

Abstract
It was unknown if a mitochondria-targeted nitroxide (JP4-039) could augment potentially lethal damage repair (PLDR) of cells in quiescence. We evaluated 32D cl 3 murine hematopoietic progenitor cells which were irradiated and then either centrifuged to pellets (to simulate PLDR conditions) or left in exponential growth for 0, 24, 48 or 72 h. Pelleted cells demonstrated cell cycle arrest with a greater percentage in the G1-phase than did exponentially growing cells. Irradiation survival curves demonstrated a significant radiation damage mitigation effect of JP4-039 over untreated cells in cells pelleted for 24 h. No significant radiation mitigation was detected if drugs were added 48 or 72 h after irradiation. Electron paramagnetic resonance spectroscopy demonstrated a greater concentration of JP4-039 in mitochondria of 24 h-pelleted cells than in exponentially growing cells. These results establish a potential role of mitochondria-targeted nitroxide drugs as mitigators of radiation damage to quiescent cells including stem cells.
Keywords: Potentially lethal damage repair, radiation mitigation, mitochondria-targeted nitroxide
The microenvironment in which cells reside after exposure to ionizing irradiation is known to modulate repair of damage that would otherwise culminate in cell death (1). This component of repair has been termed potentially lethal damage repair (PLDR) and is favored under conditions that inhibit cell division, preventing cells from entering mitosis with damaged DNA (2). Conditions which delay cell cycle progression and facilitate PLDR include incubation in balanced salt solution, hypothermia, and inhibition of protein synthesis (3–6). Little (7) demonstrated an increased survival of irradiated cells that were kept in density-inhibited stationary phase for several hours after irradiation. This technique held cells in the G1-phase, which the authors argued was a requisite for PLDR. Whether PLDR could be further enhanced through treatment with radiation-mitigating agents remained unknown.
A major mechanism of irradiation-induced cell death involves a mitochondria-dependent pathway (8, 9). Reactive oxygen species (ROS) induce lipid peroxidation in the mitochondrial membrane and initiate a cascade of events leading to increased mitochondrial membrane permeability, release of cytochrome c into the cytoplasm, and caspase-3 activation, thus culminating in apoptotic cellular death (8, 10, 11). Mechanisms to scavenge ROS and thereby prevent the caspase cascade have been explored in the development of new small molecule radiation protectors and mitigators.
Nitroxides include a class of radioprotectors which function both in vivo and in vitro by scavenging radicals created by ionizing radiation (12). Experiments with the nitroxide 2,2,6,6-tetramethyl-piperidine-N-oxyl (TEMPO) first demonstrated these radioprotective properties (13, 14); however, millimolar concentrations were required for efficacy, perhaps attributable to ineffective intracellular and mitochondrial partitioning and the limited bioavailability of this compound (15). Key to therapeutic value is the ability to deliver and concentrate the ROS scavengers at the target site, the mitochondria (16). Conjugation of a hemigramicidin S vehicle to nitroxides has been shown to increase mitochondrial localization of the drug at lower systemic concentrations (17). The hemigramicidin vehicle takes advantage of the hydrophobicity of the mitochondrial membrane to target compounds to the mitochondrial matrix. Gramicidin has also been shown to have affinity to bacterial membranes and associated lipids, which are structurally related to mitochondrial membrane constituents (18). JP4-039 is one such nitroxide compound linked to a short alkene isostere analog of hemigramicidin S that has been shown to be protective against radiation damage in vivo and in vitro (19).
In the present studies, we evaluated the mechanism by which the mitochondria-targeted nitroxide JP4-039 improved survival of 32D cl 3 murine hematopoietic progenitor cells and specifically determined its effect on cells held in conditions for PLDR.
Materials and Methods
Chemical synthesis
JP4-039 was synthesized by P. Wipf and colleagues at the University of Pittsburgh. JP4-039 was dissolved in DMSO prior to its usage.
Cell culture
The 32D cl 3 interleukin-3 (IL-3) dependent murine hematopoietic progenitor cell line was derived from a long-term bone marrow culture from a C3H/HeJ mouse and has been previously described (20). Cells were passaged in 15% WEHI-3 cell conditioned medium (as a source of IL-3), 10% fetal calf serum, and McCoy’s supplemented medium according to published methods (20).
Control non-incubation condition
32D cl 3 cells were resuspended at 1×105 cells/ml in 10×100-mm tubes which were assigned to one of three groups: radiation only, post-irradiation treatment with 10 μM JP4-039, and postirradiation treatment with 10 μM TEMPO. Cells were irradiated with doses ranging from 0 to 8 Gy and were plated immediately after irradiation.
