner and Grose, 2003). An example that illustrates the importance of inflammation during bone repair is that suppression of the inflammatory response by non-steroidal anti-inflammatory drugs (NSAIDs) causes delays in fracture healing (Altman et al., 1995; Engesaeter et al., 1992; Gerstenfeld et al., 2003; Sudmann et al., 1979). Therefore, by defining the inflammatory response to fracture, a more thorough understanding of the role that inflammation has during repair can be gained. Here, we used Ccr2−/− mutant mice to assess the role of macrophages and osteoclasts during bone fracture healing, and determined that macrophages regulate the early phases of fracture healing, possibly by directing the differentiation of chondrocytes and regulating vascularization. Interestingly, we found that these mice have osteoclasts with a reduced function, which delays the remodeling phase of fracture healing.
Role of CCR2 and macrophages during early fracture healing
Results from this study demonstrate that a lack of Ccr2 affects early fracture healing. At 3 days after fracture, Ccr2−/− mice had significantly fewer macrophages at the fracture site. At 7 days after injury, these mice exhibited smaller calluses and delayed cartilage maturation compared with the wild-type animals. Ccr2 deficiency may either delay the differentiation of chondrocytes from progenitor cells and/or delay the maturation of chondrocytes. Our RT-PCR data demonstrate that molecules related to macrophage recruitment were expressed highly at day 2, but not at day 7. In addition, immunohistochemical analysis of the fracture callus of wild-type mice revealed abundant macrophages at 3 days after injury, but fewer at day 7 after injury, and macrophages were absent from cartilage at day 7. During this time period, osteoprogenitor cells are beginning to differentiate and form the skeletal components of the fracture callus. Since we observed differences in the composition of the callus in Ccr2-null mice, we suspect that macrophages may be important during the early phases of cell differentiation after injury. We also observed a delay in endochondral ossification. Macrophages may stimulate the initial differentiation of progenitor cells, which leads to enhanced maturation at later time points. Thus, delayed cartilage maturation in Ccr2-null mice could result from inadequate stimulation of chondroprogenitor cell differentiation owing to a lack of macrophage infiltration. By contrast, Ccr2 deficiency did not appear to affect intramembranous ossification. At 7 days after fracture, Ccr2−/− mice exhibited a similar amount of new bone in the callus as the wild-type controls. In a separate study, we examined the effect of Ccr2 inactivation on the healing of stabilized fractures, which normally heal through direct intramembranous ossification. We did not detect significant differences in bone formation in mutant mice compared with wild-type animals (data not shown).
Further exploration is required to determine the exact role of macrophages during early fracture healing. However, a number of intriguing possibilities exist. First, macrophages are a major source of cytokines and other factors that are important for tissue repair. For example, macrophages could produce bone morphogenetic protein-2 (BMP-2) (Champagne et al., 2002), which stimulates the formation of skeletal tissues. Macrophages also produce interleukin (IL)-6, which is a crucial factor in the regulation of bone marrow mesenchymal stem cells (Rodriguez Mdel et al., 2004). Second, the inflammatory response involves the coordinated interaction between a variety of distinct cell lineages, including neutrophils and macrophages. Our results demonstrate that the recruitment of macrophages to a fractured bone site was significantly decreased in Ccr2−/− mice, whereas the recruitment of neutrophils to the injured site was not affected. Thus, in Ccr2 mutant mice, the composition of inflammatory cells at the fracture site was altered, and this altered ratio may directly affect the healing process. Lastly, macrophages could affect bone healing by altering vascular repair after fracture. We found that, compared with wild-type animals, the vasculature within fracture callus in Ccr2−/− mice had a reduced surface area for nutrient exchange. This observation is consistent with other reports demonstrating that impaired macrophage recruitment in Ccr2−/− mice leads to a reduction in tissue vascular endothelial growth factor (VEGF) levels and a delay in capillary formation (Capoccia et al., 2008; Ochoa et al., 2007). Macrophages are key effectors of angiogenesis that contribute to tumor angiogenesis in several carcinomas (Mazibrada et al., 2008; Ohta et al., 2002), and tumor-associated macrophages have been shown to express VEGF (Gonzalez et al., 2007).
