

Abstract
L-Kyotorphin (L-KTP), an endogenous analgesic neuropeptide, is a substrate for aminopeptidases and a proton-coupled oligopeptide transporter, PEPT2. This study examined the CSF efflux, antinociceptive response, and hydrolysis kinetics in brain of L-KTP and its synthetic diastereomer D-kyotorphin (D-KTP) in wild-type and Pept2 null mice. CSF clearance of L-KTP was slower in Pept2 null mice than in wild-type animals, and this difference was reflected in greater L-KTP-induced analgesia in Pept2 null mice. Moreover, dose-response analyses showed that the ED50 of L-KTP in Pept2-deficient animals was one-fifth of the value observed in Pept2-competent animals (4 vs. 21 nmol for null vs. wild-type mice, respectively). In contrast, the ED50 of D-KTP was very similar between the two genotypes (9–10 nmol). Likewise, there was little difference between genotypes in slope factor or baseline effects of L-KTP and D-KTP. The enhanced antinociceptive response to L-KTP in Pept2 null mice could not be explained by differences in neuropeptide degradation since Vmax and Km values did not differ between genotypes. Our results demonstrate that PEPT2 can significantly impact the analgesic response to an endogenous neuropeptide by altering CSF (and presumably brain interstitial fluid) concentrations and that it may influence the disposition and response to exogenous peptide/mimetic substrates.
Keywords: aminopeptidase, antinociception, intracerebroventricular, kyotorphin, Pept2, transgenic mice
Amino acid-based neurotransmission is truncated by the transport of transmitter from the synaptic cleft. Thus, for example, glutamate is taken up by EAAT (excitatory amino acid transporter) 3, EAAT4 and EAAT5 in neurons and EAAT1 and EAAT2 in astrocytes (Tzingounis and Wadiche 2007). In contrast, it is widely held that peptide-based neurotransmission is truncated by enzymatic degradation rather than transport (Davis and Konings 1993). However, there is evidence for the expression of several peptide transporters in brain (Shen et al. 2004; Smith et al. 2004), although their role in regulating neuropeptide levels and neurotransmission is largely unknown.
Peptide transporter 2 (PEPT2) is a member of the proton-coupled oligopeptide transporter family that includes the peptide transporters (PEPT1 and PEPT2) and the peptide/histidine transporters (PHT1 and PHT2) (Daniel and Kottra 2004; Smith et al. 2004). PEPT2 is a high-affinity, low-capacity transporter abundantly expressed in apical membranes of renal proximal tubule and choroid plexus epithelia, in astrocytes (newborns) and neuronal cells (newborn and adults) of brain parenchyma, and in lung and mammary gland (Shen et al. 1999, 2004; Brandsch et al. 2008; Rubio-Aliaga and Daniel 2008). PEPT2 transports glycylsarcosine and cefadroxil from CSF into choroid plexus, thereby limiting the CNS exposure of these peptide/mimetic substrates (Ocheltree et al. 2005; Shen et al. 2007). Moreover, PEPT2 transport of 5-aminolevulinic acid significantly impacts the neurotoxicity of this peptidomimetic substrate (Hu et al. 2007). Since PEPT2 transports many small neuropeptides found in brain, this protein is believed to regulate neuropeptide homeostasis at the blood-CSF interface (Teuscher et al. 2001, 2004; Hu et al. 2005; Thakker et al. 2008).
L-Kyotorphin (L-Tyr-L-Arg; L-KTP) is a neuropeptide first isolated from bovine brain (Takagi et al. 1979a). L-KTP exhibits significant analgesic action by inducing Met-enkephalin release from nerve terminals (Ueda et al. 1987; Ueda and Inoue 2000). Although 4.2x more potent than Met-enkephalin, centrally-administered L-KTP is 5-6x less potent than D-kyotorphin (L-Tyr-D-Arg; D-KTP) (Takagi et al. 1979a, 1979b). To account for these differences, it was suggested that L-KTP is more susceptible to aminopeptidase degradation than D-KTP, thereby reducing its analgesic potency. In support of this contention, L-, but not D-KTP, was found to be rapidly hydrolyzed by enzymes in brain homogenates (Matsubayashi et al. 1984). Moreover, L-KTP hydrolysis was inhibited by the aminopeptidase inhibitor bestatin, which potentiates analgesia (Matsubayashi et al. 1984; Ueda et al. 1985). L-, but not D-KTP, is also a competitive inhibitor of the PEPT2-mediated uptake of glycylsarcosine in choroid plexus (Teuscher et al. 2001), and L-KTP has recently been shown to be a substrate of PEPT2 (Thakkar et al. 2008). Therefore, it is possible that the PEPT2-mediated clearance of L-KTP may also be a factor in reducing its analgesic effect relative to D-KTP.
With this in mind, we hypothesized that PEPT2 could influence the pharmacological activity of neuropeptides in brain by modulating their concentrations in CSF. To test this hypothesis, we studied the CSF efflux, antinociceptive response, and hydrolysis kinetics in brain of L-and D-KTP in wild-type and Pept2 null mice.
Materials and methods
Chemicals
[3H]L-KTP (560 mCi/mmol) was obtained from Moravek Biochemicals, Inc. (Brea, CA) and [14C]mannitol (53 mCi/mmol) from American Radiolabeled Chemicals, Inc. (St. Louis, MO). Radiochemical purity, as determined by HPLC, was > 98% for both compounds. Unlabeled L-KTP and D-KTP were obtained from Bachem Americas, Inc. (Torrance, CA), and glycylsarcosine (GlySar) and bestatin from Sigma-Aldrich Corp. (St. Louis, MO). All other chemicals were obtained from standard sources and were of highest quality available.
