

Abstract
The goal of this study was to examine metabolic differences between a novel chronic myelogenous leukemic (CML) cell line, MyL, and a sub-clone, MyL-R, which displays enhanced resistance to the targeted Bcr-Abl tyrosine kinase inhibitor imatinib. 1H nuclear magnetic resonance (NMR) spectroscopy was carried out on cell extracts and conditioned media from each cell type. Both principal component analysis (PCA) and specific metabolite identification and quantification were used to examine metabolic differences between the cell types. MyL cells showed enhanced glucose removal from the media compared to MyL-R cells with significant differences in production rates of the glycolytic end-products, lactate and alanine. Interestingly, the total intracellular creatine pool (creatine + phosphocreatine) was significantly elevated in MyL-R compared to MyL cells. We further demonstrated that the MyL-R cells converted the creatine to phosphocreatine using non-invasive monitoring of perfused alginate-encapsulated MyL-R and MyL cells by in vivo 31P NMR spectroscopy and subsequent HPLC analysis of extracts. Our data demonstrated a clear difference in the metabolite profiles of drug-resistant and sensitive cells, with the biggest difference being an elevation of creatine metabolites in the imatinib-resistant MyL-R cells.
Keywords: Metabolomics, Chronic myelogenous leukemia, 1H NMR spectroscopy, Drug resistance, Imatinib, Creatine
Introduction
The presence of the Bcr-Abl fusion protein is a distinguishing characteristic of chronic myelogenous leukemia (CML) (Rowley 1973; Melo et al. 1993). Resulting from the reciprocal translocation of chromosomes 9 and 22, the Bcr-Abl protein possesses aberrant and constitutive kinase activity and is the oncogene responsible for CML development and progression (Daley et al. 1990). A major advance in CML and cancer therapy came with the introduction of imatinib (Gleevec, Novartis), a highly specific kinase inhibitor designed to inhibit the tyrosine kinase activity of the Bcr-Abl protein (Deininger et al. 1997; Druker et al. 1996). While imatinib has been extremely successful in the treatment and management of CML for many patients, the development of imatinib resistance remains a clinical problem (Apperley 2007).
Many mechanisms have been described and may contribute to imatinib resistance, including increased Bcr-Abl expression (Mahon et al. 2000; Campbell et al. 2002), multi-drug resistance protein over expression (Kuwazuru et al. 1990; Illmer et al. 2004), Bcr-Abl-independent tyrosine kinase signaling (Donato et al. 2003; Ito et al. 2007), and specific Bcr-Abl point mutations within the imatinib-binding site (Gorre et al. 2001). Additionally, the remodeling of cellular metabolism has been explored and described as a possible mechanism underlying enhanced cell proliferation and growth in imatinib resistance (Klawitter et al. 2009). Human CML cell lines with loss of imatinib sensitivity display increased glycolytic rates, as evidenced by high lactate production, and elevations in phosphocholines, metabolic events thought to support increased cell proliferation in the presence of imatinib exposure (Kominsky et al. 2009). Additionally, imatinib treatment has been shown to limit glucose uptake and metabolism for de novo nucleic acid and fatty acid synthesis through inhibition of the oxidative branch of the pentose phosphate shunt (Boren et al. 2001). Within the context of imatinib resistance, a shift from the oxidative to the non-oxidative, transketolase dependent pathway may overcome imatinib’s inhibitory effects and thereby support the sufficient generation of important macromolecules for continued cell proliferation (Boros et al. 2003).
With the development of high resolution nuclear magnetic resonance (NMR) spectroscopy, the identification and quantification of cellular metabolites, second messengers, lipids, and other small molecules within biological systems are possible (Griffin 2003). When coupled to multivariate statistical analysis methods, such as principal component analysis (PCA), processed spectral data from various biological conditions can be distinguished from each other based upon differences within their unique metabolic profiles in an unbiased manner (Reo 2002). Furthermore, use of NMR spectroscopy allows for quantification of individual metabolites (Weljie et al. 2006). These data can lead to potential “biomarker” identification and provide a better understanding of the possible role cellular metabolism contributes to a particular disease state, drug response, or alteration in biological condition.
Here we examined and compared the metabolic profiles of a recently isolated and characterized CML cell line and a subline, which displays resistance to imatinib using 1H NMR. Our data show that when maintained in imatinib-free culture conditions, cells previously made resistant to imatinib-mediated cell death, maintained this resistance and displayed a unique metabolic phenotype compared to the parental CML cell line. The resistant cells showed suppressed glycolysis, lower overall levels of choline derivatives, lower taurine levels and elevated creatine. We further demonstrated that the resistant cells maintained a portion of the creatine pool as phosphocreatine, which may provide an additional energy reserve allowing cells to escape imatinib-mediated cell death.
Materials and methods
Materials
NMR supplies were obtained from multiple vendors. Sodium alginate, calcium chloride, perchloric acid, sodium citrate, sodium chloride, potassium chloride, and 3-(trimethylsily) propionic-2,2,3,3-d4 acid (TSP) were purchased from Sigma Chemical company (St. Louis, MO). Mono and dibasic phosphate was purchased from Mallinckrodt (Paris, KY). RPMI (Gibco) and fetal bovine serum were purchased from Invitrogen (Carlsbad, CA). Deuterium oxide was from Cambridge Isotope Laboratories (Andover, MA). The 5 and 10 mm NMR tubes were purchased from Wilmad Labglass Inc. (Miamisburg, OH). Materials for the bioreactor and life support system can be found in Keshari et al. (2010) and others (R. Jeffries et al., unpublished).
Cell culture
The human CML cell line, MyL, and the imatinib-insensitive subline, MyL-R, were kindly provided by Dr. Hideo Tanaka (Department of Haematology and Oncology, Hiroshima University, Hiroshima, Japan). Each cell type was maintained at 37°C with 95% air and 5% CO2 in RPMI medium (GIBCO) with 10% fetal bovine serum (FBS) and antibiotic/antimycotic (Cellgro) and were determined to be mycoplasma free. As originally reported, MyL-R cells could be maintained in imatinib-free culture medium for up to 6 months without an alteration in their sensitivity to imatinib.
For the comparison of MyL and MyL-R by 1H NMR, 1 × 108 cells were incubated for 2 h in 20 ml of fresh RPMI medium (RPMI + 10% FBS). Following the incubation, the cells were collected by centrifugation, washed 3× with cold PBS and then extracted with cold-methanol method as described below. Approximately 1 ml of the conditioned media from each cell type was collected for 1H NMR analysis.
