

Abstract
Although diffusion theory predicts that IMS resolving power increases with the square root of the voltage applied across the drift tube, in practice there exists an optimum voltage above which resolving power decreases. This optimum voltage was determined to be both compound and initial ion pulse width-dependent. A “conditional” resolving power equation is introduced that can be used to quickly approximate realistic resolving powers for specific instrumental operating parameters and compounds. Using four common environmental contaminants [trichloroethylene (TCE), tetrachloroethylene (PCE), methyl tert-butyl ether (MTBE) and methyl iso-butyl ketone (MIBK)], diffusion-limited (theoretical), Rd, conditional, Rc, and actual (or measured), Rm, IMS resolving powers were determined and compared for a small IMS instrument designed for subsurface measurements. Detection limits determined at the optimal resolving power for the environmental contaminants ranged from 18 parts per trillion volume-to-volume (pptv) to 80 parts per billion volume-to-volume (ppbv). The maximal measured resolving power for our small, ambient-pressure stand-alone IMS ranged from 42 to 54, yielding an IMS resolving power efficiency, defined as Rm/Rc × 100%, of 56 to 74% of the maximal conditional resolving power possible.
Introduction
First introduced in 19701, stand-alone ambient pressure ion mobility spectrometry (IMS) has grown to become an important analytical separation and detection technique.2–3 It has been routinely used for detecting and monitoring explosives,4–12 illicit drugs,13–19 chemical warfare agents,20–22 environmental contaminants,23–24 biological compounds,25 industrial chemicals for process control,26 volatile organics in space shuttle cabin air27 and toxins in workplace air.28
Stand-alone IMS has several advantages for field application over other analytical methods such as chromatography and mass spectrometry. Compared to chromatographic and electrophoretic techniques, it is fast, sensitive and reproducible. At ambient pressure, gas phase ions are separated with resolving powers similar to that of chromatography in a few milliseconds. When normalized to standard temperature and pressure, arrival times can be converted to reduced ion mobility constants which are reproducible from day to day and instrument to instrument. These ion mobility constants are fundamental measurements based on the ion’s diffusion coefficient. In addition, the response from ions in an IMS can be collected and recorded efficiently. Sensitivity and detection limits of an IMS are similar to those of a flame ionization detector in gas chromatography and are usually better than UV-Vis absorption detectors in liquid chromatography. When compared to a mass spectrometer, IMS is less expensive, mechanically more robust and easier to operate.
The ion separation capabilities of an IMS are quantified using resolving power. IMS resolving power has been defined as the drift time (td) of the ion divided by the temporal peak width-at-half-height (w0.5) of the ion mobility peak.29–31 This relationship is given by Eq. 1:
| (1) |
where Rm is the measured resolving power.
Although high resolution IMS has been reported in which resolving powers exceeded some chromatographic systems,32–33 resolving powers no greater than 30 have been reported20 for most commercial and field instruments. Despite the large number of commercial IMS systems in operation as analytical instruments, surprisingly, the literature cites few investigations supported by detailed experimental data that evaluated key instrumental parameters responsible for obtaining maximal resolving powers for IMS systems. One thorough study by Siems et al.30 considered not only diffusion but also gate pulse widths, voltages, and temperature in a gate pulse width-dominated regime. Here we build on that work and provide practical examples that demonstrate the conditional effects of gate pulse width and voltage used for achieving optimal resolving powers.
In investigating resolution for an IMS tube, Rokushika et al.31 used a single peak approach to calculate resolving power. Because the measured resolving power did not match the theoretical estimates in this study, the authors suggested that this discrepancy may have been due to the difficulty in achieving a smooth electric field throughout the drift region,31 an argument supported by Siems et al.’s work. Siems et al.30 also first suggested that contrary to what is expected from diffusion theory, an increase in voltage can lead to a decrease in resolving power.
The temporal peak width-at-half-height (w0.5) is a result of the initial ion gate pulse width, diffusion, coulombic repulsion, electric field homogeneity in the drift tube, effectiveness of the aperture grid and the response time of the amplifier. Because Rm is inversely proportional to w0.5, a smaller value for w0.5 will result in a higher resolving power of the IMS cell. Two expressions for w0.5 in an IMS have been reported.30,37 Spangler used the continuity equation39 that defines ion velocity and density in a one-dimensional steady flow with control volume conditions40 to develop an analytical expression for w0.5.29 His work considered the separation between the aperture grid and the ion collector, and the effect this separation has on w0.5. He reported two approaches for calculating w0.5; the difference in error function approach and the multiplicative factor approach that used the central moment of the IMS response. According to Spangler, the major factors contributing to w0.5 are the initial width of the gate pulse (Wg) applied to the ion gate and diffusional broadening that occurs in the ion pulse as it passes through the drift tube (Wdiff).29,41 Siems et al.30 fitted their expression for w0.5 with the temperature of the drift tube, the voltage of the drift tube, and parameters that adjust for the diffusion, gate pulse width and offset contributions (i.e., instrument response time, dynamic aspects of ion distributions around opening and closing gates). Several other investigations with either uniformly inlaid ceramic resistor IMS drift tubes or the stacked ring design IMS tubes resulted in different explanations, but all made reference to factors such as initial gate width, coulombic repulsion, diffusion, electric field homogeneity and space charge, etc. as the factors contributing to w0.5. 42–47
Siems et al.’s work addressed most of the potential band broadening mechanisms and provided a means to empirically evaluate them. By varying voltage and gate width simultaneously, they developed empirical values that correctly predicted the optimal conditions for operation of a drift tube. On a routine basis, however, it is impractical to conduct all of the experiments needed to optimize resolving power. A rapid method for estimating resolving power for an IMS instrument is needed.