CCR2 regulates osteoclast activity
In addition to delayed formation of callus and cartilage in the fractures of Ccr2 mutant animals, we also observed a delay in callus remodeling during the later stages of fracture healing. This delay in callus remodeling in Ccr2−/− mice may have been the consequence of a delay in the early stage of the repair process that occurs in these animals. Alternatively, the delay could have been a direct result of altered osteoclast function in these animals, which we then confirmed with our in vitro assay of osteoclast function. We found that Ccr2−/− osteoclasts had reduced bone resorption capacity compared with wild-type cells. Similar findings have been reported by Binder et al. (Binder et al., 2009), who showed that Ccr2 mutant mice are osteopetrotic owing to the decreased function and number of osteoclasts in bone (Binder et al., 2009). In osteopetrotic animals, remodeling of the fracture callus is delayed (Marks and Schmidt, 1978; Schmidt et al., 1977), supporting our conclusion that defective osteoclasts lead to a delay in callus remodeling in Ccr2-null mice. These data indicate that the CCR2 signaling axis is required for adequate osteoclast function.
In this study, we did not detect differences in the number of osteoclasts in the fracture callus at day 21 in mutant and wild-type mice, which is inconsistent with the findings of Binder et al. who showed that Ccr2−/− mice have fewer osteoclasts (Binder et al., 2009). Binder et al. quantified mature osteoclasts with three or more nuclei, whereas we counted all TRAP-positive cells, which could include immature osteoclasts. Also, we do not know whether the osteoclasts that remodel the fracture callus are of the same population as the osteoclasts in normal bone that regulate homeostasis. Nonetheless, our results agree with those of Binder et al., in that they show defective bone remodeling in Ccr2 mutant animals.
In summary, our results indicate that CCR2 plays an important role during fracture repair. This molecule regulates macrophage recruitment and participates in callus and cartilage formation during early fracture healing. During the late stages of fracture repair, CCR2 is involved in regulating osteoclast function. Interestingly, fracture healing was only moderately affected in Ccr2−/− mice, suggesting that other mechanisms may compensate for the reduction in macrophages at the fracture site. Additionally, our data suggest that macrophages may enhance the healing process, but that these cells are not absolutely required for healing. Further experiments are required to determine these compensating mechanisms, as well as the exact role of macrophages during fracture healing.
METHODS
Animals
All procedures were approved by the Institutional Animal Care and Use Committee at University of California at San Francisco. Ccr2−/− mice were purchased from the Jackson Laboratory (Bar Harbor, Maine) and kept in a specific pathogen-free barrier facility. The production of these mice has been described previously (Boring et al., 1997). Male, 10-week-old mice were used in this study. Age- and gender-matched, congenic C57BL/6L mice (Jackson Laboratory, Bar Harbor, Maine) were used as wild-type controls.
Non-stabilized fracture
A guillotine designed to provide reproducible blunt trauma was used to create a tibial fracture by three-point bending in anesthetized mice (2% avertin) (Bonnarens and Einhorn, 1984). The fractures were not stabilized and the mice were allowed to be fully active. In this mechanical environment, fractures heal through robust endochondral ossification (Thompson et al., 2002), as well as through a small amount of intramembranous ossification. An analgesic (1% buprenex), was injected subcutaneously (100 μl per injection), twice a day, for 2 days.
Real-time PCR
Non-stabilized tibia fractures were created as described above. Animals were sacrificed at 2 days (n=4) and 7 days (n=4) after injury, and fractured tibiae were collected. Briefly, we removed the skin from all specimens, and then we dissected the fractured bone and all adjacent tissues that were located 0.5 cm distal and proximal to the edge of the callus/hematoma. A similar region of the non-fractured tibia was used as a control (n=4). RNA was extracted from these tissues and the quality of RNA was confirmed using an RNA 6000 Nano Chip kit (Agilent, Santa Clara, CA). The primers for CCL2, CCL7, CCL8, CCR2 and MSR1 were purchased from SABiosciences (Frederick, MD). cDNA synthesis and real-time PCR were performed at the Genome Analysis Core Facility at UCSF following their standard protocols. L19, a nonregulated ribosomal housekeeping gene, was used as the internal control.
Histology and histomorphometry
Mice were sacrificed at 3, 7, 14 and 21 days after fracture. Tibiae and surrounding callus tissues were collected and fixed in 4% paraformaldehyde at 4°C for 24 hours. The samples were decalcified in 19% EDTA at 4°C for 10–14 days and then cryo-embedded in OCT compound (Sakura Finetechnical Co., Tokyo, Japan). Consecutive frozen sections (20 μm or 10 μm) were cut through the entire callus and stored at −20°C.