Cold methanol cell extraction
Cells were harvested by low-speed centrifugation and metabolites extracted using the cold methanol extraction as previously described (Maharjan and Ferenci 2003). Briefly, the cells were collected by centrifugation and washed three times with cold PBS. Following removal of the last wash, 500 μl of ice-cold 50% methanol was added to the cell pellet and vortexed. The cell extracts were then incubated for 30 min on dry ice, and then allowed to thaw for 10 min on ice. The extracts were clarified by centrifugation at 16,000g for 10 min. The methanol extract (supernatant) was collected and transferred to a new microfuge tube, while an additional 500 μl of 50% methanol was added to the pellet for a second extraction. The second 50% methanol extract was collected and combined with the previous (first) extract. The total cell extract was evaporated to dryness using a Speed-Vac®.
Sample preparation and NMR spectroscopy
Prior to NMR spectroscopy, the evaporated cell extract pellet was suspended in 600 μl D2O containing 1.5 mM (final concentration) TSP and transferred to a 5 mm NMR tube for subsequent high resolution NMR analysis. For media analysis, a 400 μl aliquot of conditioned media was mixed with 200 μl of D2O containing 4.5 mM TSP. 1D 1H NMR spectra were acquired on a 16.4T Varian INOVA (700 MHz 1H, Varian Instruments) equipped with 5 mm indirect cold probe. The FIDs were acquired using a one-pulse sequence with a total repetition time (TR) of 12.65 s, number of transients (nt) of 64 and a 90° flip angle.
Spectral processing, PCA and metabolite determination
Spectral processing
All NMR spectra were processed using ACD/1D NMR Manager (version 12.0; Advanced Chemistry Development, Inc., Toronto, ON, Canada). Imported FID’s were zero filled to 64,000 points and an exponential line broadening of 0.1 Hz was applied prior to Fourier transformation. Spectra were phase and baseline corrected, and referenced to the TSP peak at 0.00 ppm. For in vivo 31P NMR spectra, the FIDs we zero filled to 32,000 and an exponential line broadening of 20 Hz was applied prior to Fourier transformation.
PCA
Following annotation (removal of spectral regions around water, >9.7 ppm, <0.4 ppm), grouped spectra were data-reduced to 250 regions, or bins, using intelligent bucketing and the integrals within each bin were determined. PCA was carried out using SIMCA-P (version 11.0; Umetrics, Umea, Sweden).
Metabolite determination
Metabolite identification and quantification were determined using Chenomx NMR Suite (version 5.1; Chenomx Inc., Edmonton, Canada), using TSP as a concentration reference.
In vivo 31P NMR spectroscopy of alginate encapsulated MyL and MyL-R cells
In vivo 31P NMR spectroscopy was performed using a fluidized-bed NMR-compatible bioreactor as described previously (Keshari et al. 2010; R. Jeffries et al., unpublished). Briefly, MyL and MyL-R cells were collected by centrifugation, mixed with 1:1 (vol:vol) of 2% alginate and electrostatically encapsulated and separately perfused in the bioreactor. The 31P NMR were obtained on a 14.1 T Varian INOVA (242 MHz 1H, Varian Instruments) equipped with a 10 mm broadband probe at 37°C. The 31P NMR time courses were acquired using a TR = 2s, nt = 2048 and a 77° flip angle, resulting in spectra of 68 min each.
HPLC analysis
To avoid phosphocreatine hydrolysis during extraction, cells were extracted using perchloric acid. Briefly, 10 × 106 cells were collected by centrifugation, washed three times with cold PBS and then extracted with 500 μl of 0.6 M HClO4 with 1.0 mM EDTA. After vortex, extracts were centrifuged for 5 min at 14,000 rpm and then the supernatant collected. Approximately 80 μl of 2 M KHCO3 was added, followed by a 20-min incubation on ice, and then a 5-min centrifugation at 12,000 rpm. The supernatants were collected and stored at −80°C. Extracts were analyzed by HPLC using a reverse phase C-18 column. Creatine and phosphocreatine standards were used to determine elution times.
Cell viability assays
For imatinib dose response experiments, 10 × 103 MyL or MyL-R cells were plated per well in 50 μl of medium in a 96-well plate. An additional 50 μl volume of medium was added with increasing concentrations of imatinib (DMSO was used as the vehicle control). After 48 h, 20 μl of MTS reagent (CellTiter 96®AQueous One Solution Reagent, Promega, Madison, WI) was added to each well and incubated for an additional 2 h according to the manufacturer’s instructions. The formation of the soluble formazon product was determined by measuring the absorbance at 490 nm on a SpectraMAX plate reader (Molecular Devices, Sunnyvale, CA) and subtracting a media + MTS only control. Percent viability was calculated from the DMSO control. Duplicate readings were obtained for each condition and each experiment was performed three times.
Statistics
Concentrations from cell extracts and media were analyzed using Prism (GraphPad Software, La Jolla, CA) and statistical comparisons were made using two-tailed Student’s t test or analysis of variance (ANOVA), and post hoc comparisons were made in pair-wise fashion using Tukey’s method as appropriate. A value of P < 0.05 was selected before the study as the level of significance.
Results
Imatinib resistant MyL-R cells display persistent resistance to imatinib
Imatinib-resistant cell lines have been generated from imatinib-sensitive CML cells by maintaining cells in suboptimal concentrations of imatinib and gradually increasing the dose over time (Mahon et al. 2000). Recently, a similar strategy was employed to generate the MyL-R cell line from the parental CML isolate, MyL. Imatinib resistance in the MyL-R cells was shown not to be a result of Bcr-Abl over-expression or Bcr-Abl kinase-domain point mutations. Importantly, the insensitivity to imatinib persisted even when maintained in the absence of imatinib (Ito et al. 2007). We verified this persistent insensitivity to imatinib by maintaining MyL and MyL-R cells in RPMI medium + 10% FBS only and then examining the effectiveness of increasing concentrations of imatinib on cell viability. As expected, the MyL-R cells showed enhanced tolerance to imatinib treatment when compared to MyL cells, even after having been maintained in imatinib-free culture medium (Fig. 1). These results suggest that the mechanisms associated with resistance are maintained even when cells are cultured in the absence of imatinib. Interestingly, MyL-R cells are also highly resistant to gemcitabine, AraC and Immucillin H treatment, demonstrating an overall increase in MyL-R cell survival (E. Zimmerman et al., unpublished observations).
Fig. 1.