The concept of “conditional resolving power” (Rc) is introduced as a means to estimate IMS resolving power and provide a practical approach for quickly evaluating the efficiency of an IMS drift tube. This work also demonstrates the relationship of Rc with voltage and initial gate pulse width.
Theoretical Considerations
The reduced mobility of an ion under normalized conditions of temperature and pressure is given as:48–49
| (2) |
where L (cm) is the length of an IMS drift tube, V is the voltage drop across L (V), td is the drift time of the ion mobility peak, T is the drift gas temperature (K) and P is the drift gas pressure (Torr) of the drift region where the ion’s mobility, K0 (cm2 V−1 s−1) was determined. Thus, by measuring the drift time of an ion in an IMS and knowing the pressure, temperature, drift tube length and voltage across the drift region, reduced mobilities can be determined. If experimental conditions are carefully controlled and accurately known, reduced mobility values for a given ion can be reproduced on a variety of IMS instruments and under a variety of operating conditions.
As described above, the width-at-half-height of an IMS ion peak can be attributed primarily to two band broadening mechanisms: the initial ion gate pulse width broadening (Wg, [s]) and diffusional broadening (Wdiff [s]).29,30 If the initial width of the ion pulse is assumed to be Gaussian in shape, the final temporal peak width (w0.5, [s]) can be expressed as a function of both Wg and Wdiff:
| (3) |
The spatial standard deviation, σs (cm), due to diffusion can be related to the ion mobility drift time, td [s], through the Brownian diffusion coefficient, D (cm2 s−1):
| (4) |
D can also be related to the ion mobility constant via the Nernst-Einstein equation:
| (5) |
where k is Boltzmann’s constant (J K−1 or C V K−1), T is the temperature (K) in the drift tube, K is the mobility (cm2 V−1 s−1) and q = e × z (C) is the ionic charge. Combining Equations 4 and 5 yields a relationship between the spatial standard deviation in terms of instrumental parameters of temperature (T), voltage across the ion drift region (V) and drift tube length (L, [cm]):
| (6) |
The spatial (σs) and temporal (σt, [s]) standard deviations are related as follows:
| (7) |
Combining Eqs. 6 and 7, and noting that the relationship between Wdiff and σt is given by Wdiff = (8ln2)1/2σt,30 the diffusion band broadening contribution can then be written as:
| (8) |
Combining Eqs. 3 and 8 gives an expression for band broadening in terms of the initial gate pulse and diffusion broadening:49
| (9) |
Combining Eqs. 9 and 1 provides an expression for resolving power which considers both the effects of the initial gate pulse width and diffusion:
| (10) |
If conditions are such that Wg ≪ td, then the resolving power reduces to a diffusion-limited resolving power, Rd, and
| (11) |
where q = e × z; e is the electronic charge and z is the charge on the ion.31
By rearranging Eq. 2 for td, and substituting into Eq. 10, a conditional resolving power, Rc, can be defined in terms of the instrumental parameters and reduced mobility constant of the ions measured:
| (12) |
While this conditional resolving power equation does not account for effects of coulombic repulsion, field homogeneity, or other instrumental effects, it does reflect the major contributions to band broadening, diffusion and initial ion pulse width and thus provides a rapid method for estimating the potential resolving power for a given ion under a specific set of operating conditions.
The separation efficiency of an IMS drift tube can be determined by measuring Rm and by using the instrumental parameters to calculate Rc. This efficiency can be defined as:
| (13) |
Experimental
Instrumentation
The small ambient pressure IMS used in this study was designed and fabricated at Boise State University in collaboration with Washington State University.23 The fundamental components of the instrument consisted of the following parts: a 15-mCi 63Ni radioactive foil; a reaction region; a Bradbury-Nielson ion gate; a counter flow ambient pressure drift region; an aperture grid; a faraday plate; and a data acquisition system. Table 1 summarizes the operating conditions used in this investigation.
Table 1.