Serial sections (10 μm) through the entire callus that were 300 μm apart were stained with modified Milligan’s trichrome to visualize the callus and the new bone in the callus. Safranin O staining was performed to visualize the cartilage in the callus. The sections were digitized using a Leica DMRB microscope and Optronics camera, and were then analyzed using Adobe Photoshop, as described (Lu et al., 2005). Briefly, the number of pixels comprising each tissue component of the callus was used to estimate the area. The number of pixels/mm2 was determined using a 1-mm scale bar. The total areas of callus, new bone or new cartilage were determined by dividing the number of pixels by the number of pixels/mm2. The volume of the callus, new bone or new cartilage was calculated using the equation for a conical frustum:
Ai and Ai+1 are the area of callus, cartilage or bone in the sequential sections; h is the distance between sections (300 μm), and n is the total number of sections that were analyzed for each specimen.
Detection of macrophages and neutrophils at the fracture site
Macrophages and neutrophils in the callus at day 3 after fracture were visualized by immunostaining using macrophage-specific (F4/80 or Mac-3; eBioscience, San Diego, CA) and neutrophil-specific (MCA771G; Serotec, Raleigh, NC) antibodies. Briefly, sections were incubated with primary antibodies (1:100 dilution for F4/80 and MCA771G; 1:50 dilution for Mac-3) at 4°C overnight or at room temperature for 1 hour. Sections were then incubated with a second antibody. The immune complex was visualized with diaminobenzidine (DAB) and sections were counterstained with hematoxylin. No antigen retrieval was required. Negative controls were either treated with isotype control antibodies or not incubated with any primary antibodies. For each sample, three to five sections were systematically and randomly selected for immunostaining. The cellular densities of macrophages and neutrophils in the fracture callus were estimated using an Olympus CAST system (Olympus, Center Valley, PA) and Visiopharm software (Visiopharm, Hørsholm, Denmark). On each section, the fracture callus was outlined and fields covering 20% of the outlined callus were randomly selected. A counting frame probe was applied to quantify the number of macrophages and neutrophils within these fields and the area of each field. Values were expressed as the number of macrophages or neutrophils per mm2 of callus tissue.
In situ hybridization
Radioactive in situ hybridization was performed on sections (10-μm thick) to visualize the expression patterns of collagen type 2 (Col2) and collagen type 10 (Col10) in the callus at day 7 after fracture. Radiolabeled sense and anti-sense probes were generated, and hybridization was performed, as described (Thompson et al., 2002). Slides were counterstained with 2 mg/ml of Hoechst dye. Dark-field microscopy was used to image the exposed silver grains, and superimposed on the fluorescent image. Color was applied linearly to the dark-field image in Adobe Photoshop.
Quantification of blood vessels in fracture callus
To examine angiogenesis in the callus at day 7 after fracture, we performed platelet endothelial cell adhesion molecule (PECAM) staining using an anti-PECAM-1 antibody (Pharmingen, San Diego, CA) on slides prepared from sections through the callus that were 600 μm apart. The length density (the length of blood vessels per unit volume of the reference space) and the surface density (the area of the outer surface of blood vessels per unit volume of the reference space) of the blood vessels within the fracture callus were analyzed using an Olympus CAST system, as described previously (Lu et al., 2008).
TRAP staining and osteoclast count
TRAP staining was performed on serial sections (20 μm) through the callus that were 600 μm apart. The number of osteoclasts in the callus at day 21 after fracture was defined as the number of TRAP-positive cells on the surface of the bone using stereology, as described above. Values were expressed as the number of osteoclasts per mm2 of callus tissue.