Post-irradiation logarithmic growth condition
32D cl 3 cells were resuspended at 5×105 cells/ml in 10×100-mm tubes which were assigned to one of three groups: radiation only, postirradiation treatment with 10 μM JP4-039, and postirradiation treatment with 10 μM TEMPO. Cells were irradiated with doses ranging from 0 to 8 Gy and were then incubated in exponentially growing colonies (flasks) for 24, 48 and 72 h in a high-humidity incubator at 37°C with 95% air/5% CO2.
Post-irradiation PLDR condition
32D cl 3 cells were resuspended at 5×105 cells/ml in 10×100-mm tubes. Cells were irradiated with doses ranging from 0 to 8 Gy and were then centrifuged at 1,500 RPM for 10 min and incubated held tight in pellets with a total of 5×105 cells/pellet in vertical tubes for 24, 48 and 72 h in a high-humidity incubator at 37°C with 95% air/5% CO2.
Clonogenic radiation survival curves
Cells incubated under either logarithmic or PLDR conditions were resuspended at 24, 48 and 72 h after irradiation, and cell viability was assessed using an automated cell viability counter (Beckman Coulter, Fullerton, CA). Subsequently, at 24, 48, 72 h, cells in pellets and flasks were resuspended and treated with 10 μM JP4-039, 10 μM TEMPO, or no treatment and were plated in triplicate in semisolid methylcellulose-containing medium at viable cell densities ranging from 500–40,000 cells/ml. Cells were incubated in a high-humidity incubator at 37°C with 95% air/5% CO2 for 7 d and colonies of greater than 50 cells were scored according to published methods (20).
Cell cycle analysis
The percentages of cells in each phase of the cell cycle were determined by flow cytometry. Briefly, 32D cl 3 cells were resuspended at 1×105 cells/ml and were irradiated with doses ranging from 0 to 8 Gy. Cells were subsequently incubated in pellet or flask conditions for 0, 24, 48 and 72 h. After the incubation period, cells were resuspended, washed three times in phosphate-buffered saline (PBS), fixed in 70% ethanol, and stored at −20°C for at least 24 h. Cells were stained with 0.1 μg/ml of propium iodide, and cell cycle analysis was performed by flow cytometry as previously described (21).
Electron paramagnetic resonance-based analysis of nitroxide distribution and partitioning
To determine nitroxide partitioning and distribution in subcellular compartments, electron paramagnetic resonance (EPR) was utilized. 32D cl 3 cells were incubated for 24 h under pelleted or flask growth conditions. After incubation, 150×106 cells were resuspended at 10×106 cells/ml and were treated with 10 μM JP4-039 or 10 μM TEMPO for 1 h (22).
Subcellular compartments were isolated by differential centrifugation. Cells were centrifuged at 1,500 RPM for 10 min and the supernatant (media) was removed. The remaining whole cell pellet was washed with cold PBS, resuspended in 0.3 ml of hypotonic buffer solution, and incubated on ice for 45 min. Cells were then lysed by three rapid freeze-thaw cycles in liquid nitrogen for 30 s and water at 25°C for 3 min. Lysed cells were centrifuged at 3,000×g for 5 min to isolate nuclei and cellular debris. Supernatant was centrifuged at 12,000×g for 45 min to isolate mitochondria from cytoplasm (22).
Each cellular fraction (media, whole cell pellet, nuclei/cellular debris, mitochondria, and cytoplasm) was mixed with dimethyl sulfoxide (DMSO) (1:1 v/v) and 2 mM of potassium ferricyanide to convert nitroxides to EPR-detectable radical forms (23). An aliquot of 70 μl of the homogenate was loaded into Teflon tubing (0.8 mm internal diameter) (Alpha Wire Corp., Elizabeth, NJ, USA) which was folded in half and placed into an open ESR quartz tube (inner diameter of 3.0 mm). EPR measurements were made in triplicate using a JEOL-RE1X EPR spectrometer (Jeol, Tokyo, Japan) under the following conditions: 334.7 mT center field, 5 mT sweep width, 0.079 mT field modulation, 20 mW microwave power, 0.1 s time constant, and 2 min scan time at 22.5°C. Utilizing signal magnitude and isolated volumes, the amount and concentration of JP4-039 or TEMPO were calculated for each sample.
