

Abstract
Fourier transform ion cyclotron resonance (FTICR) provides the highest resolving power of any commercially available mass spectrometer. This advantage is most significant for species of low mass-to-charge ratio (m/z), such as metabolites. Unfortunately, FTICR spectra contain a very large number of data points, most of which are noise. This is most pronounced at the low m/z end of spectra, where data point density is the highest but peak density low. We therefore developed a filter that offers lossless compression of FTICR mass spectra from singly charged metabolites. The filter relies on the high resolving power and mass measurement precision of FTICR and removes only those m/z channels that cannot contain signal from singly charged organic species. The resulting pseudospectra still contain the same signal as the original spectra but less uninformative background. The filter does not affect the outcome of standard downstream chemometric analysis methods, such as principal component analysis, but use of the filter significantly reduces memory requirements and CPU time for such analyses. We demonstrate the utility of the filter for urinary metabolite profiling using direct infusion electrospray ionization and a 15 tesla FTICR mass spectrometer.
Electronic supplementary material
The online version of this article (doi:10.1007/s11306-010-0202-2) contains supplementary material, which is available to authorized users.
Keywords: Data compression, Mass spectrometry, Electrospray, FTICR, Metabolites, Urine
Introduction
A wide range of metabolites have been identified and quantified in body fluids by techniques such as gas and liquid chromatography, NMR and mass spectrometry (MS). Analytical strategies are either direct, solely relying on NMR or MS, or hyphenated with one or more dimensions of GC and LC prior to NMR, MS or another detector. Examples of common hyphenations are LC-NMR, GC-MS and LC-MS. High resolution instruments such as high-field NMR and Fourier transform ion cyclotron resonance (Marshall et al. 1998) (FTICR) MS are especially powerful as they can resolve, identify and quantify a large number of metabolites by direct analysis, also of complex samples. Although MS is not as inherently quantitative as NMR for small molecules, where the signal is directly proportional to the total number of hydrogen atoms in a particular chemical environment in the sample, it is much more sensitive and provides information complementary to that from NMR for the identification of metabolites. Many types of mass spectrometry are routinely used for measuring metabolites in urine (Pasikanti et al. 2008), most often in hyphenation with other separation techniques, but also by direct infusion (Erve et al. 2008, Beckmann et al. 2008). At the low m/z range of interest in metabolomics, FTICR has the highest resolving power of any commercially available type of mass analyzer. High magnetic field FTICR also has a wide dynamic range (104–105) (Limbach et al. 1993, Palmblad et al. 2000), especially in comparison with other ion trapping instruments. As with most types of mass spectrometry, signal depends on the ionization and transmission efficiency. However, under ideal circumstances, these are nearly constant between samples for the same species, and the ion signal from a particular metabolite can thus be compared between samples.
Metabolites can be putatively identified from the elemental compositions derived from highly accurate mass measurements (Aharoni et al. 2002). Additionally, measured isotopic envelopes can be fitted to predicted distributions for confirmation, such as in the Sigma Fit™ algorithm (Bruker Daltonics, Billerica, MA) or a special case of AUTOHD (Palmblad et al. 2001). To confidently identify metabolites, selected compounds can be analyzed by MS/MS, or MS/MS spectra can be acquired in an automated and data-dependent mode during LC-MS/MS to build a database of identified metabolites. In metabolomics, urine is among the most commonly studied and clinically relevant body fluids. Urine contains a wide range of compounds, such as salts, urea, organic acids, amines, sugars, steroids, amino acids, peptides and proteins, which vary greatly in terms of physiochemical properties as well as in relative abundance (Kind and Fiehn, 2007, Park et al. 2009). For routine analysis of such biologically derived samples by mass spectrometry, we need both a robust sample preparation technique for removing interfering compounds and a simple or automated data analysis method. In our laboratory we rely on off-line solid-phase extraction (SPE) for sample cleanup. This is not necessarily a very fast technique compared to the time required for direct analysis by MS, but since it is off-line, it does not cost valuable time on the mass spectrometer. It can also be highly parallelized, for instance by using a 96-well plate format. Off-line sample cleanup also allows optimization of flow rates and solvent compositions without respect to any limitations of the electrospray ion source. To retain as much information from the spectrum as possible in the data analysis, one would ideally use the raw data files or profile spectra. As in NMR, FTICR spectra files are very large (millions of data points) which presents a challenge in handling and analyzing large numbers of spectra. Most methods to reduce data size or dimensionality, for example binning or peak picking, inherently lose information. While these techniques are certainly very useful, we investigated an alternative data processing method using theoretical knowledge of the possible m/z of singly charged metabolites. By retaining only those m/z channels from the spectra that can possibly contain useful information, data size can be reduced to facilitate analysis without loss of information.