Osteoclast differentiation and function
Osteoclasts were generated from bone marrow monocyte/macrophage (BMM) precursor cells, as described previously (Mocsai et al., 2004). Briefly, cells were isolated from femurs/tibias by flushing with PBS using a 25-gauge needle. The cells were then washed and treated with red blood cell lysis buffer (0.16 M NH4Cl, 0.17 M Tris, pH 7.65) for 5 minutes at room temperature. Cells were cultured in complete α-MEM (UCSF Cell Culture Facility) supplemented with 10% fetal bovine serum, 1% glutaminepenicillin-streptomycin, and 10–20 ng/ml of M-CSF (Sigma, St Louis, MO). After 2 days of M-CSF stimulation, non-adherent precursors were transferred to a new plate and cultured in complete α-MEM with 25 ng/ml RANKL (Peprotech, Rocky Hill, NJ) and 20 ng/ml M-CSF for an additional 4–5 days, with the addition of fresh media every 3 days. After 3–5 days in culture, cells were fixed with 3.7% formaldehyde in PBS for 10 minutes. Plates were then washed twice in PBS. Cells were stained for TRAP using a commercial kit (product 387-A; Sigma-Aldrich) according to manufacturer’s protocol. Multinucleated (>2 nuclei), TRAP-positive cells were then counted by light microscopy. To assess osteoclast function, macrophages were plated on BD BioCoat osteologic discs (BD Biosciences) at a density of 100,000 cells/well and were then treated with RANKL 25 ng/ml and M-CSF 20 ng/ml for 5–10 days. The media were changed every 3 days. BD BioCoat osteologic discs were treated with 20% bleach and agitated for 5 minutes to remove adherent cells, followed by five washes with dH2O (distilled water), and air-dried. The surface area with active bone resorption was visualized under an Olympus microscope BX51 and measured by using BioQuant OSTEO II software (BioQuant Image Analysis Corporation, Nashville, TN).
Statistical analysis
Data are presented as the mean plus or minus one standard deviation. Two-way analysis of variance (ANOVA) was performed to test the main effects of Ccr2 gene knockout and healing time, and the interaction between Ccr2 gene knockout and healing time on callus formation. Student’s t-test was performed to assess the differences in outcomes at the same time point. P<0.05 was considered significant.
Supplementary Material
Acknowledgments
This work was funded by the NIH ( R01-5R01AR053645 to T.M.). Z.X. was supported by a fellowship from Synthes and the Hellman Family Foundation. We thank the members of the Skeletal Regeneration laboratory in the Orthopaedic Trauma Institute at San Francisco General Hospital, especially Dr Xiaodong Wang, for discussions of this work and technical help. Deposited in PMC for release after 12 months.
Footnotes
COMPETING INTERESTS
The authors declare no competing financial interests.
AUTHOR CONTRIBUTIONS
Z.X. and R.S.M. conceived and designed the experiments; Z.X., C.L., D.H., Y.-y.Y., X.W. and Y.W. performed the experiments; Z.X., C.L., C.C., M.N., T.M. and R.S.M. analyzed the data and wrote the paper.
SUPPLEMENTARY MATERIAL
Supplementary material for this article is available at http://dmm.biologists.org/lookup/suppl/doi:10.1242/dmm.003186/-/DC1
REFERENCES
- Altman RD, Latta LL, Keer R, Renfree K, Hornicek FJ, Banovac K. (1995). Effect of nonsteroidal antiinflammatory drugs on fracture healing: a laboratory study in rats. J Orthop Trauma 9, 392–400 [DOI] [PubMed] [Google Scholar]
- Binder NB, Niederreiter B, Hoffmann O, Stange R, Pap T, Stulnig TM, Mack M, Erben RG, Smolen JS, Redlich K. (2009). Estrogen-dependent and C-C chemokine receptor-2-dependent pathways determine osteoclast behavior in osteoporosis. Nat Med. 15, 417–424 [DOI] [PubMed] [Google Scholar]
- Bonnarens F, Einhorn TA. (1984). Production of a standard closed fracture in laboratory animal bone. J Orthop Res. 2, 97–101 [DOI] [PubMed] [Google Scholar]