O2 measurement in pelleted or logarithmic-phase growth 32D cl 3 cell cultures
To determine if pelleted cells became hypoxic, the oxygen content in cells was measured. 32D cl 3 cells were resuspended at 1×105 cells/ml and were centrifuged to a pellet or left in exponential growth and incubated for 24 h. After the incubation period, cells in flasks were also centrifuged to a pellet. Media were decanted from both groups and nitrogen gas was forced into a plugged microcentrifuge tube to remove oxygen from the headspace above the pellet. Cell lysis buffer (Qiagen, Hilden, Germany) and headspace was also nitrated in a separate plugged microcentrifuge tube. Pelleted 32 D cl 3 cells were resuspended in 50 μl of nitrated cell lysis buffer and the oxygen concentration was measured using an OM-4 Oxygen Meter (Microelectrodes Inc., Bedford, NH, USA) (24).
Statistical analysis
Data from radiation survival curves were analyzed using the linear-quadratic and single-hit, multitarget models and were compared using D0 (final slope representing multiple-event killing) and Ñ (extrapolation number measuring width of the shoulder on the radiation survival curve) (2, 25). Results are presented as the mean±standard error of the mean from at least three measurements. Student’s t-test and two-way ANOVA were used to compare means of different groups where appropriate.
Results
Pelleting of cells for 24 h simulates PLDR conditions
Centrifuging cells to pellets after irradiation was first confirmed to facilitate the conditions for measurement of PLDR. Radiation survival curves were measured on cells held in pellets compared to exponentially growing cells after 0, 24, 48 and 72 h post-irradiation (Table I). Cells pelleted for 24 h demonstrated significant radiation mitigation (D0=1.69±0.17) over cells in exponential growth for 24 h (D0=1.25±0.09) and cells plated immediately after irradiation (D0=0.91±0.11) (p=0.0489 and 0.0151 respectively). Cells pelleted for 48 and 72 h did not demonstrate significant radiation mitigation compared to exponentially growing cells. The radiation mitigation at 24 h coupled with a subsequent cell cycle analysis in pelleted cells demonstrated G1-phase arrest and confirmed that this model fulfills the criteria for PLDR (26).
Table I.
Radiosensitivity of 32D cl 3 cells treated with JP4-039 or TEMPO.
| Treatment | Ñ | D0 | |
|---|---|---|---|
| 0 hours | Control | 6.85±0.49 | 1.04±0.05 |
| JP4-039 | 37.43±7.50* | 0.91±0.11 | |
| TEMPO | 8.23±4.35 | 1.04±0.07 | |
| 24 hours (Flask) | Control | 1.78±0.78 | 1.25±0.09 |
| JP4-039 | 10.35±0.33* | 1.22±0.16 | |
| TEMPO | 8.86±6.69 | 1.26±0.11 | |
| 24 hours (Pellet) | Control | 1.38±0.23 | 1.69±0.17† |
| JP4-039 | 18.49±3.90* | 1.12±0.27 | |
| TEMPO | 7.59±1.71‡ | 1.13±0.19 |
32D cl 3 cells incubated for 0, 24, 48, and 72 h in exponential growth colonies (flasks) or pellets were treated with JP4-039, TEMPO or no treatment. Cells pelleted for 24 h simulated PLDR conditions as evidenced by accumulation in G1-phase and increased Ñ (shoulder on radiation survival curve) as compared to the 0 h control and 24 h exponential growth cells (†p=0.0489 and 0.0151, respectively). JP4-039 demonstrated significant radiation mitigation over untreated cells at 0 h and at 24 h under both flask and pellet post-irradiation conditions as evidenced by increased Ñ (extrapolation number) (*p=0.0353, 0.0184 and 0.0118, respectively). TEMPO added after 24 h to pelleted cells demonstrated significant radiation mitigation compared to control cells (‡p=0.0219). No significant radiation damage mitigation was found at 48 h or 72 h.
JP4-039 improves survival of irradiated 32D cl 3 cells both in logarithmic growth and in cell pellets
To demonstrate the effect of mitochondria-targeted nitroxide compounds in protecting cells from irradiation death, clonogenic survival curves were determined (Figure 1).
Figure 1.

Clonogenic irradiation survival of 32D cl 3 cells treated with JP4-039, TEMPO or no treatment. Cells were plated immediately after irradiation (A–B), after post-irradiation incubation for 24 h in exponential growth (C–D), or after post-irradiation incubation for 24 h in pelleted condition (E–F). Data are plotted using the linear-quadratic (A, C, E) and single-hit multitarget (B, D, F) models. JP4-039 increased the shoulder on the survival curve as compared to that of untreated cells in the immediately plated group (p=0.0353), after 24 h under exponential growth conditions (p=0.0184), and after 24 h incubation under PLDR conditions (p=0.0118).