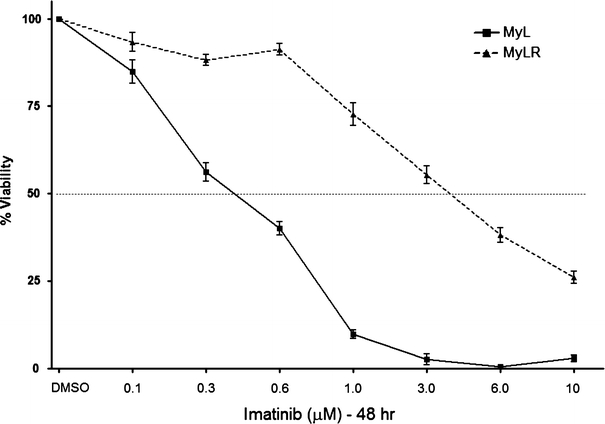
Sensitivity of MyL and MyL-R cells to imatinib treatment. MyL or MyL-R cells (10 × 103) in 100 μl of medium were incubated for 48 h with DMSO (vehicle control) or the concentrations of imatinib shown (μM). Percent viability was determined as described in Materials and methods (n = 3)
Metabolic phenotype associated with MyL and MyL-R cells as examined by 1H NMR
We examined the soluble metabolite profiles of MyL and MyL-R cells maintained under the conditions described above by 1H NMR analysis of both cell extracts and cell-conditioned media. A representative 1H NMR spectra (primarily the aliphatic region) of a cell extract from MyL or MyL-R cells is shown in Fig. 2. By applying PCA to the NMR spectra generated from MyL and MyL-R cells, metabolic differences associated with each cell type were investigated. The scores plot revealed a separation of MyL from MyL-R in the first principal component (PC1) (Fig. 3a). The corresponding loadings plot is shown in Fig. 3b and identifies spectral regions responsible for the separation observed in the scores plot. The metabolites most likely associated with these spectral regions are shown.
Fig. 2.
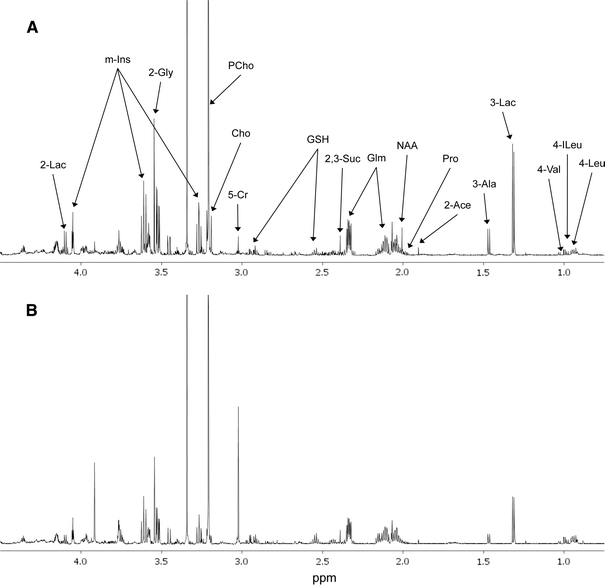
1H NMR spectra from MyL and MyL-R cells extracts. MyL (a) and MyL-R (b) cells were expanded and maintained in T175 flasks containing RPMI culture medium with 10% FBS in the absence of imatinib. The expanded cells were collected by low speed centrifugation, counted and then plated at a density 5.0 × 106 cells/ml in 20 ml of fresh medium. After incubation for 2 h at 37°C, the cells were collected, washed three times with cold PBS, and extracted as described in Sect. 2. Cell extracts were evaporated to dryness followed by suspension in D2O containing TSP (internal standard), and analyzed by 1H NMR. Spectra are representative for each cell type and highlight major metabolites identified. Lac lactate, m-Ins myo-inositol, Gly glycine, Glm glutamine, PCho phosphocholine, Cho choline, Cr creatine, GSH glutathione, Suc succinate, Pro proline, Ace acetate, Ala alanine, Val valine, Ileu isoleucine, Leu leucine, NAA n-acetylaspartate
Fig. 3.
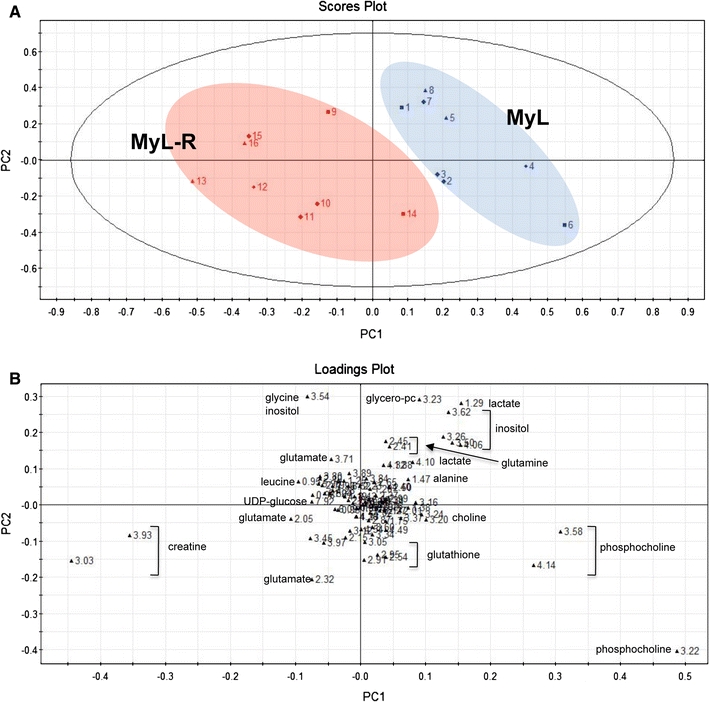
Metabolic distinction of MyL from MyL-R cells. a Scores plot (PC1/PC2) based on 1H NMR spectra of cell extracts from MyL and MyL-R cells grown in the absence of imatinib. b Corresponding loadings plot. Values given correspond to chemical shift values (ppm) from 1H spectra (n = 8)
From the 1D spectra of cell extracts a total of 56 metabolites were identified and quantified using Chenomx software, based upon the known concentration of TSP standard added to each sample. A comparison of selected metabolites from the cell extracts of MyL and MyL-R cells is shown in Fig. 4a. We observed a statistically significant decrease in several metabolites in MyL-R cells compared to MyL cells including alanine, glutamine, lysine, choline, phosphocholine, myo-inositol, AXP, lactate, acetate, acetylaspartate, formate, fumarate, pyroglutamate, succinate, and taurine. Interestingly, the only metabolite found to be elevated in MyL-R cells compared to MyL cells was creatine. This was most evident when the MyL-R to MyL ratio of each metabolite in the cell extracts was determined (Fig. 4b). Many metabolites, mostly amino acids, showed little difference between the two cell lines, as shown by a MyL-R/MyL ratio close to 1.0 and included isoleucine, leucine, methionine, tyrosine, valine and glutathione.
Fig. 4.