Animals
Wild-type (Pept2+/+) and null (Pept2−/−) mice were generated in-house (Shen et al. 2003) and used for the proposed studies. For all study designs, the mice were gender- and weight-matched (8 to 10 weeks of age), and congenic (> 99% C57BL/6 genetic background). The animals were kept in a temperature-controlled environment with a 12-hour light, 12-hour dark cycle and given ad libitum access to food and water. Animal studies were conducted in accordance with the guidelines from the National Institutes of Health for the care and use of animals, and were approved by the Institutional Animal Care and Use Committee.
CSF efflux studies
Animals were anesthetized with sodium pentobarbital (50 mg/kg ip) and their heads fixed in a stereotaxic apparatus. A 1.0 μl syringe (SGE 26 gauge-0.47 mm, 70 mm length, 8.0 mm OD; World Precision Instruments, Sarasota, FL) was then inserted into right lateral ventricle through a hole drilled in the skull (2.5 mm in depth, 1.2 mm lateral from midline, 0.4 mm caudal from bregma). A 5 nmol dose of [3H]L-KTP (1.0 μCi), dissolved in 1 μl of artificial CSF buffer, was given by intracerebroventricular (icv) injection over 30 sec and the needle kept in place until sampling. At designated times (e.g., 2, 5, 10, 15 min after the injection), a single sample of CSF (~5 μl) was withdrawn by cisternal puncture from each mouse. [14C]Mannitol (0.2 μCi), which is transported in the CNS by passive diffusion, was co-injected with radiolabeled L-KTP to assess adequacy and comparability of the injections. Inhibition studies were also performed with GlySar (200 nmol) and unlabeled L-KTP (200 nmol) in which CSF samples were obtained 15 min after the icv injection.
Dose-response studies
Mice were lightly anesthetized with isoflurane and administered a single icv injection L-KTP or D-KTP (administered over 30 sec and the needle kept in place for another 30 sec). The period of anesthesia was kept very short, just long enough to allow administration of the antinociceptive agent, so that the animals could recover rapidly (i.e., within a minute after removing the needle). The time of peak effect (tpeak) was determined by analyzing the time course of antinociception at a given dose. Thus, the responses were determined serially in each mouse at 2, 5, 10, 15, 20, 25, 30 and 40 min after the needle had been removed. Mice were then studied at L-KTP and D-KTP doses of 0.1, 1, 2.5, 5, 10, 20, 30, 50, 100 and 200 nmol, and the antinociceptive response measured at 5 min (i.e., the tpeak). Vehicle control doses (i.e., artificial CSF buffer and no dipeptide) were also administered and the responses measured.
Assessment of antinociceptive response
The hot-plate test, a measure of supraspinal antinociception, was used to determine the analgesic activity of L-KTP and D-KTP in wild-type and Pept2 null mice. The animals were placed individually on a Columbus Instruments Hot-Plate Analgesia Meter (Columbus, OH) that was thermostatically set at 55 ± 0.1°C. Latency was then measured as the interval between placement of each mouse on the hot plate and the onset of hind paw licking or shaking, or escape jump. Only mice displaying baseline latencies between 5 and 10 sec were used for these studies. The maximal cutoff latency was 30 sec to avoid possible tissue damage (i.e., the mouse was taken off the hot plate and scored as being fully analgesic). Data are presented as percent of the maximum possible effect (% MPE), which is calculated as:
Hydrolysis kinetics
Following sodium pentobarbital anesthesia (50 mg/kg ip), the mouse was decapitated and the cerebral cortex and choroid plexuses rapidly harvested. The tissues were then homogenized (4°C) with tris-HCl buffer (50 mM, pH 7.5) to make a 0.2% homogenate. Protein content was measured using the Pierce® BCA Protein Assay Kit (Thermo Fisher Scientific Inc., Rockford, IL). The cerebral cortex and choroid plexus homogenates were then further diluted to 120 μg protein/ml. After 6 min of preincubation (37°C), a 0.2-ml aliquot of diluted tissue homogenate was incubated (37°C) for 8 min with 20 μl of mixed substrate, containing unlabelled L-KTP, [3H]L-KTP and trace amounts of [14C]mannitol as internal standard to make final dipeptide concentrations of 36.5, 50, 80, 150 and 200 μM. The reaction mixture was terminated by adding 20 μl of 20% trichloroacetic acid to each tube. The mixture was then centrifuged at 15,000 g × 10 min under ambient conditions, and the supernatants collected for HPLC analysis. The Michaelis-Menten parameters (Vmax and Km) were determined by Lineweaver-Burk double reciprocal plots.
Analytical
CSF samples from the efflux experiments were diluted with 10 μl of 1% trichloroacetic acid and centrifuged at 15,000 g × 10 min under ambient conditions. The resultant supernatants were injected into the HPLC system, which consisted of a Waters 515 pump (Waters, Milford, MA), a Rheodyne® injector port (Rohnert Park, CA), and a Packard 500TR Radiochemical detector (Packard Instrument Company, Downers Grove, IL). Radiolabeled L-KTP and mannitol were separated on a Hypersil 5-μm (C18) column, 250 mm × 4.6mm (Thermo Fisher Scientific Inc., Rockford, IL). The mobile phase consisted of 20 mM KH2PO4 - 0.1% trifluoroacetic acid (v/v) and 3.0% acetonitrile (adjusted to pH 7.0 with 1.0 M NaOH), pumped isocratically at 1.0 ml/min under ambient conditions. Peaks were recorded and integrated using FLO-ONE software for Windows Analysis (Packard Instrument Company, version 3.61). The retention times of [3H]L-KTP, [3H]L-Tyr (a hydrolysis product of L-KTP) and [14C]mannitol were 8.3, 4.7 and 3.1 min, respectively. The percent of radiolabeled L-KTP remaining in CSF is expressed as the [3H]/[14C] ratio in that fluid divided by the same ratio in injectate, such that:
The [14C]mannitol served as an internal reference control to account for differences in injection volume and diffusion away from the CSF space (Suzuki et al. 1989; Matsushita et al. 1991). Tissue homogenate supernatants from cerebral cortex and choroid plexus were also analyzed by HPLC, as described previously. The concentration of L-KTP during the enzymatic reaction was calculated by the ratio of [3H]/[14C] in the final incubation divided by the same ratio in the initial incubation, multiplied by the concentration of L-KTP in the initial incubation.