Figure 1 demonstrates representative irradiation survival curves using both the linear quadratic and single-hit multi-target models for 32D cl 3 cells plated immediately after irradiation and after 24 h incubation in flasks or pellets. 32D cl 3 cells treated immediately after irradiation with JP4-039, but not TEMPO, demonstrated significant radiation mitigation by an increased shoulder on the radiation survival curve (Ñ=37.43±7.50 or 8.23±4.35, respectively) compared to Ñ=6.85±0.49 for control irradiated cells (p=0.0353 or 0.0228, respectively) (Table I). These data indicate that in the immediate post-irradiation setting, JP4-039 enhanced cell repair and increases clonogenic cell survival. A radiation-mitigating effect of delayed post-irradiation addition of JP4-039 over untreated cells was also observed at 24 h in exponentially growing cells (Ñ=10.35±0.33, compared to non-treated cells, 1.78±0.78, respectively, p=0.0184).
In cells incubated as pellets for 24 h (Figure 1), both JP4-039 and TEMPO demonstrated greater radiation mitigation (Ñ=18.49±3.90 and 7.59±1.71, respectively) compared to Ñ=1.38±0.23 for control cells (p=0.0118 and 0.0219, respectively). No significant radiation mitigation was detected in cell pellets held for 48 or 72 h after irradiation before the drug was added. These data demonstrate that nitroxide compounds enhanced clonogenic cell survival when applied to cells held as pellets for 24 h after irradiation. After 48 and 72 h in pellets there was no mitigation detected.
32D cl 3 cells in pellets are quiescent after 24 h
To further confirm that pelleting of cells simulated PLDR conditions, the percentage of cells in each phase of the cell cycle was determined. Under two post-irradiation incubation conditions, as exponentially growing cells in flasks and in pellets across the full radiation dose spectrum, cells were resuspended, fixed, stained with propidium iodide and analyzed by flow cytometry.
After 24 h, pelleted cells demonstrated a significantly higher proportion of cells in the G1-phase as compared to cells in exponential growth across the dose range shown (p<0.0001) (Figure 2A). At 48 and 72 h, this difference was still present at lower doses but at 6 Gy and above the two conditions did not have significantly different percentages of cells in the G1-phase (p=0.95, 0.06 respectively) (Figure 2B–C).
Figure 2.

G1-phase cell cycle analysis of irradiated 32D cl 3 cells. The percentage of G1-phase 32D cl 3 cells after irradiation and incubation was quantified for pellets and flasks after 24 h (A), 48 h (B), and 72 h (C). At 24 h, cells that had been pelleted after irradiation demonstrated a greater percentage of cells in the G1-phase than did cells in exponential growth across the full dose range (p<0.0001). The accumulation in G1 persisted at 48 and 72 h for lower doses (0–5 Gy, p<0.0001); however, at 6 Gy and above there was no statistical difference between the two post-irradiation growth conditions.
The percentage of cells in the S-phase was significantly lower in pelleted cells than in exponentially growing cells across the radiation dose range at 24 h (Figure 3A) (p<0.0001). At 48 and 72 h, the difference persisted, except at 6 Gy and above, where again the percentage of cells in the S-phase was not statistically different between the two conditions (p=0.052, 0.20 respectively) (Figure 3B–C).
Figure 3.

S-phase cell cycle analysis of irradiated 32D cl 3 cells. The percentage of S-phase 32D cl 3 cells after irradiation with full dose range and incubation was quantitated for pellets or flasks after 24 h (A), 48 h (B), or 72 h (C). At 24 h, cells that had been pelleted after irradiation demonstrated a significantly lower percentage of cells in S-phase than cells in exponential growth at most doses across the full dose range (p<0.0001). At 48 h and 72 h, this difference persisted for lower doses (0–5 Gy, p<0.0001), however, at 6 Gy and above there was no statistically significant difference between the two post-irradiation conditions.
Thus for cells kept in pellets for 24 h, there was a significant difference in the percentage of cells in the G- and S-phases across the dose spectrum (0–8 Gy). However, with the 48 and 72 h post-irradiation incubation times, these differences in the cell cycle were no longer significant at 6 Gy and above.
Apoptosis is greater in pelleted 32D cl 3 cells compared to cells in logarithmic growth
The percentage of apoptotic cells also differed based on the condition and duration of incubation. Regarding the condition, cell cycle analysis revealed that pelleted cells undergo a greater degree of apoptosis than cells in flasks at all doses for cells incubated for 24 and 48 h (Table II) (p<0.0001). At 72 h, pelleted cells demonstrated increased apoptosis compared to cells in flasks for most doses below 6 Gy (p=0.011).
Table II.