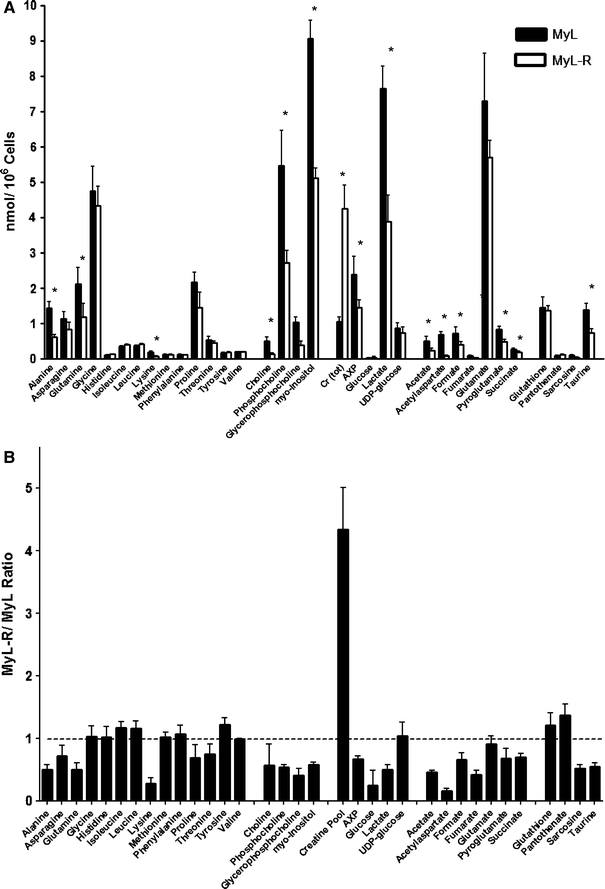
Quantification of the intracellular metabolites in MyL and MyL-R cells. a Individual metabolite concentrations from 1H NMR processed spectra were determined using Chenomx software and calculated as nmol/106 cells. * P < 0.05 for comparison between MyL and MyL-R by two-tailed student’s t test (n = 8). b MyL-R to MyL ratio for each metabolite shown in (a)
Similar to cell extracts, 1H NMR spectra were acquired from Total Medium (RPMI + 10% FBS only) and MyL and MyL-R cell-conditioned medium. Table 1 shows the μmol quantity of selected metabolites from each media condition. Significant differences comparing each cell-conditioned media to the Total Medium were observed for the following metabolites: arginine, glucose, glutamine, histidine, isoleucine, leucine, phenylalanine, proline, pyroglutamate, fructose and lactate. We observed a significant difference in glucose consumption (removal from the media) where MyL cells consumed 475.36 ± 19.71 nmol/h/106 glucose compared to only 297.13 ± 37.81 nmols/h/106 in the MyL-R cells. This was accompanied by a significant difference in lactate release, where MyL cells produced 176.77 ± 9.55 nmols/h/106 cells compared to only 93.44 ± 10.61 nmols/h/106 MyL-R cells. We further observed a significant decrease in glutamine and phenylalanine removal from the media by the MyL-R cells. MyL cell acetate and formate production, 3.25 ± 0.66 and 15.68 ± 3.99 nmol/h/106 cells, respectively, were statistically different and elevated compared to MyL-R formate production. Interestingly, MyL cells showed a release or production of alanine into the media compared to the MyL-R cell, which displayed overall consumption of this metabolite. Lastly, glutamate release into the media was elevated in the MyL-R cells compared to the MyL cells, however this difference was not statistically significant (Table 1).
Table 1.
Change in selected metabolites from Total Medium following incubation with MyL and MyL-R cells
| Metabolite | Total medium (μmols) | Conditioned (μmols) | Consumption or production (nmols/h/106 cells) | ||
|---|---|---|---|---|---|
| MYL | MYL-R | MyL | MyL-R | ||
| Arginine | 17.60 ± 0.55 | 12.84 ± 0.24a | 12.99 ± 1.61 | 23.62 ± 1.31 | 21.12 ± 8.05 |
| Aspartate | 3.42 ± 0.38 | 2.02 ± 0.22 | 2.51 ± 0.30 | 7.59 ± 1.19 | 4.53 ± 1.50 |
| Choline | 0.62 ± 0.04 | 0.39 ± 0.04 | 0.45 ± 0.06 | 1.21 ± 0.18 | 0.83 ± 0.29 |
| Glucose | 201.33 ± 6.19 | 113.29 ± 3.66a | 141.81 ± 7.56a | 475.36 ± 19.71 | 297.13 ± 37.81b |
| Glutamate | 3.51 ± 0.14 | 3.43 ± 0.22 | 4.82 ± 0.99 | 0.44 ± 1.20 | 6.55 ± 4.69 |
| Glutamine | 24.34 ± 0.61 | 15.63 ± 0.60a | 20.07 ± 0.42a | 34.21 ± 3.22 | 9.44 ± 2.10b |
| Glycine | 3.87 ± 0.09 | 4.80 ± 0.86 | 4.06 ± .75 | 6.00 ± 4.64 | 1.81 ± 3.73 |
| Histidine | 2.03 ± 0.07 | 1.37 ± 0.07a | 1.71 ± 0.27 | 3.59 ± 0.36 | 1.61 ± 1.33 |
| Isoleucine | 7.21 ± 0.74 | 4.34 ± 0.21a | 4.53 ± 0.55a | 16.44 ± 1.13 | 14.24 ± 2.77 |
| Leucine | 7.06 ± 0.59 | 3.88 ± 0.07a | 4.47 ± 0.59a | 17.69 ± 0.40 | 13.44 ± 3.00 |
| Phenylalanine | 2.03 ± 0.14 | 1.17 ± 0.02a | 1.49 ± 0.11 | 4.68 ± 0.13 | 2.74 ± 0.54b |
| Proline | 4.00 ± 0.19 | 2.26 ± 0.07a | 2.99 ± 0.58 | 9.23 ± 0.40 | 5.16 ± 2.90 |
| Pyroglutamate | 9.03 ± 0.67 | 2.64 ± 0.12a | 2.92 ± 0.16a | 38.43 ± 0.63 | 34.18 ± 0.81 |
| Valine | 3.77 ± 0.06 | 2.57 ± 0.25 | 2.48 ± 0.35 | 6.54 ± 1.33 | 6.49 ± 1.75 |
| Acetate | 0.41 ± 0.05 | 1.01 ± 0.12 | 0.69 ± 0.20 | 3.25 ± 0.66 | 1.42 ± 1.00 b |
| Alanine | 2.06 ± 0.09 | 3.05 ± 0.22 | 1.52 ± 0.29 | 5.32 ± 1.21 | 2.70 ± 1.44b |
| Formate | 0.69 ± 0.02 | 3.59 ± 0.74 | 2.68 ± 0.73 | 15.68 ± 3.99 | 9.95 ± 3.66 b |
| Fructose | 6.96 ± 0.57 | 4.33 ± 0.09a | 5.31 ± 0.95 | 14.23 ± 0.47 | 8.27 ± 4.77 |
| Lactate | 33.70 ± 0.46 | 68.24 ± 1.76 a | 54.23 ± 2.12 a | 176.77 ± 9.55 | 93.44 ± 10.61 b |
Total medium (RPMI + 10% FBS) or medium incubated with either MyL or MyL-R cells for 2 h were analyzed by 1H NMR. Individual metabolites were identified and quantified using Chenomx software as described in Sect. 2. The relative consumption or production rates were calculated by subtracting each cell condition from the mean Total Medium value and dividing by incubation time and total number of cells. Results are expressed as mean ± SEM, n = 3. Values in Bold represent metabolites that accumulated (production) in medium
a p < 0.05 for comparison with Total Medium by ANOVA with post hoc test using Tukey’s method
b p < 0.05 for comparison between MyL and MyL-R by Student’s t test
MyL-R cells maintain creatine as the high energy phosphagen, phosphocreatine