- Boring L, Gosling J, Chensue SW, Kunkel SL, Farese RV, Jr, Broxmeyer HE, Charo IF. (1997). Impaired monocyte migration and reduced type 1 (Th1) cytokine responses in C-C chemokine receptor 2 knockout mice. J Clin Invest. 100, 2552–2261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capoccia BJ, Gregory AD, Link DC. (2008). Recruitment of the inflammatory subset of monocytes to sites of ischemia induces angiogenesis in a monocyte chemoattractant protein-1-dependent fashion. J Leukoc Biol. 84, 760–768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Champagne CM, Takebe J, Offenbacher S, Cooper LF. (2002). Macrophage cell lines produce osteoinductive signals that include bone morphogenetic protein-2. Bone 30, 26–31 [DOI] [PubMed] [Google Scholar]
- Chang JK, Li CJ, Wu SC, Yeh CH, Chen CH, Fu YC, Wang GJ, Ho ML. (2007). Effects of anti-inflammatory drugs on proliferation, cytotoxicity and osteogenesis in bone marrow mesenchymal stem cells. Biochem Pharmacol. 74, 1371–1382 [DOI] [PubMed] [Google Scholar]
- Contreras-Shannon V, Ochoa O, Reyes-Reyna SM, Sun D, Michalek JE, Kuziel WA, McManus LM, Shireman PK. (2007). Fat accumulation with altered inflammation and regeneration in skeletal muscle of CCR2−/− mice following ischemic injury. Am J Physiol Cell Physiol. 292, C953–C967 [DOI] [PubMed] [Google Scholar]
- Dimmen S, Nordsletten L, Engebretsen L, Steen H, Madsen JE. (2008). Negative effect of parecoxib on bone mineral during fracture healing in rats. Acta Orthop. 79, 438–444 [DOI] [PubMed] [Google Scholar]
- Engesaeter LB, Sudmann B, Sudmann E. (1992). Fracture healing in rats inhibited by locally administered indomethacin. Acta Orthop Scand. 63, 330–333 [DOI] [PubMed] [Google Scholar]
- Gerstenfeld LC, Thiede M, Seibert K, Mielke C, Phippard D, Svagr B, Cullinane D, Einhorn TA. (2003). Differential inhibition of fracture healing by non-selective and cyclooxygenase-2 selective non-steroidal anti-inflammatory drugs. J Orthop Res. 21, 670–675 [DOI] [PubMed] [Google Scholar]
- Gonzalez FJ, Vicioso L, Alvarez M, Sevilla I, Marques E, Gallego E, Alonso L, Matilla A, Alba E. (2007). Association between VEGF expression in tumour-associated macrophages and elevated serum VEGF levels in primary colorectal cancer patients. Cancer Biomark. 3, 325–333 [DOI] [PubMed] [Google Scholar]
- Ito S, Suzuki N, Kato S, Takahashi T, Takagi M. (2007). Glucocorticoids induce the differentiation of a mesenchymal progenitor cell line, ROB-C26 into adipocytes and osteoblasts, but fail to induce terminal osteoblast differentiation. Bone 40, 84–92 [DOI] [PubMed] [Google Scholar]
- Kalfas IH. (2001). Principles of bone healing. Neurosurg Focus 10, E1. [DOI] [PubMed] [Google Scholar]
- Kellinsalmi M, Parikka V, Risteli J, Hentunen T, Leskela HV, Lehtonen S, Selander K, Vaananen K, Lehenkari P. (2007). Inhibition of cyclooxygenase-2 down-regulates osteoclast and osteoblast differentiation and favours adipocyte formation in vitro. Eur J Pharmacol. 572, 102–110 [DOI] [PubMed] [Google Scholar]
- Kuziel WA, Morgan SJ, Dawson TC, Griffin S, Smithies O, Ley K, Maeda N. (1997). Severe reduction in leukocyte adhesion and monocyte extravasation in mice deficient in CC chemokine receptor 2. Proc Natl Acad Sci USA 94, 12053–12058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloberas J, Celada A. (2002). Effect of aging on macrophage function. Exp Gerontol. 37, 1325–1331 [DOI] [PubMed] [Google Scholar]
- Lu C, Miclau T, Hu D, Hansen E, Tsui K, Puttlitz C, Marcucio RS. (2005). Cellular basis for age-related changes in fracture repair. J Orthop Res. 23, 1300–1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C, Hansen E, Sapozhnikova A, Hu D, Miclau T, Marcucio RS. (2008). Effect of age on vascularization during fracture repair. J Orthop Res. 26, 1384–1389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma M, Wei T, Boring L, Charo IF, Ransohoff RM, Jakeman LB. (2002). Monocyte recruitment and myelin removal are delayed following spinal cord injury in mice with CCR2 chemokine receptor deletion. J Neurosci Res. 68, 691–702 [DOI] [PubMed] [Google Scholar]