Materials and methods
Sample preparation
The samples used to develop the data filter were all aliquots of second morning mid-stream urine from healthy male volunteers. Within 5 h of collection, the samples where filtered with a 45 μm syringe filter and H3BO3 was added to a concentration of 10 mM as preservative (Thongboonkerd 2007, Lee et al. 2008). The samples where stored at −80°C until analysis. After thawing, the samples were acidified by adding formic acid to 0.5% v/v. For desalting and removal of urea, a Spark Symbiosis™ (Spark Holland BV, Emmen, The Netherlands) system with 20 mg HD-C18 cartridges where used for solid phase extraction. First, the C18 material was wetted using 2 ml HPLC grade methanol with 0.5% formic acid and equilibrated with 2 ml 2% HPLC grade methanol in water with 0.5% formic acid. Each 2 ml sample was applied to the column material by the Symbiosis™ autosampler. Samples were then washed by 2 ml of equilibration solution and eluted in 200 μl 50% methanol in water with 0.5% formic acid, using the autosampler as fraction collector, as previously described by Balog et al. (2009). In each step the mobile phase was pumped trough the column material under high pressure in the Symbiosis™ system. During the procedure and following measurements, samples where kept at 4°C or on ice. All samples were diluted tenfold with a 50% methanol, 0.5% formic acid for optimal electrospray performance.
Mass spectrometry
All samples were injected into a Bruker 15 tesla solariX™ FTICR mass spectrometer (Bruker Daltonics, Billerica, MA) using the built-in syringe pump with a 100 μl syringe and a flow rate of 120 μl/h in the standard electrospray source assembly with a capillary voltage of 4500 V. A mass spectrum of 221 (~2 million) data points in the m/z range 200−600 was acquired for each sample by adding 64 individual spectra. The syringe, all tubing and source were cleaned between each sample, first with 50% methanol and then 80% methanol in water with 0.5% formic acid. All acquired spectra where checked by superposition to see if internal calibration was necessary and then exported to ASCII “.xy” files using DataAnalysis version 4.0.234 (Bruker) using an automation script.
Data analysis
To remove noise from the data, a filter program, msfilter, was designed and implemented in C. The filter calculates all possible masses of singly charged ions of organic compounds using nested for-loops, one each for the elements H, C, N, O, S and P, taking into account the nitrogen rule (Sparkman 2007), allowing a few double bonds or aromatic systems (Kind and Fiehn 2007) and finally adding the mass of a charge carrier (H+, Na+ or K+). This results in a filter representing those m/z channels that could theoretically contain signal from singly charged metabolites. The filter was then applied to real spectra allowing a relative peak width of 10 ppm (of the m/z) and keeping a data point only if its m/z corresponded to a mass allowed by the filter, that is within the allowed peak width from a calculated ion mass. The resulting filtered spectrum is referred to as a pseudospectrum, similarly to the data compression method described by Lange and Senko (2008). The filter is lossless in the sense that no real (chemical) signal is likely to be lost in the filtering process. In the strictest sense, the filter is still lossy as it is theoretically possible that signals from rare species not considered in the design of the filter (i.e. species that contain halogens or transition metals) are lost. However, for simplicity we refer to this filter as “lossless”. Analysis of the lossless filter and pseudospectra revealed that more compression can be gained by applying a more stringent filter. Therefore we also constructed an alternative minimal filter, which removes all the channels outside the ion m/z observed in urine samples. However, this filter is not guaranteed to be lossless, as some previously unobserved species could fall outside the region defining the filter.