IMS operating conditions summary.
| Parameter | Values | Units |
|---|---|---|
| Total IMS length | 113 | mm |
| Drift tube length | 68 | mm |
| Voltage applied at ionization regions | 1044–6518 | V |
| Voltage applied to ion gate (drift voltage) | 800–5000 | V |
| Gate voltage | 100–5000 | V |
| Aperture (last ring) voltage | 32–197(a) | V |
| Drift tube pressure | 698 ± 7 | Torr |
| Drift gas temperature | 295.5 ± 1.2 | K |
| Carrier gas flow | 10 | mL min−1 |
| Drift gas flow | 200 | mL min−1 |
| Gas | Air | |
| IMS gate pulse frequency | 40 | Hz |
| IMS scan time for 100 scans per spectrum | 25 | ms |
| IMS gate pulse width | 100, 200, 300, 400 & 500 | μs |
Both the reaction and drift regions were constructed with metal electrodes and ceramic (Macor) insulators (Dow Corning, Midland, MI) in a stacked, interlocking design50 with terminating end caps for a total overall length of 113 mm. Each Macor insulator ring was machined to an O.D. of 38 mm, and a thickness of 5 mm. A counter-bore was machined into each face of an insulator to provide a pocket for the mating electrode. In the drift region, these Macor insulating rings had a constant I.D. of 22 mm whereas those of the reaction region varied from 11 mm to 22 mm forming a tapered transition region from the ion source to the ion gate entrance. The reaction and drift regions contained 5 and 12 rings, respectively. To generate a uniform electric field down the tube, the stainless steel guard rings were connected to each other through 1 MΩ high-temperature resistors (Caddock Electronics Inc., ±1%).
Carrier gas (that gas which “carried” the analyte to the ionization region) flowed into the IMS through the ionization region where the 63Ni radioactive foil was positioned. High-energy beta particles emitted from the 63Ni radioactive foil initiated the ionization process through collisions with ambient pressure gas. This process resulted in the formation of positive low-energy reactant ions. These low-energy reactant ions interacted with neutral analyte vapors to produce product ions.
At the interface between the reaction and drift region a Bradbury-Nielson ion gate was positioned and operated electronically to admit periodic pulses of ions into the drift tube. This design was constructed using parallel Alloy 46 (California Fine Wire Co., Grove Beach, CA) wires (75 μm in diameter) with 0.25 mm spacing as described previously.50 The gate was “closed” by applying ± 24 V to adjacent wires so that a ~ 441 V cm−1 closing field was placed orthogonal to the drift tube of the IMS. As positive or negative ions approach the gate, they were collected on the negative and positive wires, thereby preventing them from passing through the gate. When the gate was “opened” all gate wires were referenced to a single voltage appropriate to the gate’s position in the drift electric field.
Under the influence of the applied electric field, ions in the drift region were directed towards a faraday plate collector electrode against a counter-flow of atmospheric pressure air drift gas at a flow rate of 200 mL min−1. An aperture grid, located just prior to the faraday plate, served to filter out electronic noise generated from opening and closing of the gate and to shield the effect of induced current on the faraday plate from the gas phase ions as they approached the plate.
The electronic components for the IMS system included a high-voltage power supply and software controlled gate drivers designed and constructed at Washington State University. Responses from the faraday detector were processed with a 109 V A−1 amplifier (Model 427, Keithley Instruments, Cleveland, OH). The resulting signal was sent to a personal computer (operating LabVIEW code developed at WSU) through a SHC68-68-EPM noise-rejecting, shielded cable (National Instruments, Austin, TX). The resulting text file data was then processed using Igor Pro 5.0.3 (WaveMetrics, Portland, OR).
Methods
Samples were introduced into the IMS using the exponential dilution approach, as described previously.23,51–52 In this investigation, the exponential dilution device was constructed from a 3.9-liter stainless steel metal canister with a height and width of 20 and 18 cm, respectively. The canister had two openings; one was used as the inlet, and the other as the outlet. The inlet was fitted with a ¼“ stainless steel T-union (Swagelok, Tri-Cities Valve & Fitting Co., Richland, WA, USA). One end of the T-union was connected to the canister, the “T” end was used as the carrier gas inlet, and the third end of the union was sealed with a 10-mm Thermogreen™ septum (Supelco, Bellefonte, PA) to create a gas-tight injection inlet. A one-way valve obtained from Swagelok at the canister outlet was used to move the exiting carrier gas into the IMS or the laboratory exhaust hood. Exponential dilution mixing at 50 °C was initiated by injecting 100 ppbv to 1 ppmv of the analyte studied into the canister via the injection inlet. Diluted analytes then flowed through the one-way valve into the IMS reaction region. The effluent mixing ratio declined exponentially and the concentration at each time step predicted as described in the next section. Figure 1 is a side view of the exponential diluter interfaced with the IMS.
Figure 1.

Side view of the exponential dilution canister used for sample introduction and interfaced with the ambient pressure IMS.
Four compounds: trichloroethylene (TCE), tetrachloroethylene (PCE), methyl tert-butyl ether (MTBE) and methyl iso-butyl ketone (MIBK) (Sigma Aldrich, St. Louis, MO) were employed to measure resolving powers. Sample introduction was achieved by injecting headspace gas aliquots for MTBE & MIBK and liquid aliquots for TCE & PCE into the exponential dilution canister using a gas syringe (Sigma Aldrich Chemical Company). Gate voltages were varied from 100 to 5,000 V in 100-V increments. Each voltage setting was evaluated with gate pulse widths of 100, 200, 300, 400 and 500 μs. Five replicate IMS spectra were obtained at each voltage. The spectra used as replicates to calculate measured resolving powers were taken from successive runs of the same dilution, and it was assumed that the concentrations for all replicates were the same. For each of the four test compounds, Rm was measured and Rc calculated as a function of voltage and gate pulse width. The maximal resolving powers obtained at the optimum conditions were then compared. Detection limits, defined as the quantity of sample required to produce a response equal to three times the root-mean-square noise level, were determined at the conditions for optimum resolving power for each compound.