Statistics
Data are expressed as mean ± SEM. Statistical comparisons between wild-type and Pept2 null mice were performed by a two sample t-test (Prism version 5.0; GraphPad Software, Inc., La Jolla, CA) in which a probability of p ≤ 0.05 was considered statistically significant. Statistical differences between multiple treatment groups (in the same genotype) were determined by ANOVA and pairwise comparisons with the control group were made using Dunnett’s test. Lineweaver-Burk analyses were also performed with Prism software. Dose-response analyses were performed by nonlinear least-squares regression (WinNonlin version 5.0.1; Pharsight Corp., Mountain View, CA) and a weighting factor of unity, according to the expression:
where the ED50 is the dose that causes 50% of the maximum effect due to L-KTP or D-KTP, N is the slope factor, and Eo is the baseline effect in the absence of dipeptide. The quality of fit was determined by the standard error of parameter estimates, by the coefficient of determination (r2), and by the residual plots.
Results
CSF efflux of L-KTP is faster in wild-type than in Pept2null mice
A comparison of remaining [3H]L-KTP levels between genotypes revealed that PEPT2 had a dramatic effect on the removal of this neuropeptide from CSF. As shown in Fig. 1, the half-life of L-KTP was over 2-fold longer in Pept2 null mice compared to wild-type animals. These differences reflect the PEPT2-mediated apical uptake of L-KTP from CSF into choroid plexus epithelia or parenchymal cells. Any potential differences in diffusion are corrected by mannitol activity. The specificity of this uptake process was supported by experiments in wild-type mice, where the remaining [3H]L-KTP in CSF was 3.5-fold greater in the presence of GlySar, a PEPT2 substrate (p < 0.05), and 7.9-fold greater during self-inhibition studies with unlabeled L-KTP(p < 0.001) (Fig. 2). In contrast, GlySar did not significantly alter the exposure of [3H]L-KTP in CSF of Pept2 null mice, although the remaining [3H]L-KTP in CSF was 4.7-fold greater in the presence of unlabeled L-KTP (p < 0.001). The significant changes caused by unlabeled L-KTP in both genotypes probably reflects the dual actions of this dipeptide, first as an inhibitor of [3H]L-KTP uptake by PEPT2 and second as an inhibitor of [3H]L-KTP hydrolysis by aminopeptidasein brain (see discussion for more details).
Fig. 1.
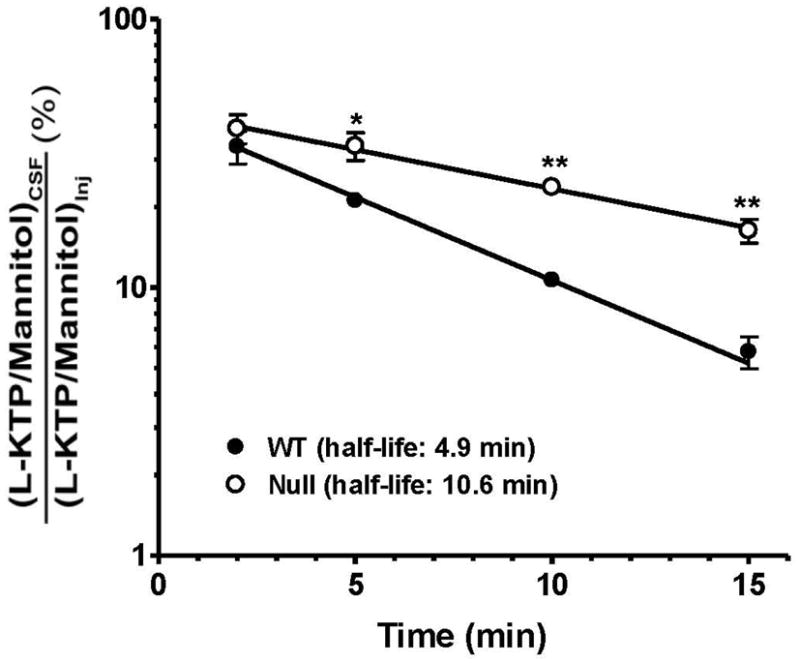
CSF efflux of [3H]L-KTP from wild-type and Pept2 null mice after an icv injection of 1 μCi (5 nmol) of neuropeptide. L-KTP levels are expressed as fraction remaining in CSF relative to that of injectate. [14C]Mannitol (0.2 μCi), which is transported by passive diffusion, was co-injected to assess adequacy and comparability of the injections. Data are expressed as mean ± SEM (n=3–6 per genotype). *p < 0.05 and **p < 0.01, as compared to wild-type mice.
Fig. 2.