To assess the performance of the filters and whether they interfere with or alter the outcome of subsequent data analysis, we used a common first step in chemometric data processing: principal component analysis (PCA), performed in R version 2.8.1. We compared the filtered data with a robust ‘peak picking’ method using integration of predefined (peak) m/z regions using simple spectrum integration (see Appendix in Supplementary Material). The vendor data analysis software and most publicly available peak picking routines are designed to provide peak lists containing only peaks above some peak shape and signal thresholds. This results in peak lists for different samples with different peaks and different numbers of peaks, which in turn requires reformatting the peak lists and adding in missing peaks before applying standard chemometric methods requiring well-defined data matrices as input. The filter software as well as the peak integration routine can be downloaded from www.ms-utils.org/msfilter. At the moment, the programs are only available as command-line tools. The filter program was compiled and tested with GCC version 3.4.4 under Cygwin and 32-bit Windows XP as well as 64-bit Debian 4.0 Linux. The software currently supports only tabulated ASCII data, such as the “.xy” format exported from DataAnalysis, but future versions will also contain support for the open standard mzXML (Pedrioli et al. 2004) or mzML (Deutsch 2008) formats. Unlike most vendor formats, the open formats encode the abscissa (m/z axis) explicitly for each spectrum, making it possible to store pseudospectra in the same file format as the original spectra.
Results and discussion
Data compression
The compression obtained with the lossless filter was approximately 50% for singly charge species at m/z 250 and >80% at m/z 100. The data point density or digital resolution in FTICR mass spectrometry is inversely proportional to m/z, as m/z is inversely proportional to the ion cyclotron resonance frequency (which is sampled uniformly). Filtering data therefore results in a significant file size reduction for spectra covering low m/z. The acquired single raw spectra containing 221 (2,097,152) data points, are about 8 MB in binary or 60 MB in easily readable ASCII format. The rationale behind the minimal (but potentially lossy) filter is illustrated in Fig. 1 with the distribution of masses around the integer mass as a function of mass. Figure 2 shows the effect of both filters (green and red) on a raw data (blue) as a function of data channel (a) and m/z (b). When the lossless filter is applied to a complete spectrum covering m/z 200–600, the size is reduced to 40% of the original size, or 23 MB file in ASCII format, containing exactly the same information. Most of the channels removed by the filter are between the distributions of peaks near each integer mass. However, for high-resolution spectra such as obtained from a 15 tesla FTICR system, the filter also removes some channels within these distributions. The minimal filter provides even more compression, down to 26% of the original file size. The compressed ASCII files are still larger than the original binary files in their proprietary format. However, the binary format can not be read by most third party software, so the ASCII format is needed as an intermediary. The ASCII format is only used to transfer data between vendor and third party data analysis software and never for storage. Importantly, in third party software such as MATLAB and R the internal storage requirement is the same whether data is read in binary or ASCII format. The need to compress data is more obvious when one considers a larger number of samples or spectra. For instance, a matrix of 25 spectra covering m/z 200–600 is about 1.7 GB in ASCII format, whereas the lossless filtered matrix is 530 MB, about a third of the original and small enough to be handled by a standard desktop computer. The absolute gains will be smaller for mass spectrometers that yield data at lower resolution and with fewer data points per spectrum, but the demonstration of the filter on data from a high-field FTICR is relevant since such instruments are frequently used for these types of direct analyses.
Fig. 1.