Calculations
The decay of analyte concentration in an exponential dilution flask is given by:
| (14) |
where Ct was the concentration in units of parts per million volume-to-volume of the analyte remaining in the flask after time, t, Co is the initial concentration of sample in the flask, f is the carrier gas flow (10 mL min−1), and v is the volume of exponential dilution canister (3,900 mL). The procedures used for calculating concentrations have been described previously.23
Detection limits were determined from the spectra; Because these measurements were made near the detection limit a linear response was assumed to estimate the detection limits as follows:23
| (15) |
where R is the detector response (nano-ampere, nA), S is the sensitivity at the detection limit (nA ppmv−1 or nA ppbv−1), Ci is the mixing ratio (ppmv or ppbv) of A and B is the background current (nA). Given that the response of an unknown A (RA) is RA − B, and RA− B = SCi from Eq. 15, then at the detection limit where the detector response of A, is RA = 3σ, (σ is the root mean square of the noise above the background current), it follows that:
| (16) |
Rearranging Eq. 16 for mixing ratio yields:
| (17) |
where Ci(D) is the minimum detectable concentration, and S = RA/C where C is the mixing ratio of A at which the detection limit was determined.
Results and Discussion
Figure 2 shows typical ion mobility spectra obtained with this spectrometer. This investigation demonstrated the effect of gate width on mobility, resolving power and sensitivity. The drift voltage was held constant at 5,000 V and the gate pulse width was varied from 100 to 500 μs. Figures 2a & b show the ion mobility spectra obtained when MTBE and MIBK were introduced into the IMS, respectively.
Figure 2.

Representative IMS spectra for (a) MTBE and (b) MIBK as a function of initial gate pulse widths. Pulse widths of 100, 200, 300, 400, and 500 μs were used. As the initial ion pulse width increases, IMS resolving power decreases with a corresponding increase in the SNR. The drift gas for these experiments was air and the electrical potential across the drift tube was 5,000 V.
In theory, mobility in IMS is not a function of the gate pulse width or voltage but is a function of the size, shape, and charge on the ion. The reduced mobility, Ko, is the ion’s measured mobility adjusted for the operating temperature and pressure. In these studies the reduced mobility of MTBE was found to be 1.63 cm2s−1V−1 and 1.48 cm2s−1V−1 for MIBK. As expected, these mobilities did not change as a function of the gate pulse width.
The signal-to-noise ratio (SNR), however, did change as a function of the gate pulse width. As the ion gate pulse width was decreased, SNR also decreased. For the 500-μs gate pulse width, the SNR was about 92 for both the MTBE and the MIBK, but when the gate pulse width was reduced to 100 μs, the SNR was reduced to 55 for both compounds. This reduction in SNR was not proportional to the reduction in the gate width. When the gate pulse width was decreased by a factor of 5, the SNR was decreased only by a factor of 1.8. In fact, for the wider gate widths, there was little loss in SNR with gate width reduction. Reducing the gate width from 500 μs to 200 μs only decreased the SNR about 11% to 83 while, as we shall see, significantly increasing the resolving power.
With the wide gate pulse width of 500 μs, the measured resolving power, Rm, was relatively low, 9 for MTBE and 11 for MIBK. When the gate pulse width was decreased to 200 μs, the Rm doubled. A further decrease in the gate pulse width to 100 μs increased the Rm to 35. This enhancement in resolving power relative to a minor loss in the SNR implies that gates should be operated with as short gate pulse width for which the decrease in the SNR is not significant. Increasing the gate pulse width does not always equate to improving the SNR but it does reduce resolving power. In addition, it is interesting to note that when the gate pulse width was decreased by a factor of 5, the resolving power was only improved by a factor of 3, indicating that other peak broadening mechanisms besides the initial gate pulse width are important and are contributing to the overall peak width.
The diffusion-limited resolving power, Rd, calculated for 5000 V (Eq. 11) was 133, which is significantly higher than Rm at the same voltage. One reason for this discrepancy between Rm and Rd is that Rd assumes an infinitely small initial gate pulse width. However, the initial gate pulse width as well as the voltage affects the resolving power of an IMS. To more closely predict the resolving power that can be expected under a given set of IMS operating conditions, “conditional” resolving power values were calculated that not only considered voltage, but also the initial peak width of the ions and reduced ion mobility. The conditional IMS resolving powers, Rc, are shown in Figure 3 for a variety of operational voltages and gate pulse widths and compared to Rm for the reactant ion peak (RIP).
Figure 3.