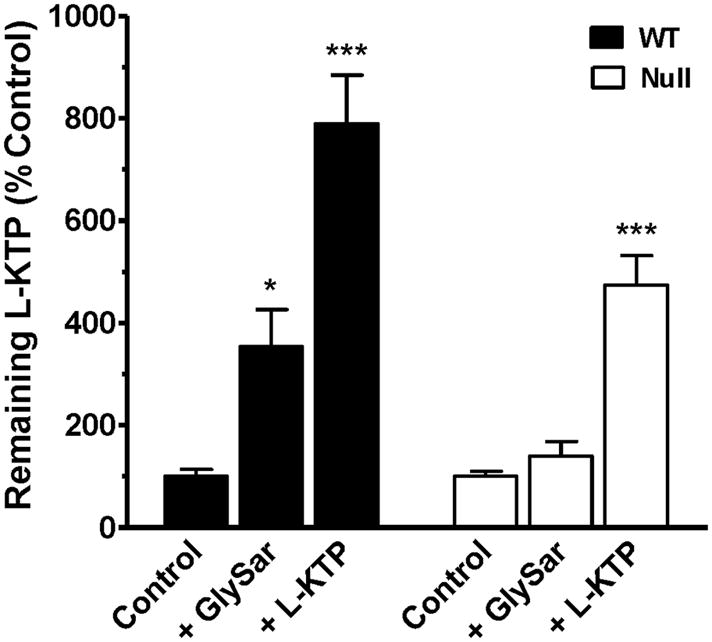
Effect of GlySar (200 nmol) and unlabeled L-KTP (200 nmol) on L-KTP remaining in the CSF of wild-type and Pept2 null mice 15 min after an icv injection of 1 μCi (5 nmol) of neuropeptide. L-KTP levels are expressed as fraction remaining in CSF relative to that of injectate, and then displayed as percent of the control value. [14C]Mannitol (0.2 μCi), which is transported by passive diffusion, was co-injected to assess adequacy and comparability of the injections. Data are expressed as mean ± SEM (n=3–7 per genotype). *p < 0.05 and ***p < 0.001, as compared to the control group for each genotype.
Enhanced antinociceptive response to L-KTP but not D-KTP in Pept2 null mice
Given the significant effect of PEPT2 ablation on the CSF efflux of L-KTP in transgenic mice, we hypothesized that we should observe differences in the analgesic effect of L-KTP between wild-type and Pept2 null mice. Since D-KTP is not a substrate for PEPT2, no differences in analgesia should be observed between the two genotypes. As shown in Fig. 3, the time of maximum analgesia occurred 5 min after the icv injection of either L-KTP or D-KTP with the response generally waning over time. Moreover, it is clear that the L-KTP-induced antinociception is much greater in Pept2 null mice as compared to wild-type animals (Fig. 3A). In contrast, there does not appear to be a difference between genotypes in the response to D-KTP (Fig. 3B). It is also interesting to note that Pept2-competent and Pept2-deficient animals exhibited a maximum analgesia (100% MPE) at 100 nmol doses of either neuropeptide. However, the response to 100 nmol L-KTP subsided in both genotypes about 20 min after dosing while the 100 nmol dose of D-KTP was fully analgesic throughout the 40 min experiment. These differences probably reflect the upper limit of dose effect for both dipeptides, with D-KTP being cleared from the CSF at a slower rate than L-KTP because of its lack of affinity for PEPT2.
Fig. 3.


Antinociceptive response (hot-plate method, 55°C) at different doses of L-KTP (A) and D-KTP (B) as a function of time. % MPE is the maximum possible effect (see Methods section, Assessment of antinociceptive response). Data are expressed as mean ± SEM (n=10–20 mice per measurement for L-KTP in both genotypes; n=5–10 mice per measurement for D-KTP in both genotypes).
To examine antinociception more quantitatively, dose-response curves were constructed for both L-KTP and D-KTP over a dose range of 0.1 to 200 nmol. As shown in Fig. 4A, wild-type mice demonstrated a substantial shift to the left in the % MPE versus dose relationship of L-KTP. In contrast, the % MPE versus dose relationships of D-KTP were superimposable between the two genotypes (Fig. 4B). The ED50 of L-KTP in Pept2 null mice was one-fifth of the value observed in wild-type animals (Table 1). In contrast, the ED50 of D-KTP was very similar between genotypes (~ 10% difference; Table 1). Likewise, there was little difference between genotypes in the slope factor or baseline effect of L-KTP and D-KTP (< 10%). Comparing all four curves on the same plot (Fig. 4C), it appears the potency of these dipeptides can be ranked as: L-KTP in Pept2 null mouse > D-KTP in wild-type mice = D-KTP in Pept2 null mice > L-KTP in wild-type mice. These findings reveal that L-KTP-induced analgesia can be modified by the presence of PEPT2 activity in brain and that differences in the affinity of L- and D-KTP for PEPT2 may contribute to differences in the potency of these two peptides.
Fig. 4.



Dose-response curves for L-KTP (A), D-KTP (B) and both compounds (C) following icv doses of 0.1 to 200 nmol of neuropeptide, where antinociception was measured by the hot-plate method (55°C). % MPE is the maximum possible effect (see Methods section, Assessment of antinociceptive response). The responses to vehicle control doses (aCSF only) are shown by horizontal lines for each genotype. Data are expressed as mean ± SEM (n=10–20 mice per measurement for L-KTP in both genotypes; n=10 mice per measurement for D-KTP in both genotypes).
Table 1.
Dose-response parameters of L-KTP and D-KTP in wild-type and Pept2 null mice
| L-KTP | D-KTP | |||
|---|---|---|---|---|
| Wild-type | Null | Wild-type | Null | |
| ED50 (nmol) | 20.8 ± 1.7 | 4.0 ± 0.4 | 10.3 ± 1.0 | 9.1 ± 0.8 |
| N | 1.7 ± 0.2 | 1.6 ± 0.2 | 2.0 ± 0.3 | 2.0 ± 0.3 |
| Eo (% MPE) | 26.5 ± 2.1 | 24.4 ± 3.3 | 25.7 ± 2.8 | 23.7 ± 2.7 |
| r2 | 0.991 | 0.989 | 0.989 | 0.991 |
Dose-response parameters are expressed as estimate ± standard error (n=164 mice per genotype for L-KTP; n=100 mice per genotype for D-KTP). ED50 is the dose that causes 50% of the maximum effect due to dipeptide, N is the slope factor, Eo is the baseline effect in the absence of dipeptide, and r2is the coefficient of determination.