Interestingly, one of the most significant differences observed between MyL and MyL-R cells was the total intracellular creatine pool, which was 5-fold higher in the MyL-R cells. From the 1H NMR data we determined that the total intracellular creatine pool (creatine + phosphocreatine) was 0.601 ± 0.078 nmol/106 cells in the MyL as compared to 2.93 ± 0.43 nmol/106 in the MyL-R (Fig. 4a). Creatine kinase (CK) catalyzes both the forward and reverse reaction conversion of creatine to the high-energy phosphagen, phosphocreatine utilizing or generating ATP (Wyss and Kaddurah-Daouk 2000). Because the 1H NMR peaks of creatine and phosphocreatine are nearly identical, it is difficult to distinguish between creatine and phosphocreatine by this method. We therefore generated perchloric acid extracts of MyL and MyL-R cells and analyzed them by reverse phase HPLC (C18) as described in Sect. 2. We observed an increase in the total intracellular creatine pool (creatine + phosphocreatine) in MyL-R compared to MyL cells and the values determined from HPLC in nmols/106 cells were comparable to those obtained by 1H NMR (Fig. 5a). Interestingly, MyL-R cells maintained a creatine/phosphocreatine ratio close to 1.0, whereas in the MyL cells more of the total intracellular creatine pool was in the form of creatine (Fig. 5b).
Fig. 5.
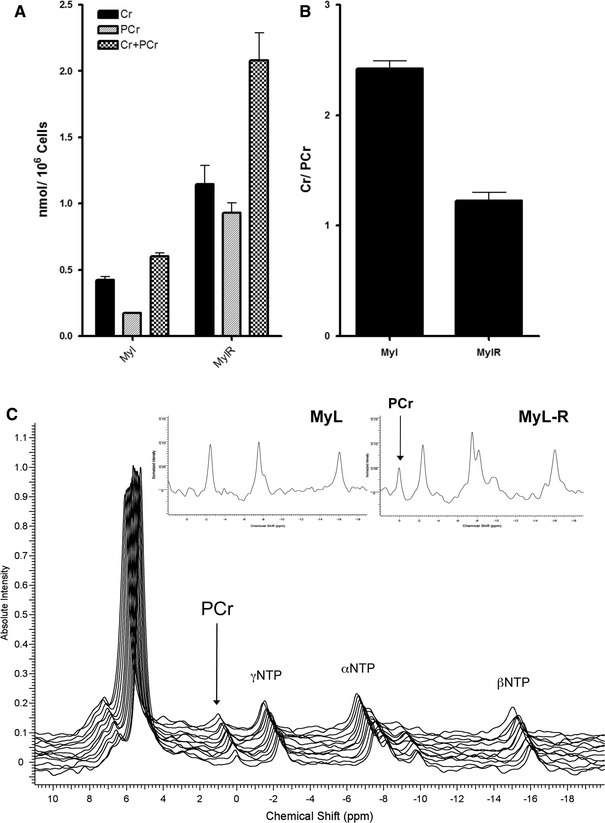
In vivo detection of phosphocreatine in MyL-R cells. Perchloric acid extracts from 10 × 106 MyL and MyL-R cells from normal growth conditions were analyzed by HPLC to determine intracellular creatine (Cr) and phosphocreatine (PCr) concentrations (a) and the Cr/PCr ratio (b). Stacked 31P spectra of viable MyL-R cells in an NMR compatible bioreactor (c). Inset in (c) is the comparison of 31P spectra from perfused alginate-encapsulate MyL and MyL-R cells
We further confirmed the ability of the MyL-R cells to convert creatine to phosphocreatine using a recently designed NMR-compatible bioreactor. Alginate encapsulated MyL-R cells were loaded into the lower chamber of the bioreactor NMR tube and in vivo 31P NMR spectra were acquired every 30 min over an 8-h time period. We observed a clear peak representing phosphocreatine during the entire duration of the experiment (Fig. 5c) confirming that under in vivo conditions MyL-R cells maintain a portion of their intracellular creatine pool in the form of phosphocreatine. Inset in Fig. 5c is the comparison of the in vivo 31P NMR spectra from MyL and MyL-R cells perfused in the bioreactor demonstrating that the primary difference between the two spectra is the greatly reduced phosphocreatine peak in the MyL cells.
Discussion
We used 1H NMR based metabolomic analysis to compare MyL, a Bcr-Abl+ human CML cell line, to a subline that displays resistance to imatinib. By comparing the overall metabolic profiles of these cells we hoped to determine if imatinib resistant cells displayed a unique metabolic signature different from the non-resistant cell line. Previous work has demonstrated that Bcr-Abl expression results in elevated cellular levels of Glut-1 protein and imatinib treatment resulted in decreased glucose uptake due to Glut-1 internalization (Barnes et al. 2005). Interestingly, more recent evidence has shown that Glut-1 internalization did not occur when imatinib resistant cell lines were cultured in the presence of imatinib and thus, glucose uptake and metabolism is maintained (Kominsky et al. 2009). Here our data demonstrate that when maintained in culture in the absence of imatinib, the MyL-R cells display a different glycolytic profile when compared to both the imatinib sensitive MyL cells and previously reported data. We observed lower glucose removal from the media by MyL-R in comparison to the MyL cells and both the intracellular lactate concentration and release into the culture medium was lower in MyL-R cells compared to MyL (Fig. 4a, Table 1). If for every mole of glucose metabolized by glycolysis two moles of lactate are produced, then the percent of glucose used for glycolysis for MyL and MyL-R cells is 36 and 18%, respectively. The two major metabolic pathways that utilize glucose in transformed cells are the pentose phosphate pathway for generation of ribose in RNA and DNA synthesis, and pyruvate entry into mitochondrial TCA cycle. Significant alterations in diversion of glucose into the pentose phosphate pathway have been described in response to imatinib treatment (Boren et al. 2001). Future experiments employing 13C-labeled glucose could be done to determine relative glucose flux into these other pathways and to more accurately define the difference in percent glucose generated lactate between MyL and MyL-R cells.