Conditional resolving powers, Rc, calculated for drift potentials from 0 to 5000 V and for initial gate pulse widths from 100 to 500 μs for the RIP (a). The results demonstrate that there is an optimal voltage for each initial width used for the RIP. Contrary to that predicted by the diffusion limited resolving power equation, resolving power does not continue to increase with higher voltages. Measured resolving power, Rm, as a function of drift voltage obtained from IMS spectra for the RIP (b). The data demonstrate that decreasing the gate width increases resolving power and that increasing the drift voltage produces a resolving power maximum at each gate pulse width for the RIP. Note the points at which the response ion disappeared into the background for Rm.
Figure 3a shows a plot of Rc versus voltage for the reactant ion peak. Initially, as the electrical potential across the drift region is increased, resolving power increases as predicted from the diffusion-limited equation. However, a maximal conditional resolving power is reached, and beyond this voltage, resolving power decreases with increasing voltage. For the RIP, the maximal resolving power occurred at 600V when the initial gate pulse width was 500 μs. As the gate pulse width was decreased, the voltage, at which the maximal resolving power occurred, increased. For example, the voltage at which the maximal resolving power for the RIP occurred was at 600, 700, 800, 1100, to 1,700V for gate pulse widths of 500, 400, 300, 200, and 100 μs, respectively.
In practice however, as shown in Figure 3b, true resolving power maxima were only observed for gate pulse widths of 100 and 200 μs. The measured resolving power at the maximum of 100 μs gate pulse width was 51 while the conditional resolving power was a little higher at 64. For the reactant ion peak (RIP), the efficiency of this IMS tube was 80%. Thus there is still some room for improvement even after the contribution of the initial gate width is considered; however the conditional resolving power matched the measured resolving power much closer than did the diffusion-limited resolving power to the measured value.
Similar results were obtained for product ions as shown in Figure 4. In this experiment, conditional resolving powers for product ions of MTBE at a K0 = 1.63 cm2V−1s−1 and MIBK at K0 = 1.48 cm2V−1s−1 were calculated for various initial gate pulse widths as a function of drift voltage. In all cases, conditional resolving power maxima, shown in Figures 4a and 4b, were observed and occurred at voltages that depended on the initial gate pulse width and the mobility of the ion investigated. For the product ions of MTBE and MIBK, conditional resolving powers ranged from a low of 11 at a gate width of 500 μs and a voltage of 5,000 V, to a high of 72 at a gate width of 100 μs and a voltage of 2,200 V. This result demonstrates the major point of this paper. For many IMS instruments developed for field applications, the practical operating conditions are such that the resolving power is inversely proportional to the drift voltage, rather than proportional to the square of the drift voltage as is predicted from diffusion theory.
Figure 4.


Conditional resolving powers, Rc, calculated for drift potentials from 0 to 5,000 V and for initial gate pulse widths from 100 to 500 μs for the product ion peak for (a) MTBE; and (b) MIBK. These results demonstrate that there is an optimal voltage for each initial width used and for each compound. Contrary to that predicted by the diffusion limited resolving power equation, resolving power does not continue to increase with higher voltages. Measured resolving power, Rm, as a function of drift voltage obtained from IMS spectra for the product ion peaks for (c) MTBE, and (d) MIBK. These data demonstrate that decreasing the gate width increases resolving power and the voltage limit of detection, and that increasing the drift voltage produces a resolving power maximum which is unique for each compound. Note the points in (c) and (d) at which the response ions disappeared into the background.
Figures 4c and 4d show the measured resolving power values for the product ions of MTBE and MIBK. While they are lower than the calculated conditional values shown in Figures 4a and 4b, the trends with respect to gate pulse widths and drift voltage are similar. Narrower initial gate pulse widths and lower drift voltages provide higher resolving powers. In addition, as predicted by the conditional resolving power equation (Eq. 12), there was a unique drift voltage for each ion and gate pulse width which produced a resolving power maximum. For the RIP, the maximum was predicted to occur at 1,700 V, but the actual measured maximum occurred at 1,800 V. Maximal conditional resolving powers, however, for MIBK, and MTBE were predicted from Eq. 12 to be 2,200 V and 2,100 V, respectively, and also measured to be 2,200 and 2,100 V, respectively. Despite slight variations with the optimal voltage for the reactant ion peak, our results showed that the Rc can accurately predict the voltage at which one should operate an IMS for the highest resolving power.
It is important to note that for the measured values, resolving power maxima were noticeable only for the 100 μs and 200 μs initial gate width conditions. As the voltage is lowered and the drift time of the ion increases, diffusion broadens the ion peak and reduces the signal to noise ratio. Figure 5 shows a comparison of two mobility spectra of MIBK taken at drift voltages of 3,000 V and the resolving power optimal of 2,100 V. For the lower voltage, the peak width is significantly broadened and the SNR is reduced. Figure 6 shows a series of spectra for MIBK & MTBE at various voltages. The spectrum at the bottom of the graph for MIBK (6a) was taken with a drift voltage of 5,000 V. The subsequent stacked spectra were taken at decreasing voltage increments of 100 V to a minimum of 1,900 V, where the peak for the response ion disappeared into the background. A similar result is shown for MTBE (6b) where the response ion disappeared into the background at 1,700 V.