No difference between genotypes in the hydrolysis kinetics of L-KTP
Substantial differences between the antinociceptive effect of L-KTP in wild-type and Pept2 null mice make a strong case for the importance of this peptide transporter in modulating neuropeptides in the brain. However, to make sure this was not an artifact of genotypic differences in metabolism (via aminopeptidase), hydrolysis studies were performed in homogenates prepared from the cerebral cortex and choroid plexus tissues of wild-type and Pept2 null mice. As shown in Table 2, there were no significant differences between genotypes in both the Vmax and Km values for L-KTP degradation in either tissue. In particular, the Vmax was consistent between genotypes and tissues, averaging about 88 nmol/mg protein/min. The Km was consistent between genotypes, averaging about 0.21 mM in cerebral cortex and about 0.13 mM in choroid plexus. Thus, it appears that the enhanced antinociceptive response to L-KTP in Pept2 null mice cannot be explained by genotypic differences in neuropeptide degradation. This contention was supported by a targeted study in which an aminopeptidase inhibitor, bestatin (160 nmol), was co-administered by icv injection with 4 nmol L-KTP in Pept2 null mice (n=10 mice per treatment). Under these conditions, bestatin had little effect on the MPE of L-KTP in Pept2-deficient animals (46 ± 5% for L-KTP vs. 54 ± 5% for L-KTP plus bestatin; p=0.2931). Still, it appears that L-KTP was metabolized more efficiently in the choroid plexus as compared to cerebral cortex tissue (e.g., Vmax/Km values were about 2-fold greater for both genotypes).
Table 2.
Hydrolysis kinetics of L-KTP in tissue homogenates from wild-type and Pept2 null mice
| Cerebral cortex | Choroid plexus | |||
|---|---|---|---|---|
| Wild-type | Null | Wild-type | Null | |
| Vmax (nmol/mg protein/min) | 79 ± 13 | 84 ± 14 | 88 ± 6 | 99 ± 6 |
| Km (mM) | 0.22 ± 0.06 | 0.20 ± 0.03 | 0.11 ± 0.01 | 0.14 ± 0.02 |
| Vmax/Km (μl/mg protein/min) | 384 ± 40 | 413 ± 45 | 788 ± 44** | 714 ± 69* |
| r2 | 0.984±0.017 | 0.977±0.005 | 0.986±0.008 | 0.949±0.013 |
Values are expressed as mean ± SEM (n=3). Vmax is the maximum rate of enzymatic degradation, Km is the Michaelis constant, and r2 is the coefficient of determination. Vmax and Km were not different between genotypes for either tissue.
p < 0.05 between tissues in null mice
p < 0.01 between tissues in wild-type mice.
Discussion
Studies using transgenic mice offer a unique opportunity to examine the role and relevance of a particular protein under physiological in vivo conditions. In this study, we evaluated the CSF efflux, antinociceptive response, and hydrolysis kinetics in brain of L-KTP and D-KTP in wild-type and Pept2 null mice. Our findings were novel in establishing, for the first time, a pharmacologic phenotype for the proton-coupled oligopeptide transporter PEPT2. Specifically, we demonstrated that: 1) the CSF clearance of L-KTP was slower in Pept2 null mice than in wild-type mice, 2) the antinociceptive response to L-KTP was 5.2-fold more potent in Pept2 null mice as compared to wild-type animals, 3) the antinociceptive response to D-KTP was unaltered between genotypes, 4) the hydrolysis kinetics of L-KTP in cerebral cortex or choroid plexus were similar between the Pept2-competent and Pept2-deficient mice, and 5) in the absence of PEPT2 and aminopeptidase activity in brain, L-KTP was a more potent neuropeptide analgesic than D-KTP. The results demonstrate that PEPT2 can significantly impact the analgesic response to endogenous neuropeptides by altering their CSF (and presumably brain interstitial fluid) concentrations and that it may also influence the disposition and response to exogenous peptide/mimetic substrates.
Previous studies have evaluated the analgesic response to L-KTP and its synthetic diastereomer D-KTP in mice (Takagi et al. 1979a, 1979b). Using the tail-pinch method, in which 10 μl injections of neuropeptide were injected into the cisterna magna, these investigators reported an ED50 = 34.6 nmol for L-KTP and an ED50 = 6.2 nmol for D-KTP. Using the hot-plate method (temperature and injection volume not given), an ED50 = 15.7 nmol for L-KTP was reported by the same group. These estimates are very similar to values reported in the wild-type mice of our study using the hot-plate method (i.e., 20.8 nmol for L-KTP and 10.3 nmol for D-KTP). At that time, Takagi et al. (1979b) attributed the greater potency of D-KTP to its greater resistance against hydrolyzing enzyme(s). In support of this contention, Takagi and coworkers (1984 (1985) found that 1.3 μM bestatin was a potent inhibitor of L-KTP degradation in mouse and rat brain homogenates (Ki = 0.1 μM) and that co-administration of 50 μg bestatin potentiated the analgesic effects to intracisternally administered L-KTP in mice by 4.8 times. However, in addition to being an aminopeptidase inhibitor, bestatin is also a PEPT2 inhibitor (Ki = 18 μM), as demonstrated in LLC-PK1 cells stably transfected with rat PEPT2 (Terada et al. 2000). Therefore, it is possible that bestatin had a dual role in potentiating the analgesic effect of L-KTP.