Recent evidence has demonstrated the importance of glutamine in cancer cells undergoing high rates of aerobic glycolysis (DeBerardinis et al. 2007). Due to a high efflux rate of substrates of the TCA cycle, particularly citrate, which is used for fatty acid synthesis, glutamine anapleurosis replenishes TCA substrates through conversion of glutamine to α-ketoglutarate (DeBerardinis et al. 2007). In this study MyL cells, which displayed higher rates of glycolysis also consumed more glutamine from the culture media compared to the MyL-R cells (Table 1). Moreover, we observed an accumulation of glutamate in the MyL-R media. As glutamine must first be converted to glutamate, and then subsequently to α-ketoglutarate for entry into the TCA cycle, the diminished role of glutamine anapleurosis in the MyL-R cells may be reflected by an accumulation of glutamate. These data suggest that mitochondrial metabolism in the MyL-R cells may be in an alternative metabolic/respiratory state when compared to the highly glycolytic MyL cells. Indeed, succinate and adenosine phosphates (AXP) levels were significantly lower in MyL-R cells extracts, with the later being possibly related to the elevations observed in the creatine/phosphocreatine ratio (Fig. 4). This altered mitochondrial state may simply be due to the reduction in glycolytic flux observed in the MyL-R cells. However, a recent study demonstrated that glutamine-dependent anapleurosis regulated MondoA, a nutrient-dependent transcription factor, which consequently controlled glucose uptake (Kaadige et al. 2009). Whether these mitochondrial alterations in metabolism are critical to drug resistance remain to be elucidated.
Choline and metabolic pathways associated with choline metabolism are often elevated in cancerous tissues (Ackerstaff et al. 2003). Moreover, previous work has demonstrated an overall increase in phosphocholine (PC) and glycerophosphocholine (GPC) in chronic lymphocytic leukemia cells compared to normal human lymphocytes (Franks et al. 2002). More recent evidence has demonstrated that the ratio of PC/GPC can be used as a measure of enhanced cell proliferation in cancerous tissues (Glunde et al. 2006). A significant rise in PC and loss of GPC was observed in imatinib resistant CML cell when compared to their sensitive counterparts (Serkova and Boros 2005). In this study, comparing drug sensitive to resistant cells we observed an overall decrease in choline and choline derivatives (PC and GPC) in the imatinib resistant MyL-R cell compared the MyL line (Fig. 4a). These data suggest the possibility that the proliferation rate of MyL-R cells differs and may be slightly slower than that of the MyL cells. However, it should be noted that while the overall PC and GPC levels were lower in the MyL-R cell presented here, there was only a slight increase in the PC/GPC ratio compared to the MyL cells. Compared to previous reports demonstrating a significant change in the PC/GPC ratio in resistant cell, the modest increase in the PC/GPC ratio observed here in imatinib resistance cells could be due to the absence of imatinib in their growth media, a selective pressure promoting enhanced cell proliferation to overcome the cytostatic effects of the drug.
Taurine is a conditionally essential amino acid, not utilized for protein synthesis but found free in the cytoplasm or in short peptides. The biological importance and physiological functions of cellular taurine are numerous and highly cell type specific (Huxtable 1992). We observed taurine in both MyL and MyL-R cell extracts, however intracellular concentrations were significantly lower in the MyL-R imatinib resistant subline (Fig. 4a). Other work has observed similar differences in vinblastine-sensitive and resistant T-lymphoid leukaemic cell lines where taurine was significantly lower in the resistant cell line (Jiang et al. 1993). A more recent report provides evidence that increased taurine levels correlated with apoptosis in glioma cells (Opstad et al. 2009), however, in contrast, taurine can act as a cellular anti-oxidant and has been shown to protect against high-glucose induced apoptosis in vascular endothelial cells (Wu et al. 1999).
From our data and analysis of cell extracts the elevation of the total creatine pool in the MyL-R cells was the most distinguishing feature comparing this drug resistant cell model from the parental non-resistant cells (Fig. 4b). A rise in the high-energy compound phosphocreatine and its precursor, creatine, has been observed in drug resistant human breast cancer cells (Kaplan et al. 1990). The mechanism for the elevation of total intracellular creatine in the imatinib-resistant cells is unknown, however other works have demonstrated that increased intracellular levels of creatine are associated with cell differentiation and were due to enhanced uptake (Loike et al. 1984, 1986). Alternatively, de novo creatine synthesis could be accounting for the elevated levels. However, the primary tissues involved and expressing key enzymes required for creatine synthesis are kidney and liver, thus making de novo synthesis in leukemic cells unlikely.
The significance of the elevated creatine in the MyL-R cells and its possible role in imatinib-resistance is not known at this time. However, modulation of the mitochondrial permeability transition and cytochrome c release leading to apoptotic induction, an event thought to be induced by imatinib treatment, has been shown to be affected by mitochondrial phosphocreatine levels (O’Gorman et al. 1997; Dolder et al. 2003). Imatinib treatment has been shown to decrease glycolysis, an ATP generating process (Boren et al. 2001; Gottschalk et al. 2004) and moreover, prolonged imatinib treatment has been shown to deplete phosphocreatine in K562 cells (Klawitter et al. 2009). Thus cells possessing an alternative high-energy source may ultimately stave off loss of cell viability better than ones lacking or contain less of that alternative energy supply.
This work further suggests that metabolic differences may exist between parental CML cell line models. As mentioned above, the presence of phosphocreatine has been reported in the K562 cell line, a commonly described imatinib sensitive CML cell line model (Gottschalk et al. 2004). Here, only low levels of phosphocreatine were observed in the parental MyL cell line. This difference could be due to lineage specific cell differences as K562 represent have an erythrocytic lineage, whereas the MyL line has been described as more monocytic. Additionally, K562 represent a late stage blast crisis CML, while the MyL cell line model may better represent an earlier stage of CML.
Concluding remarks
In this study we performed a metabolic comparison of a recently characterized CML cell line, MyL, and its imatinib resistant counterpart, MyL-R. The original work describing this novel cell line model demonstrated that as opposed to other human CML cell lines displaying imatinib resistance, the MyL-R cells show no Bcr-Abl kinase domain mutations, lack elevated Bcr-Abl expression, and did not express multi-drug transport channels. Using NMR based metabolomic analysis, we observed that the imatinib resistant CML cell line displayed a noticeably different metabolite profile compared to the drug sensitive parental cell line. Most notable were significantly higher creatine and phosphocreatine levels and an overall decrease in glycolytically related endpoints. Thus, similar to previous works, our data support the idea that CML cells undergoing high rates of glycolytic metabolism (i.e. the Warburg effect) in order to support high rates of cellular proliferation tend to be more sensitive to imatinib treatment. In this study however, our data suggest that the inherent metabolic differences, that the MyL-R cells are less dependent upon glycolytic metabolism and have a possible alternative energy store in the form of phosphocreatine, may make the MyL-R cells less susceptible to imatinib treatment and therefore contribute to their drug resistance. Further works will be required to examine the relevance of these metabolic differences in relation to imatinib drug resistant.