Figure 5.

The effect of voltage on the response of MIBK. Increase voltage resulted in increased SNR for the MIBK product ions.
Figure 6.
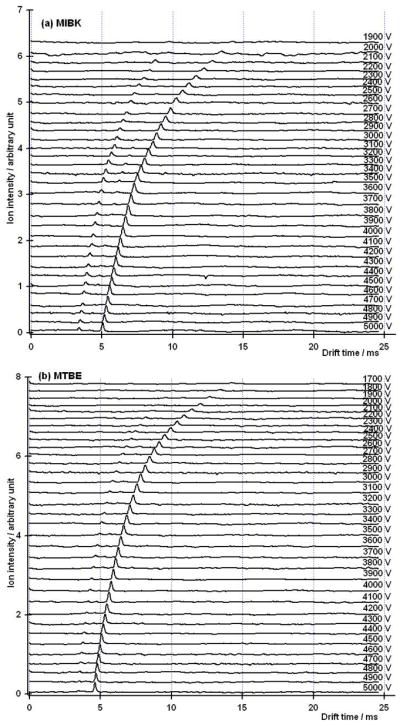
Ion mobility spectra at a gate pulse width of 100 μs and voltages ranging from 1,900 to 5,000 V for the product ions of MIBK (a), 1,700–5,000 V for the product ions of MTBE (b). Mixing ratios for MTBE and MIBK were 100 ppbv.
Thus, as with all analytical instruments, the selection of the best operating conditions is a compromise between sensitivity and resolving power. Figure 7 is a conditional resolving power plot similar to those in Figures 3 and 4, with the exceptions that this plots Rc as a function of the V0.5 for TCE and PCE. From this investigation the optimal resolving power was obtained at 1,200 V but the operating voltage chosen for field work was 3,100 V, due to the increased sensitivity of the signal at that voltage.
Figure 7.

Plot of conditional resolving power vs. the square root of the operating voltage for the environmental contaminants TCE (diamonds) & PCE (triangles). Both plots were obtained with a 200 μs gate pulse width (tg). The result showed that the optimum voltage for the two contaminants was 1,200 V, but the voltage chosen for operation in the field was 3,100 V to the increase sensitivity of the signal.
Nevertheless, detection limits at the optimal resolving power settings can be impressive. Figure 8 shows representative ion mobility spectra near the detection limit at the resolving power maxima for (a) MTBE and (b) MIBK. The first peak is the reactant ion peak and peak 1 in Figure 8a is 18 pptv of MTBE and peak 1 in Figure 8b is 24 pptv of MIBK. While these values were reproduced several times, they are estimates of the mixing ratio from the exponential dilution flask experiment. Table 2 provides a detailed summary of the results from our investigations, including calculated diffusion-limited resolving powers, Rd, calculated conditional resolving powers, Rc, and measured resolving powers, Rm.
Figure 8.

Ion mobility spectra of the minimum detectable amount at the optimum voltage and gate pulse width of 100 μs for (a) MTBE, and (b) MIBK. The reduced mobility value for peak 1 in (a) and (b) were 1.91 and 1.77 cm2 V−1 s−1, respectively, indicating that they were the monomer ions of both MTBE and MIBK.
Table 2.
Table summarizing experimental results.
| Compound | Reduced Mobility (cm2/Vs), K0 | Optimal Drift Voltage (V) | Calculated Diffusion- limited Resolving Power, Rd | Calculated Conditional Resolving power, Rc | Measured Resolving power, Rm | Efficiency (%), Rm/Rc(100%) | Detection limit at Rmax |
|---|---|---|---|---|---|---|---|
| MTBE | 1.63 | 2100 | 86 | 70 | 52 ± 0.12 | 74 | 18 pptv |
| MIBK | 1.48 | 2200 | 88 | 72 | 52 ± 0.11 | 72 | 24 pptv |
| PCE | 1.33 | 2400 | 92 | 75 | 42 ± 0.33 | 56 | 80 ppbv |
| TCE | 1.65 | 2100 | 86 | 70 | 47 ± 0.21 | 67 | 60 ppbv |
| RIP | 2.14 | 1800 | 80 | 64 | 51 ± 0.11 | 80 | - |
These experiments were designed to demonstrate how conditional resolving power calculations can be used to optimize IMS instruments. Eq. 13 enables the estimation of the efficiency of the drift tube. Our calculated efficiency fell between 56–74% of the maximal conditional resolving power possible. Thus, Rc, as introduced in this paper, provides a way to rapidly predict where the optimal operating condition of an IMS will be and what it will be (or at least the best it can be). The efficiency is a measure of how well the instrument is constructed. In our case it was 74%, so there was still room for improvement. We suspect that the field homogeneity may be the reason why the efficiency did not equal 100%, but it may also be due to the construction of the detector or coulombic repulsion. Smaller instruments are more prone to additional band spreading mechanisms than larger instruments.