L-KTP exhibits high-affinity interactions with PEPT2 (Ki values of 8–30 μM), as shown by its competitive inhibition of GlySar in rat synaptosomes (Fujita et al. 1999, 2004) and isolated choroid plexus (Teuscher et al. 2001). In contrast, D-KTP had no effect on the uptake of GlySar in rat choroid plexus even at concentrations 1,000 times in excess (Teuscher et al. 2001). Marked inwardly-directed currents were subsequently observed for L-KTP in Xenopus oocytes expressing human PEPT2, demonstrating that the neuropeptide was indeed a transportable substrate (Thakker et al. 2008). These same authors also reported that the opioid peptide transport system in SK-N-SH cells (a human neuronal cell line) was stimulated 2.5-fold by L-KTP but that little stimulation was observed by D-KTP. The authors concluded that since activity of the opioid transport system is modulated by extracellular L-KTP and since PEPT2 is an important determinant of neuropeptide homeostasis in CSF, the activity of PEPT2 may be a critical factor in the in vivo modulation of opioidergic neurotrans mission in the brain.
In the present study, the CSF efflux of L-KTP was substantially reduced by a PEPT2 substrate, GlySar, in wild-type mice but not in Pept2 null animals, confirming the specificity of the neuropeptide for PEPT2. Excess concentrations of unlabeled L-KTP reduced the CSF efflux of radiolabeled L-KTP in both genotypes although the effect was more dramatic in Pept2-competent mice (i.e., 7.9-fold vs. 4.7 reduction, respectively, in wild-type vs. null animals). These results demonstrate that, at 15 min after the icv injection, excess L-KTP could inhibit both its PEPT2-mediated transport and degradation (Fig. 2). However, our bestatin results (i.e., no significant change in % MPE of L-KTP ± bestatin in Pept2 null mice) suggest that, at 5 min after the icv injection, little if any hydrolysis of L-KTP actually occurred during the antinociception studies. Thus, the KTP-induced analgesic effects shown in Fig. 4 reflect the influence of PEPT2 and not enzymatic degradation. Even if some metabolism was occurring in our dose-response experiments, it is important to recognize that no differences were observed between genotypes in the maximal hydrolysis rate and affinity of L-KTP for aminopeptidase enzymes in either mouse choroid plexus or cerebral cortex tissue. Interestingly, we did see some differences between the two tissues in Km but not Vmax values. In this regard, the Km estimates were 130 μM for L-KTP in choroid plexus and 210 μM for L-KTP in cerebral cortex. A previous study has reported two kyotorphin-hydrolyzing peptidases in rat brain, KTPase I and KTPase II, with Km values of 22 μM and 100 μM, respectively (Akasaki et al. 1991). It is unclear, however, whether or not the Km values in our mice reflects differences in the tissue expression of these two isoforms of KTP.
Scant information is currently available on the physiological concentrations of L-KTP in various brain compartments. In one study (Ueda et al. 1987), tissue levels of L-KTP in rat ranged from a low of 0.1 μM in the cerebellum to a high of 2.2 μM in the midbrain. In another study (Nishimura et al. 1991), L-KTP levels in human CSF were reported as 1.2 nM for healthy subjects and as 0.24 nM for patients with persistent pain. However, it is unlikely that L-KTP would be uniformly distributed in the brain since the neuropeptide is highly concentrated in the synaptosomal fraction of rat brain (Ueda et al. 1982). As a result, L-KTP levels are expected to be several times higher in specific regions of the brain, approximating that achieved during the antinociception studies. For example, assuming a value of 40 μl for mouse CSF (Rudick et al. 1982), we estimate the initial concentrations of L-KTP at 0.0025 to 5 mM following the 0.1 to 200 nmol icv doses of neuropeptide, with much lower levels of L-KTP occurring over time as it is cleared rapidly from CSF. In addition to the physiological relevance of L-KTP levels during the antinociception experiments, our studies have pharmacological relevance by providing insight into D-KTP-induced analgesia and the role that PEPT2 might play in developing strategies for the exogenous targeting of analgesic dipeptide analogs.
A number of oligopeptides (2-3 amino acids) have physiological activity in the brain. These oligopeptides may be synthesized directly in the brain (e.g., carnosine by carnosine synthetase) (Margolis et al. 1987) or produced during the degradation of larger neuropeptides (e.g., glycine-proline-glutamate, an N-terminal fragment of insulin-like growth factor-1) (Ikeda et al. 1995). There is a growing realization that peptide fragments may impact parent neuropeptide signaling although, as yet, most attention has focused on larger fragments (Nyberg and Hallberg 2007). The results presented here suggest that PEPT2 may affect the CNS activity of oligopeptides (synthesized or metabolically produced). In addition, it should be noted that another proton-coupled oligopeptide transporter, the peptide/histidine transporter PHT1, is highly expressed in brain (Yamashita et al. 1997). Whether it has a function in neuropeptide regulation has not been examined.
In conclusion, we have provided definitive evidence that PEPT2 can significantly modulate the antinociceptive response to neuropeptides (i.e., L-KTP) in the brain. Our results show that Pept2 null mice cleared L-KTP more slowly from CSF, thereby, resulting in a 5-fold greater L-KTP-induced analgesia as compared to equivalent doses of this neuropeptide in wild-type animals. Moreover, it appears that L-KTP is “intrinsically” more potent than D-KTP in the absence of PEPT2 activity. Taken as a whole, our findings suggest that PEPT2 might function in the control of analgesia through regulation of endogenous neuropeptides (and exogenous drug candidates) in the CSF or other brain compartments. These results show that peptide transport can affect the activity of a neuropeptide. Whether PEPT2 and other peptide transporters present in brain have similar effects on the activity of other neuropeptides merits further investigation.