Acknowledgments
We thank NIH grants (GM075941, NIEHS T32 EX007126) for funding the research.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Footnotes
Lee M. Graves and Jeffrey M. Macdonald are co-senior authors.
References
- Ackerstaff E, Glunde K, Bhujwalla ZM. Choline phospholipid metabolism: A target in cancer cells? Journal of Cellular Biochemistry. 2003;90:525–533. doi: 10.1002/jcb.10659. [DOI] [PubMed] [Google Scholar]
- Apperley JF. Part I: Mechanisms of resistance to imatinib in chronic myeloid leukaemia. Lancet Oncology. 2007;8:1018–1029. doi: 10.1016/S1470-2045(07)70342-X. [DOI] [PubMed] [Google Scholar]
- Barnes K, McIntosh E, Whetton AD, Daley GQ, Bentley J, Baldwin SA. Chronic myeloid leukaemia: An investigation into the role of Bcr-Abl-induced abnormalities in glucose transport regulation. Oncogene. 2005;24:3257–3267. doi: 10.1038/sj.onc.1208461. [DOI] [PubMed] [Google Scholar]
- Boren J, Cascante M, Marin S, Comin-Anduix B, Centelles JJ, Lim S, Bassilian S, Ahmed S, Lee WN, Boros LG. Gleevec (STI571) influences metabolic enzyme activities and glucose carbon flow toward nucleic acid and fatty acid synthesis in myeloid tumor cells. Journal of Biological Chemistry. 2001;276:37747–37753. doi: 10.1074/jbc.M105796200. [DOI] [PubMed] [Google Scholar]
- Boros LG, Brackett DJ, Harrigan GG. Metabolic biomarker and kinase drug target discovery in cancer using stable isotope-based dynamic metabolic profiling (SIDMAP) Current Cancer Drug Targets. 2003;3:445–453. doi: 10.2174/1568009033481769. [DOI] [PubMed] [Google Scholar]
- Campbell LJ, Patsouris C, Rayeroux KC, Somana K, Januszewicz EH, Szer J. BCR/ABL amplification in chronic myelocytic leukemia blast crisis following imatinib mesylate administration. Cancer Genetics and Cytogenetics. 2002;139:30–33. doi: 10.1016/S0165-4608(02)00615-5. [DOI] [PubMed] [Google Scholar]
- Daley GQ, Van Etten RA, Baltimore D. Induction of chronic myelogenous leukemia in mice by the P210bcr/abl gene of the Philadelphia chromosome. Science. 1990;247:824–830. doi: 10.1126/science.2406902. [DOI] [PubMed] [Google Scholar]
- DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, Thompson CB. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:19345–19350. doi: 10.1073/pnas.0709747104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deininger MW, Goldman JM, Lydon N, Melo JV. The tyrosine kinase inhibitor CGP57148B selectively inhibits the growth of BCR-ABL-positive cells. Blood. 1997;90:3691–3698. [PubMed] [Google Scholar]
- Dolder M, Walzel B, Speer O, Schlattner U, Wallimann T. Inhibition of the mitochondrial permeability transition by creatine kinase substrates. Requirement for microcompartmentation. Journal of Biological Chemistry. 2003;278:17760–17766. doi: 10.1074/jbc.M208705200. [DOI] [PubMed] [Google Scholar]
- Donato NJ, Wu JY, Stapley J, Gallick G, Lin H, Arlinghaus R, Talpaz M. BCR-ABL independence and LYN kinase overexpression in chronic myelogenous leukemia cells selected for resistance to STI571. Blood. 2003;101:690–698. doi: 10.1182/blood.V101.2.690. [DOI] [PubMed] [Google Scholar]
- Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM, Fanning S, Zimmermann J, Lydon NB. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nature Medicine. 1996;2:561–566. doi: 10.1038/nm0596-561. [DOI] [PubMed] [Google Scholar]
- Franks SE, Smith MR, Arias-Mendoza F, Shaller C, Padavic-Shaller K, Kappler F, Zhang Y, Negendank WG, Brown TR. Phosphomonoester concentrations differ between chronic lymphocytic leukemia cells and normal human lymphocytes. Leukemia Research. 2002;26:919–926. doi: 10.1016/S0145-2126(02)00035-8. [DOI] [PubMed] [Google Scholar]
- Glunde K, Ackerstaff E, Mori N, Jacobs MA, Bhujwalla ZM. Choline phospholipid metabolism in cancer: Consequences for molecular pharmaceutical interventions. Molecular Pharmaceutics. 2006;3:496–506. doi: 10.1021/mp060067e. [DOI] [PubMed] [Google Scholar]
- Gorre ME, Mohammed M, Ellwood K, Hsu N, Paquette R, Rao PN, Sawyers CL. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001;293:876–880. doi: 10.1126/science.1062538. [DOI] [PubMed] [Google Scholar]
- Gottschalk S, Anderson N, Hainz C, Eckhardt SG, Serkova NJ. Imatinib (STI571)-mediated changes in glucose metabolism in human leukemia BCR-ABL-positive cells. Clinical Cancer Research. 2004;10:6661–6668. doi: 10.1158/1078-0432.CCR-04-0039. [DOI] [PubMed] [Google Scholar]
- Griffin JL. Metabonomics: NMR spectroscopy and pattern recognition analysis of body fluids and tissues for characterisation of xenobiotic toxicity and disease diagnosis. Current Opinion in Chemical Biology. 2003;7:648–654. doi: 10.1016/j.cbpa.2003.08.008. [DOI] [PubMed] [Google Scholar]
- Huxtable RJ. Physiological actions of taurine. Physiological Reviews. 1992;72:101–163. doi: 10.1152/physrev.1992.72.1.101. [DOI] [PubMed] [Google Scholar]
- Illmer T, Schaich M, Platzbecker U, Freiberg-Richter J, Oelschlagel U, von Bonin M, Pursche S, Bergemann T, Ehninger G, Schleyer E. P-glycoprotein-mediated drug efflux is a resistance mechanism of chronic myelogenous leukemia cells to treatment with imatinib mesylate. Leukemia. 2004;18:401–408. doi: 10.1038/sj.leu.2403257. [DOI] [PubMed] [Google Scholar]
- Ito T, Tanaka H, Kimura A. Establishment and characterization of a novel imatinib-sensitive chronic myeloid leukemia cell line MYL, and an imatinib-resistant subline MYL-R showing overexpression of Lyn. European Journal of Haematology. 2007;78:417–431. doi: 10.1111/j.1600-0609.2007.00835.x. [DOI] [PubMed] [Google Scholar]