Conclusions
Small, field-deployable IMS instruments do not normally operate in the diffusion-limited mobility region but rather in the gate-controlled region. This means that for most instruments, resolving power can be improved (at the expense of sensitivity) by reducing the drift voltage. The most important IMS instrumental parameters for determining maximal resolving power are voltage and gate pulse width. There is a specific optimum voltage, gate pulse width and resolving power for each ion. Diffusion-limited resolving powers indicate the maximal resolving power possible that are not normally achievable under field conditions. Conditional resolving power predicts the maximal resolving power possible for a given set of instrument conditions, while the measured resolving power reports the actual resolving power of an ion mobility spectrometer. The ratio of the measured resolving power to the conditional resolving power provides a practical measure of the efficiency of an IMS tube. As the efficiency approaches 100%, peak broadening effects of field non-homogeneity, coulombic repulsion and detector response time are diminished. Given that IMS determines the collision cross-section of ions, one must also consider that peak broadening might also be due to the presence of structural isomers; this is particularly true when the resolving power is very high. The maximal measured resolving power of the small subsurface IMS used in this study ranged from 42–54, producing IMS resolving power efficiency that ranged from 56 to 74% of the maximal conditional resolving power. Detection limits determined at the maximal resolving power conditions ranged from 18 pptv to 80 ppbv for the soil-gas contaminants tested.
Acknowledgments
We gratefully acknowledge the support of EPA Awards X-97031101-0 and X-97031102-0. The authors also thank Kevin Ryan, formerly a research assistant at Boise State University, for his support in this investigation.
References
- 1.Cohen MJ, Karasek FW. J Chromatogr Sci. 1970;8:330. [Google Scholar]
- 2.Collins DC, Lee ML. Anal Bioanal Chem. 2002;372:66. doi: 10.1007/s00216-001-1195-5. [DOI] [PubMed] [Google Scholar]
- 3.Eiceman GA, Karpas Z. Ion Mobility Spectrometry. 2. CRC Press; Boca Raton, FL: 2005. [Google Scholar]
- 4.Luong J, Gras R, Van Meulebroeck R. J Chromatogr Sci. 2006;44:276. doi: 10.1093/chromsci/44.5.276. [DOI] [PubMed] [Google Scholar]
- 5.Baumbach JI. Anal Bioanal Chem. 2006;384:1059. doi: 10.1007/s00216-005-3397-8. [DOI] [PubMed] [Google Scholar]
- 6.Buryakov IA. Technical Physics Letters. 2006;32:67. [Google Scholar]
- 7.Buryakov IA. NATO Science Series, II: The analytical characteristics of ion mobility increment spectrometer during the detection of explosive vapors and products of their degradation. Mathematics, Physics and Chemistry. 2004;167:113. (Vapor and Trace Detection of Explosives for Anti-Terrorism Purposes) [Google Scholar]
- 8.Gruznov VM, Baldin MN, Filonenko VG. High-speed gas analysis for explosives detection, NATO Science Series, II. Mathematics, Physics and Chemistry. 2004;167:87. (Vapor and Trace Detection of Explosives for Anti-Terrorism Purposes) [Google Scholar]
- 9.Marr AJ, Groves DM. IJIMS. 2003;6:59. [Google Scholar]
- 10.Neves JL, Haigh PE, Wu C, McGann WJ. IJIMS. 2003;6:1. [Google Scholar]
- 11.Perr JM, Furton KG, Almirall JR. Journal of Separation Science. 2005;28:177. doi: 10.1002/jssc.200401893. [DOI] [PubMed] [Google Scholar]
- 12.Asbury GR, Klasmeier J, Hill HH., Jr Talanta. 2000;50:1291. doi: 10.1016/s0039-9140(99)00241-6. [DOI] [PubMed] [Google Scholar]
- 13.Wu C, Siems WF, Hill HH. Anal Chem. 2000;72:396. doi: 10.1021/ac9907235. [DOI] [PubMed] [Google Scholar]
- 14.Miki A, Tatsuno M, Katagi M, Nishikawa M, Tsuchihashi H. Japanese Journal of Forensic Toxicology. 1997;15:142. [Google Scholar]
- 15.Carroll John J, Le Tri, DeBono R. Am Lab. 2004;36:32. [Google Scholar]
- 16.Debono R, Stefanou S, Davis M, Walia G. Pharmaceutical Technology North America. 2002;26:72. [Google Scholar]