Acknowledgments
This work was supported by NIH Grants R01 GM035498 (DES) and R01 NS034709 (RFK), and by the Vahlteich Research Endowment Fund from the University of Michigan College of Pharmacy. H.J. was supported by a fellowship from Zhejiang University and a scholarship from the China Scholarship Council.
Abbreviations
- EAAT
excitatory amino acid transporter
- icv
intracerebroventricular
- D-KTP
D-kyotorphin
- L-KTP
L-kyotorphin
- PEPT
peptide transporter
- PHT
peptide/histidine transporter
Footnotes
There are no conflicts of interest to disclose.
References
- Akasaki K, Nakamura A, Shiomi H, Tsuji H. Identification and characterization of two distinct kyotorphin-hydrolyzing enzymes in rat brain. Neuropeptides. 1991;20:103–107. doi: 10.1016/0143-4179(91)90059-r. [DOI] [PubMed] [Google Scholar]
- Brandsch M, Knütter I, Bosse-Doenecke E. Pharmaceutical and pharmacological importance of peptide transporters. J Pharm Pharmacol. 2008;60:543–585. doi: 10.1211/jpp.60.5.0002. [DOI] [PubMed] [Google Scholar]
- Daniel H, Kottra G. The proton oligopeptide cotransporter family SLC15 in physiology and pharmacology. Pflügers Arch. 2004;447:610–618. doi: 10.1007/s00424-003-1101-4. [DOI] [PubMed] [Google Scholar]
- Davis TP, Konings PN. Peptidases in the CNS: formation of biologically active, receptor-specific peptide fragments. Crit Rev Neurobiol. 1993;7:163–174. [PubMed] [Google Scholar]
- Fujita T, Kishida T, Okada N, Ganapathy V, Leibach FH, Yamamoto A. Interaction of kyotorphin and brain peptide transporter in synaptosomes prepared from rat cerebellum: implication of high affinity type H+/peptide transporter PEPT2 mediated transport system. Neurosci Lett. 1999;271:117–120. doi: 10.1016/s0304-3940(99)00540-6. [DOI] [PubMed] [Google Scholar]
- Fujita T, Kishida T, Wada M, Okada N, Yamamoto A, Leibach FH, Ganapathy V. Functional characterization of brain peptide transporter in rat cerebral cortex: identification of the high-affinity type H+/peptide transporter PEPT2. Brain Res. 2004;997:52–61. doi: 10.1016/j.brainres.2003.10.049. [DOI] [PubMed] [Google Scholar]
- Hu Y, Ocheltree SM, Xiang J, Keep RF, Smith DE. Glycyl-L-glutamine disposition in rat choroid plexus epithelial cells in primary culture: role of PEPT2. Pharm Res. 2005;22:1281–1286. doi: 10.1007/s11095-005-5261-0. [DOI] [PubMed] [Google Scholar]
- Hu Y, Shen H, Keep RF, Smith DE. Peptide transporter 2 (PEPT2) expression in brain protects against 5-aminolevulinic acid neurotoxicity. J Neurochem. 2007;103:2058–2065. doi: 10.1111/j.1471-4159.2007.04905.x. [DOI] [PubMed] [Google Scholar]
- Ikeda T, Waldbillig RJ, Puro DG. Truncation of IGF-1 yields two mitogens for retinal Müller glial cells. Brain Res. 1995;686:87–92. doi: 10.1016/0006-8993(95)00473-4. [DOI] [PubMed] [Google Scholar]
- Margolis FL, Grillo M, Hempstead J, Morgan JI. Monoclonal antibodies to mammalian carnosine synthetase. J Neurochem. 1987;48:593–600. doi: 10.1111/j.1471-4159.1987.tb04134.x. [DOI] [PubMed] [Google Scholar]
- Matsubayashi K, Kojima C, Kawajiri S, Ono K, Takegoshi T, Ueda H, Takagi H. Hydrolytic deactivation of kyotorphin by the rodent brain homogenates and sera. J Pharmacobiodyn. 1984;7:479–484. doi: 10.1248/bpb1978.7.479. [DOI] [PubMed] [Google Scholar]
- Matsushita H, Suzuki H, Sugiyama Y, Sawada Y, Iga T, Kawaguchi Y, Hanano M. Facilitated transport of cefodizime into the rat central nervous system. J Pharmacol Exp Ther. 1991;259:620–625. [PubMed] [Google Scholar]
- Nishimura K, Kaya K, Hazato T, Ueda H, Satoh M, Takagi H. Kyotorphin like substance in human cerebrospinal fluid of patients with persistent pain. Masui. 1991;40:1686–1690. [PubMed] [Google Scholar]
- Nyberg F, Hallberg M. Peptide conversion – a potential pathway modulating G-protein signaling. Curr Drug Targets. 2007;8:147–154. doi: 10.2174/138945007779315597. [DOI] [PubMed] [Google Scholar]
- Ocheltree SM, Shen H, Hu Y, Keep RF, Smith DE. Role and relevance of peptide transporter 2 (PEPT2) in the kidney and choroid plexus: in vivo studies with glycylsarcosine in wild-type and PEPT2 knockout mice. J Pharmacol Exp Ther. 2005;315:240–247. doi: 10.1124/jpet.105.089359. [DOI] [PubMed] [Google Scholar]
- Rubio-Aliaga I, Daniel H. Peptide transporters and their roles in physiological processes and drug disposition. Xenobiotica. 2008;38:1022–1042. doi: 10.1080/00498250701875254. [DOI] [PubMed] [Google Scholar]