- Jiang XR, Yang M, Morris CJ, Newland AC, Naughton DP, Blake DR, Zhang Z, Grootveld MC. High field proton NMR investigations of the metabolic profiles of multidrug-sensitive and -resistant leukaemic cell lines: Evidence for diminished taurine levels in multidrug-resistant cells. Free Radical Research Communications. 1993;19:355–369. doi: 10.3109/10715769309056526. [DOI] [PubMed] [Google Scholar]
- Kaadige MR, Looper RE, Kamalanaadhan S, Ayer DE. Glutamine-dependent anapleurosis dictates glucose uptake and cell growth by regulating MondoA transcriptional activity. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:14878–14883. doi: 10.1073/pnas.0901221106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan O, van Zijl PC, Cohen JS. Information from combined 1H and 31P NMR studies of cell extracts: Differences in metabolism between drug-sensitive and drug-resistant MCF-7 human breast cancer cells. Biochemical and Biophysical Research Communications. 1990;169:383–390. doi: 10.1016/0006-291X(90)90343-L. [DOI] [PubMed] [Google Scholar]
- Keshari KR, Kurhanewicz J, Jeffries RE, Wilson DM, Dewar BJ, Van Criekinge M, Zierhut M, Vigneron DB, Macdonald JM. Hyperpolarized (13)C spectroscopy and an NMR-compatible bioreactor system for the investigation of real-time cellular metabolism. Magnetic Resonance in Medicine. 2010;63:322–329. doi: 10.1002/mrm.22225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klawitter J, Anderson N, Christians U, Leibfritz D, Eckhardt SG, Serkova NJ. Time-dependent effects of imatinib in human leukaemia cells: A kinetic NMR-profiling study. British Journal of Cancer. 2009;100:923–931. doi: 10.1038/sj.bjc.6604946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klawitter J, Kominsky DJ, Brown JL, Christians U, Leibfritz D, Melo JV, Eckhardt SG, Serkova NJ. Metabolic characteristics of imatinib resistance in chronic myeloid leukaemia cells. British Journal of Pharmacology. 2009;158:588–600. doi: 10.1111/j.1476-5381.2009.00345.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kominsky DJ, Klawitter J, Brown JL, Boros LG, Melo JV, Eckhardt SG, Serkova NJ. Abnormalities in glucose uptake and metabolism in imatinib-resistant human BCR-ABL-positive cells. Clinical Cancer Research. 2009;15:3442–3450. doi: 10.1158/1078-0432.CCR-08-3291. [DOI] [PubMed] [Google Scholar]
- Kuwazuru Y, Yoshimura A, Hanada S, Ichikawa M, Saito T, Uozumi K, Utsunomiya A, Arima T, Akiyama S. Expression of the multidrug transporter, P-glycoprotein, in chronic myelogenous leukaemia cells in blast crisis. British Journal of Haematology. 1990;74:24–29. doi: 10.1111/j.1365-2141.1990.tb02533.x. [DOI] [PubMed] [Google Scholar]
- Loike JD, Kozler VF, Silverstein SC. Creatine kinase expression and creatine phosphate accumulation are developmentally regulated during differentiation of mouse and human monocytes. Journal of Experimental Medicine. 1984;159:746–757. doi: 10.1084/jem.159.3.746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loike JD, Somes M, Silverstein SC. Creatine uptake, metabolism, and efflux in human monocytes and macrophages. American Journal of Physiology. 1986;251:C128–C135. doi: 10.1152/ajpcell.1986.251.1.C128. [DOI] [PubMed] [Google Scholar]
- Maharjan RP, Ferenci T. Global metabolite analysis: The influence of extraction methodology on metabolome profiles of Escherichia coli. Analytical Biochemistry. 2003;313:145–154. doi: 10.1016/S0003-2697(02)00536-5. [DOI] [PubMed] [Google Scholar]
- Mahon FX, Deininger MW, Schultheis B, Chabrol J, Reiffers J, Goldman JM, Melo JV. Selection and characterization of BCR-ABL positive cell lines with differential sensitivity to the tyrosine kinase inhibitor STI571: Diverse mechanisms of resistance. Blood. 2000;96:1070–1079. [PubMed] [Google Scholar]
- Melo JV, Gordon DE, Cross NC, Goldman JM. The ABL-BCR fusion gene is expressed in chronic myeloid leukemia. Blood. 1993;81:158–165. [PubMed] [Google Scholar]
- O’Gorman E, Beutner G, Dolder M, Koretsky AP, Brdiczka D, Wallimann T. The role of creatine kinase in inhibition of mitochondrial permeability transition. FEBS Letters. 1997;414:253–257. doi: 10.1016/S0014-5793(97)01045-4. [DOI] [PubMed] [Google Scholar]
- Opstad KS, Bell BA, Griffiths JR, Howe FA. Taurine: A potential marker of apoptosis in gliomas. British Journal of Cancer. 2009;100:789–794. doi: 10.1038/sj.bjc.6604933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reo NV. NMR-based metabolomics. Drug and Chemical Toxicology. 2002;25:375–382. doi: 10.1081/DCT-120014789. [DOI] [PubMed] [Google Scholar]
- Rowley JD. Letter: A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature. 1973;243:290–293. doi: 10.1038/243290a0. [DOI] [PubMed] [Google Scholar]
- Serkova N, Boros LG. Detection of resistance to imatinib by metabolic profiling: Clinical and drug development implications. American Journal of Pharmacogenomics. 2005;5:293–302. doi: 10.2165/00129785-200505050-00002. [DOI] [PubMed] [Google Scholar]
- Weljie AM, Newton J, Mercier P, Carlson E, Slupsky CM. Targeted profiling: Quantitative analysis of 1H NMR metabolomics data. Analytical Chemistry. 2006;78:4430–4442. doi: 10.1021/ac060209g. [DOI] [PubMed] [Google Scholar]
- Wu QD, Wang JH, Fennessy F, Redmond HP, Bouchier-Hayes D. Taurine prevents high-glucose-induced human vascular endothelial cell apoptosis. American Journal of Physiology. 1999;277:C1229–C1238. doi: 10.1152/ajpcell.1999.277.6.C1229. [DOI] [PubMed] [Google Scholar]
- Wyss M, Kaddurah-Daouk R. Creatine and creatinine metabolism. Physiological Reviews. 2000;80:1107–1213. doi: 10.1152/physrev.2000.80.3.1107. [DOI] [PubMed] [Google Scholar]