- 17.Lawrence AH. Anal Chem. 1989;61:343. doi: 10.1021/ac00179a012. [DOI] [PubMed] [Google Scholar]
- 18.Keller T, Keller A, Tutsch-Bauer E, Monticelli F. Forensic Science International. 2006;161:130. doi: 10.1016/j.forsciint.2006.03.032. [DOI] [PubMed] [Google Scholar]
- 19.Gillen G, Mahoney C, Wight S, Lareau R. Rapid Commun Mass Spectrom. 2006;20:1949. doi: 10.1002/rcm.2531. [DOI] [PubMed] [Google Scholar]
- 20.Kanu AB, Haigh PH, Hill HH., Jr Anal Chim Acta. 2005;553:148. [Google Scholar]
- 21.Tarver Edward E. Sensors. 2004;4:1. [Google Scholar]
- 22.Steiner WE, Haigh PH, Clowers BH, Hill HH. Anal Chem. 2003;75:6068. doi: 10.1021/ac034349r. [DOI] [PubMed] [Google Scholar]
- 23.Kanu AB, Hill HH, Jr, Gribb M, Walters R. J Environ Monit. 2007;9:51. doi: 10.1039/b610493b. [DOI] [PubMed] [Google Scholar]
- 24.Clowers BH, Steiner WE, Dion HM, Matz LM, Tam M, Tarver EE, Hill HH., Jr Field Anal Chem Techno. 2001;5:302. [Google Scholar]
- 25.Hoaglund-Hyzer CS, Lee YJ, Counterman AE, Clemmer DE. Anal Chem. 2002;74:992. doi: 10.1021/ac010837s. [DOI] [PubMed] [Google Scholar]
- 26.Reategui J, Bacon T. Vapors Proc Control Qual. 1992;3:209. [Google Scholar]
- 27.Palmer PT, Limero TF. J Am Soc Mass Spectrom. 2001;12:656. doi: 10.1016/S1044-0305(01)00249-5. [DOI] [PubMed] [Google Scholar]
- 28.Gordon G, Pacey G, Bubnis B, Laszewski S, Gaines J. Chem Oxid. 1997;4:23. [Google Scholar]
- 29.Spangler GE. Int J Mass Spectrom. 2002;220:399. [Google Scholar]
- 30.Siems WF, Wu C, Tarver EE, Hill HH, Larsen PR, McMinn DG. Anal Chem. 1994;66:4195. [Google Scholar]
- 31.Rokushika S, Hatano H, Baim MA, Hill HH., Jr Anal Chem. 1985;57:1902. [Google Scholar]
- 32.Dugourd Ph, Hudgins HH, Clemmer DE, Jarrold MF. Rev Sci Instrum. 1997;68:1122. [Google Scholar]
- 33.Wu C, Siems WF, Klasmeier J, Hill HH., Jr Anal Chem. 2000;72:391. doi: 10.1021/ac990601c. [DOI] [PubMed] [Google Scholar]
- 34.Hill HH, Jr, Siems WF, St Louis RH, McMinn DG. Anal Chem. 1990;62:1201A. doi: 10.1021/ac00222a001. [DOI] [PubMed] [Google Scholar]
- 35.Carrico JP, Sickenberger DW, Spangler GE, Vora KN. J Phys E Sci Instrum. 1983;16:1058. [Google Scholar]
- 36.Spangler GE, Vora KN, Carrico JP. J Phys E Sci Instrum. 1986;19:191. [Google Scholar]
- 37.Spangler GE, Collins CI. Anal Chem. 1975;47:403. [Google Scholar]
- 38.Moseley JT, Gatland IR, Martin DW, McDaniel EW. Phy Rev. 1969;178:234. [Google Scholar]
- 39.Spangler GE, Miller RA. Int J Mass Spectrom. 2002;214:95. [Google Scholar]
- 40.Watts P, Wilders A. Int J Mass Spectrom Ion Process. 1992;112:179. [Google Scholar]
- 41.Schummers JH, Thomson GM, James DR, Gatland IR, McDaniel EW. Phy Rev. 1973;66:683. [Google Scholar]
- 42.Mason EA, McDaniel EW. Transport Properties of Ions in Gases. Wiley; New York: 1988. [Google Scholar]
- 43.Spangler GE. Anal Chem. 1992;64:1312. [Google Scholar]
- 44.Spangler GE. Anal Chem. 1993;65:3010. [Google Scholar]
- 45.Albritton DL, Martin DW, McDaniel EW, Miller TM, Mosely JT. Technical Report. Georgia Institute of Technology; Atlanta, GA: May 10, 1967. Measurement of the Low-Energy Transport Parameters of Mass Identified Ions in Gases; Mobilities of H3+ and H1+ Ions in Hydrogen. [Google Scholar]
- 46.Wu C, Steiner WE, Tornatore PS, Matz LM, Siems WF, Atkinson DA, Hill HH., Jr Talanta. 2002;57:123. [PubMed] [Google Scholar]
- 47.Carr TW. Plasma Chromatography. Plenum Publication Corporation; New York: 1984. [Google Scholar]
- 48.Spangler GE, Vora KN, Carrico JP. J Phys E Sci Instrum. 1986;19:191. [Google Scholar]
- 49.Wu C, Siems WF, Asbury GR, Hill HH., Jr Anal Chem. 1998;70:4929. doi: 10.1021/ac980414z. [DOI] [PubMed] [Google Scholar]
- 50.Eatherton RL. PhD Thesis. Washington State University; 1987. Development in Ion Mobility Detection for Capillary Chromatography. [Google Scholar]
- 51.Munro AM, Thomas CLP, Langford ML. Anal Chim Acta. 1996;375:49. [Google Scholar]
- 52.Thomas CLP, Rezgui ND, Kanu AB, Munro AM. IJIMS. 2002;5:31. [Google Scholar]