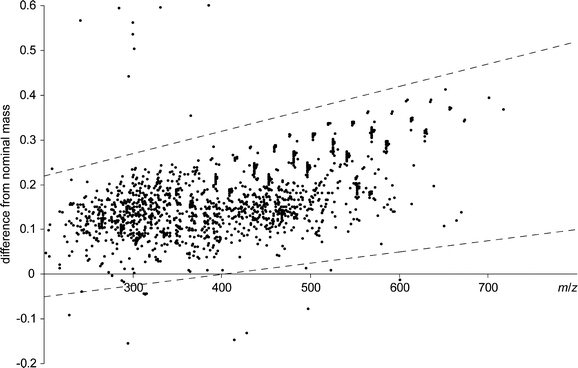
Distribution of differences from nominal mass in an FTICR mass spectrum of urine. The minimal filter is defined by the boundaries indicated by the dashed lines and only masses between these boundaries are kept. The minimal filter is more restrictive than the lossless filter, but may remove species with exotic elemental composition not taken into account when defining the filter
Fig. 2.
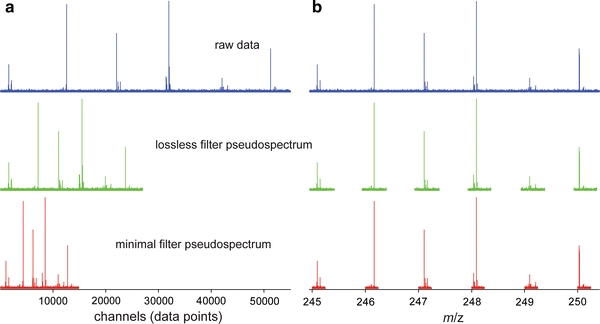
Compressed pseudospectra generated by the lossless filter (middle) and the minimal filter (bottom) relative to the raw urine spectrum (top) as functions of data point or channel (a) and m/z (b). The compression in this m/z region is about twofold for the lossless filter and threefold when using the minimal filter. The channels removed by the lossless filter cannot contain signal from singly charged organic molecules containing only C, H, O, N, S, P and a charge carrier (H+, Na+ or K+)
Data processing and chemometrics
Filtered files lead to a significant increase in processing speed. For example, 100 PCA analyses on a matrix of 25 spectra on a 3.0 GHz Intel Xeon X5365 CPU took 138 min for unfiltered data but only 35 min on filtered data. The filtering itself takes about 8 s per spectrum (2 s for calculations and 6 s for file I/O) on the same computer. This is done once and the filtered spectrum can be used repeatedly during data analysis. The filter should not influence downstream data analysis methods. To test this, we used PCA as it is a popular unsupervised method for finding patterns in multidimensional data, such as NMR and mass spectra, and commonly used to study variability in large-scale studies (Takahashi et al. 2008). Figure 3a shows that it is possible to separate the raw spectra from two sets of urine and blanks using PCA. The first principal component accounts for 51% of the variation, while the second accounts for another 16%. The separation between the sample groups in first two principal components is similar for unfiltered (Fig. 3a), lossless (b) and minimal (c) filtered spectra. This shows that the filtering does not interfere with multivariate methods such as PCA. For comparison, Fig. 3d shows the PCA for the same spectra after extracting 351 predefined peaks found by DataAnalysis in a representative urine spectrum acquired with the same instrument parameters. As peak picking inherently removes some of the information, it results in a partial representation of the data rather than a compact representation of the raw data with all valuable information preserved. Analysis of filtered raw calibrated data may be a feasible and robust alternative to peak picking, even if many highly correlated and therefore redundant variables are still present, e.g. the different data points across a peak or different isotopic peaks. A more subtle but practically important advantage of the filter method over intensity-based peak lists or culling of the spectra is that the resulting filtered pseudospectra are of uniform dimensionality regardless of spectral content and therefore ready for use with standard chemometrics software. Good calibration or at least high measurement precision is critical. In the urine spectra used to evaluate the filter program the m/z drift between spectra was much smaller than the distance between data points and therefore does not play a significant role, but if necessary, internal calibration is easily automated using a few compounds present in all samples or by adding ‘lock mass’ compounds to the electrospray solution or interface. In combination with the filter, PCA and related chemometric methods enable simple, robust and unbiased analysis of FTICR mass spectrometry data of small organic molecules such as metabolites. The filter can in principle be extended also to negative ions, accounting for elements and species that preferentially form adducts with negatively charged species.