- Marks SC, Jr, Schmidt CJ. (1978). Bone remodeling as an expression of altered phenotype: studies of fracture healing in untreated and cured osteopetrotic rats. Clin Orthop Relat Res. 137, 259–264 [PubMed] [Google Scholar]
- Mazibrada J, Ritta M, Mondini M, De Andrea M, Azzimonti B, Borgogna C, Ciotti M, Orlando A, Surico N, Chiusa L, et al. (2008). Interaction between inflammation and angiogenesis during different stages of cervical carcinogenesis. Gynecol Oncol. 108, 112–120 [DOI] [PubMed] [Google Scholar]
- Mocsai A, Humphrey MB, Van Ziffle JA, Hu Y, Burghardt A, Spusta SC, Majumdar S, Lanier LL, Lowell CA, Nakamura MC. (2004). The immunomodulatory adapter proteins DAP12 and Fc receptor gamma-chain (FcRgamma) regulate development of functional osteoclasts through the Syk tyrosine kinase. Proc Natl Acad Sci USA 101, 6158–6163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochoa O, Sun D, Reyes-Reyna SM, Waite LL, Michalek JE, McManus LM, Shireman PK. (2007). Delayed angiogenesis and VEGF production in CCR2−/− mice during impaired skeletal muscle regeneration. Am J Physiol Regul Integr Comp Physiol. 293, R651–R661 [DOI] [PubMed] [Google Scholar]
- Ohta M, Kitadai Y, Tanaka S, Yoshihara M, Yasui W, Mukaida N, Haruma K, Chayama K. (2002). Monocyte chemoattractant protein-1 expression correlates with macrophage infiltration and tumor vascularity in human esophageal squamous cell carcinomas. Int J Cancer 102, 220–224 [DOI] [PubMed] [Google Scholar]
- Oshina H, Sotome S, Yoshii T, Torigoe I, Sugata Y, Maehara H, Marukawa E, Omura K, Shinomiya K. (2007). Effects of continuous dexamethasone treatment on differentiation capabilities of bone marrow-derived mesenchymal cells. Bone 41, 575–583 [DOI] [PubMed] [Google Scholar]
- Pountos I, Georgouli T, Blokhuis TJ, Pape HC, Giannoudis PV. (2008). Pharmacological agents and impairment of fracture healing: what is the evidence? Injury 39, 384–394 [DOI] [PubMed] [Google Scholar]
- Rodriguez Mdel C, Bernad A, Aracil M. (2004). Interleukin-6 deficiency affects bone marrow stromal precursors, resulting in defective hematopoietic support. Blood 103, 3349–3354 [DOI] [PubMed] [Google Scholar]
- Schmidt CJ, Marks SC, Jr, Jordan CA, Hawes LE. (1977). A radiographic and histologic study of fracture healing in osteopetrotic rats. Radiology 122, 517–519 [DOI] [PubMed] [Google Scholar]
- Schober A, Zernecke A, Liehn EA, von Hundelshausen P, Knarren S, Kuziel WA, Weber C. (2004). Crucial role of the CCL2/CCR2 axis in neointimal hyperplasia after arterial injury in hyperlipidemic mice involves early monocyte recruitment and CCL2 presentation on platelets. Circ Res. 95, 1125–1133 [DOI] [PubMed] [Google Scholar]
- Simon AM, O’Connor JP. (2007). Dose and time-dependent effects of cyclooxygenase-2 inhibition on fracture-healing. J Bone Joint Surg Am. 89, 500–511 [DOI] [PubMed] [Google Scholar]
- Sudmann E, Dregelid E, Bessesen A, Morland J. (1979). Inhibition of fracture healing by indomethacin in rats. Eur J Clin Invest. 9, 333–339 [DOI] [PubMed] [Google Scholar]
- Thompson Z, Miclau T, Hu D, Helms JA. (2002). A model for intramembranous ossification during fracture healing. J Orthop Res. 20, 1091–1098 [DOI] [PubMed] [Google Scholar]
- Tsou CL, Peters W, Si Y, Slaymaker S, Aslanian AM, Weisberg SP, Mack M, Charo IF. (2007). Critical roles for CCR2 and MCP-3 in monocyte mobilization from bone marrow and recruitment to inflammatory sites. J Clin Invest. 117, 902–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner S, Grose R. (2003). Regulation of wound healing by growth factors and cytokines. Physiol Rev. 83, 835–870 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}