- Rudick RA, Zirretta DK, Herndon RM. Clearance of albumin from mouse subarachnoid space: a measure of CSF bulk flow. J Neurosci Methods. 1982;6:253–259. doi: 10.1016/0165-0270(82)90088-7. [DOI] [PubMed] [Google Scholar]
- Shen H, Ocheltree SM, Hu Y, Keep RF, Smith DE. Impact of genetic knockout of PEPT2 on cefadroxil pharmacokinetics, renal tubular reabsorption, and brain penetration in mice. Drug Metab Dispos. 2007;35:1209–1216. doi: 10.1124/dmd.107.015263. [DOI] [PubMed] [Google Scholar]
- Shen H, Smith DE, Keep RF, Brosius FC., 3rd Immunolocalization of the proton-coupled oligopeptide transporter PEPT2 in developing rat brain. Mol Pharm. 2004;1:248–256. doi: 10.1021/mp049944b. [DOI] [PubMed] [Google Scholar]
- Shen H, Smith DE, Keep RF, Xiang J, Brosius FC., 3rd Targeted disruption of the PEPT2 gene markedly reduces dipeptide uptake in choroid plexus. J Biol Chem. 2003;278:4786–4791. doi: 10.1074/jbc.M207397200. [DOI] [PubMed] [Google Scholar]
- Shen H, Smith DE, Yang T, Huang YG, Schnermann JB, Brosius FC., 3rd Localization of PEPT1 and PEPT2 proton-coupled oligopeptide transporter mRNA and protein in rat kidney. Am J Physiol. 1999;276:F658–665. doi: 10.1152/ajprenal.1999.276.5.F658. [DOI] [PubMed] [Google Scholar]
- Smith DE, Johanson CE, Keep RF. Peptide and peptide analog transport systems at the blood-CSF barrier. Adv Drug Deliv Rev. 2004;56:1765–1791. doi: 10.1016/j.addr.2004.07.008. [DOI] [PubMed] [Google Scholar]
- Suzuki H, Sawada Y, Sugiyama Y, Iga T, Hanano M, Spector R. Transport of imipenem, a novel carbapenem antibiotic, in the rat central nervous system. J Pharmacol Exp Ther. 1989;250:979–984. [PubMed] [Google Scholar]
- Takagi H, Shiomi H, Ueda H, Amano H. A novel analgesic dipeptide from bovine brain is a possible Met-enkephalin releaser. Nature. 1979a;282:410–412. doi: 10.1038/282410a0. [DOI] [PubMed] [Google Scholar]
- Takagi H, Shiomi H, Ueda H, Amano H. Morphine-like analgesia by a new dipeptide, L-tyrosyl-L-arginine (kyotorphin) and its analogue. Eur J Pharmacol. 1979b;55:109–111. doi: 10.1016/0014-2999(79)90154-7. [DOI] [PubMed] [Google Scholar]
- Terada T, Sawada K, Irie M, Saito H, Hashimoto Y, Inui KI. Structural requirements for determining the substrate affinity of peptide transporters PEPT1 and PEPT2. Pflügers Arch. 2000;440:679–684. doi: 10.1007/s004240000339. [DOI] [PubMed] [Google Scholar]
- Teuscher NS, Keep RF, Smith DE. PEPT2-mediated uptake of neuropeptides in rat choroid plexus. Pharm Res. 2001;18:807–813. doi: 10.1023/a:1011088413043. [DOI] [PubMed] [Google Scholar]
- Teuscher NS, Shen H, Shu C, Xiang J, Keep RF, Smith DE. Carnosine uptake in rat choroid plexus primary cell cultures and choroid plexus whole tissue from PEPT2 null mice. J Neurochem. 2004;89:375–382. doi: 10.1111/j.1471-4159.2004.02333.x. [DOI] [PubMed] [Google Scholar]
- Thakkar SV, Miyauchi S, Prasad PD, Ganapathy V. Stimulation of Na+/Cl−-coupled opioid peptide transport system in SK-N-SH cells by L-kyotorphin, an endogenous substrate for H+-coupled peptide transporter PEPT2. Drug Metab Pharmacokinet. 2008;23:254–262. doi: 10.2133/dmpk.23.254. [DOI] [PubMed] [Google Scholar]
- Tzingounis AV, Wadiche JI. Glutamate transporters: confining runaway excitation by shaping synaptic transmission. Neuroscience. 2007;8:935–947. doi: 10.1038/nrn2274. [DOI] [PubMed] [Google Scholar]
- Ueda H, Inoue M. In vivo signal transduction of nociceptive response by kyotorphin (tyrosine-arginine) through Gαi- and inositol triphosphate-mediated Ca2+ influx. Mol Pharmacol. 2000;57:108–115. [PubMed] [Google Scholar]
- Ueda H, Ming G, Hazato T, Katayama T, Takagi H. Degradation of kyotorphin by a purified membrane-bound-aminopeptidase from monkey brain: potentiation of kyotorphin-induced analgesia by a highly effective inhibitor, bestatin. Life Sci. 1985;36:1865–1871. doi: 10.1016/0024-3205(85)90160-2. [DOI] [PubMed] [Google Scholar]
- Ueda H, Tatsumi K, Shiomi H, Takagi H. Analgesic dipeptide, kyotorphin (Tyr-Arg), is highly concentrated in the synaptosomal fraction of the rat brain. Brain Res. 1982;231:222–224. doi: 10.1016/0006-8993(82)90023-3. [DOI] [PubMed] [Google Scholar]
- Ueda H, Yoshihara Y, Fukushima N, Shiomi H, Nakamura A, Takagi H. Kyotorphin (tyrosine-arginine) synthetase in rat brain synaptosomes. J Biol Chem. 1987;262:8165–8173. [PubMed] [Google Scholar]
- Yamashita T, Shimada S, Guo W, Sato K, Kohmura E, Hayakawa T, Takagi T, Tohyama M. Cloning and functional expression of a brain peptide/histidine transporter. J Biol Chem. 1997;272:10205–10211. doi: 10.1074/jbc.272.15.10205. [DOI] [PubMed] [Google Scholar]