Fig. 3.
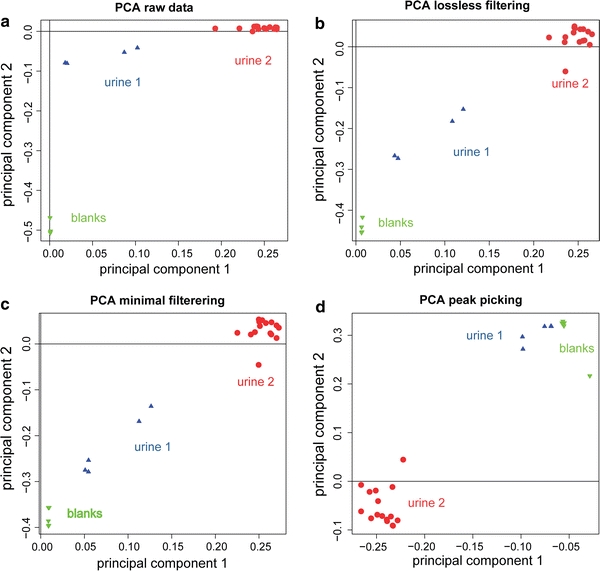
Principal component analysis of two sets of urine samples (from two different healthy male volunteers) and a blank for raw, unfiltered (a), lossless filtered (b), minimal filtered (c) and peak picked (d) spectra. The separation between the groups is apparently less good in d, which could be explained by the dominance of common background peaks in the peak list at the expense of informative, sample-dependent peaks
Conclusions
The simple idea of filtering out m/z channels that cannot contain signal from singly charged species from high resolution FTICR mass spectra of metabolites or other small molecules can lead to two- to four-fold reduction in memory requirement and processing times for common multivariate methods in chemometrics such as PCA without compromising the results. This enables datasets to be processed on a regular desktop computer with a standard 32-bit operating system, that would otherwise require a more powerful computer and a 64-bit operating system. We speculate that similar filters can be applied in other types of mass spectrometry, including hyphenated methods, although the advantage is not likely to be as significant as for high-field FTICR where the data point density and resolving power is highest at low m/z where the peak density typically is the lowest. The filter and general method described here fit well into high-throughput robust analysis pipelines based on high resolution FTICR mass spectrometry.
Acknowledgements
The authors wish to thank Dr. Alex Henneman and Dr. Rico Derks for technical assistance and Dr. Oleg Mayboroda for helpful comments on the manuscript.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Appendix
Below is the link to the electronic supplementary material.
References
- Aharoni A, Ric De Vos CH, Verhoeven HA, et al. Nontargeted metabolome analysis by use of Fourier transform ion cyclotron mass spectrometry. OMICS. 2002;6:217–234. doi: 10.1089/15362310260256882. [DOI] [PubMed] [Google Scholar]
- Balog CI, Hensbergen PJ, Derks R, et al. Novel automated biomarker discovery work flow for urinary peptidomics. Clinical Chemistry. 2009;55:117–125. doi: 10.1373/clinchem.2008.108795. [DOI] [PubMed] [Google Scholar]
- Beckmann M, Parker D, Enot DP, Duval E, Draper J. High-throughput, nontargeted metabolite fingerprinting using nominal mass flow injection electrospray mass spectrometry. Nature Protocol. 2008;3:486–504. doi: 10.1038/nprot.2007.500. [DOI] [PubMed] [Google Scholar]
- Deutsch E. mzML: a single, unifying data format for mass spectrometer output. Proteomics. 2008;8:2776–2777. doi: 10.1002/pmic.200890049. [DOI] [PubMed] [Google Scholar]
- Erve JC, Demaio W, Talaat RE. Rapid metabolite identification with sub parts-per-million mass accuracy from biological matrices by direct infusion nanoelectrospray ionization after clean-up on a ZipTip and LTQ/Orbitrap mass spectrometry. Rapid Communications in Mass Spectrometry. 2008;22:3015–3026. doi: 10.1002/rcm.3702. [DOI] [PubMed] [Google Scholar]
- Kind T, Fiehn O. Seven Golden Rules for heuristic filtering of molecular formulas obtained by accurate mass spectrometry. BMC Bioinformatics. 2007;8:105. doi: 10.1186/1471-2105-8-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange, O., & Senko, M. W. (2008). Patent number: EP1950690 (A1).
- Lee RS, Monigatti F, Briscoe AC, Waldon Z, Freeman MR, Steen H. Optimizing sample handling for urinary proteomics. Journal of Proteome Research. 2008;7:4022–4030. doi: 10.1021/pr800301h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limbach PA, Grosshans PB, Marshall AG. Experimental determination of the number of trapped ions, detection limit, and dynamic range in Fourier transform ion cyclotron resonance mass spectrometry. Analytical Chemistry. 1993;65:135–140. doi: 10.1021/ac00050a008. [DOI] [Google Scholar]
- Marshall AG, Hendrickson CL, Jackson GS. Fourier transform ion cyclotron resonance mass spectrometry: A primer. Mass Spectrometry Reviews. 1998;17:1–35. doi: 10.1002/(SICI)1098-2787(1998)17:1<1::AID-MAS1>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Palmblad M, Buijs J, Håkansson P. Automatic analysis of hydrogen/deuterium exchange mass spectra of peptides and proteins using calculations of isotopic distributions. Journal of American Society for Mass Spectrometry. 2001;12:1153–1162. doi: 10.1016/S1044-0305(01)00301-4. [DOI] [PubMed] [Google Scholar]
- Palmblad M, Håkansson K, Håkansson P, et al. A 9.4 T fourier transform ion cyclotron resonance mass spectrometer—description and performance. European Journal of Mass Spectrometry. 2000;6:267–275. doi: 10.1255/ejms.348. [DOI] [Google Scholar]
- Park EM, Lee E, Joo HJ, Oh E, Lee J, Lee JS. Inter- and intra-individual variations of urinary endogenous metabolites in healthy male college students using H-1 NMR spectroscopy. Berlin: Walter De Gruyter & Co; 2009. [DOI] [PubMed] [Google Scholar]
- Pasikanti KK, Ho PC, Chan EC. Development and validation of a gas chromatography/mass spectrometry metabonomic platform for the global profiling of urinary metabolites. Rapid Communications in Mass Spectrometry. 2008;22:2984–2992. doi: 10.1002/rcm.3699. [DOI] [PubMed] [Google Scholar]
- Pedrioli PG, Eng JK, Hubley R, et al. A common open representation of mass spectrometry data and its application to proteomics research. Nature Biotechnology. 2004;22:1459–1466. doi: 10.1038/nbt1031. [DOI] [PubMed] [Google Scholar]
- Sparkman DO. Mass spectrometry desk reference. Pittsburgh: Global View Pub; 2007. [Google Scholar]
- Takahashi H, Kai K, Shinbo Y, et al. Metabolomics approach for determining growth-specific metabolites based on Fourier transform ion cyclotron resonance mass spectrometry. Analytical and Bioanalytical Chemistry. 2008;391:2769–2782. doi: 10.1007/s00216-008-2195-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thongboonkerd V. Practical points in urinary proteomics. Journal of Proteome Research. 2007;6:3881–3890. doi: 10.1021/pr070328s. [DOI] [PubMed] [Google Scholar]