Post-irradiation apoptosis of 32D cl 3 cells (% ) grown in flasks or held in pellets.
| 24 h |
48 h |
72 h |
||||
|---|---|---|---|---|---|---|
| Dose (Gy) | Flask | Pellet | Flask | Pellet | Flask | Pellet |
| 0 | 4.47±1.14 | 35.66±11.4 | 10.30±4.33 | 38.66±9.72 | 45.84±20.42 | 68.91±20.30 |
| 0.5 | 8.52±4.75 | 35.11±17.61 | 7.03±2.21 | 51.08±15.19 | 48.91±12.80 | 69.13±21.55 |
| 1 | 4.89±1.92 | 33.07±8.18 | 5.68±4.31 | 62.44±19.88 | 27.88±13.52 | 73.33±21.01 |
| 2 | 5.19±2.41 | 34.60±8.48 | 9.07±6.43 | 67.04±16.93 | 40.78±19.60 | 89.33±0.27 |
| 3 | 8.73±3.31 | 42.89±10.69 | 22.65±14.88 | 71.67±14.67 | 38.22±26.44 | 71.76±18.42 |
| 4 | 10.47±1.93 | 41.93±1.73 | 22.15±7.82 | 73.84±12.83 | 62.16±26.62 | 74.04±14.43 |
| 5 | 15.02±2.88 | 44.90±0.96 | 29.49±13.97 | 75.40±12.12 | 58.26±13.43 | 76.50±13.79 |
| 6 | 15.32±2.21 | 43.11±5.78 | 29.80±6.46 | 76.56±14.01 | 54.00±27.05 | 93.26±1.74 |
| 7 | 20.22±5.41 | 50.27±5.94 | 38.02±7.76 | 79.17±11.36 | 85.89±4.97 | 87.76±5.44 |
| 8 | 22.84±7.50 | 50.56±10.9 | 43.60±5.12 | 87.75±5.18 | 92.52±4.42 | 95.41±0.78 |
The percentage of apoptotic 32D cl 3 cells after irradiation with full dose range and incubation was quantified for pellets and flasks at 24 h, 48 h, and 72 h. Pelleted cells had greater apoptosis than corresponding cells held in logarithmic growth (flasks) for all doses at 24 h (p<0.0001) and 48 h (p<0.0001) and most doses at 72 h (p=0.0039).
The longer incubation times induced a significantly greater percentage of cells to undergo apoptosis independent of the post-irradiation condition (p<0.0001). Figure 4 demonstrates that the percentage of apoptotic cells across the entire dose range was greater as the time of incubation increased for cells in flasks (Figure 4A) and in pellets (Figure 4B). These data are consistent with prior studies showing that differences between nutrient-deprived and exponentially growing cells in G1- and S-phase that are evident at 24 h do not persist at 48 and 72 h as cells die in apoptosis.
Figure 4.

Apoptosis and cell cycle analysis of 32D cl 3 cells. The percentage of apoptotic 32D cl 3 cells after irradiation was quantified for exponentially growing cells (flasks) (A) and pelleted cells (B) at 0, 24, 48 and 72 h. Under both conditions, prolonged post-irradiation incubation led to a significant increase in the percentage of apoptotic cells compared to cells at 0 h (p<0.0001, p<0.0001).
There was no statistically significant difference in the percentage of cells in the G2/M-phase between the two post-irradiation incubation conditions at 24, 48 or 72 h (data not shown). These results establish that cells in pellets were arrested at the G1-phase in contrast to cells in flasks which actively cycled. There was both a radiation dose-dependent and time-dependent effect on apoptosis and cell cycle arrest. The high rate of apoptosis at 48 and 72 h correlated to a relative lack of radiation mitigation by JP4-039 and TEMPO in cells in pellets and flasks.
32D cl 3 cells in pellets are not significantly hypoxic compared to cells in logarithmic growth
Since the cells pelleted for 24 h and treated with JP4-039 demonstrated the greatest radiation mitigation and cell cycle arrest, the level of hypoxia was measured.
The measured oxygen percentage in 50 μl of lysis buffer was 5.85±1.00% for pelleted cells and 6.62±1.73% for exponentially growing cells (n=6). There was no statistically significant difference between cells in these two conditions (p=0.41). This result establishes that the radiation mitigation effect of JP4-039 was not mediated by hypoxic conditions in pelleted cells and supports a role of PLDR conditions, including contact inhibition and nutrient deprivation.
JP4-039 partitions into mitochondria of cells held under PLDR conditions
The partitioning of the nitroxide signal to subcellular compartments was next evaluated using EPR spectroscopy (Figures 5 and 6).
Figure 5.
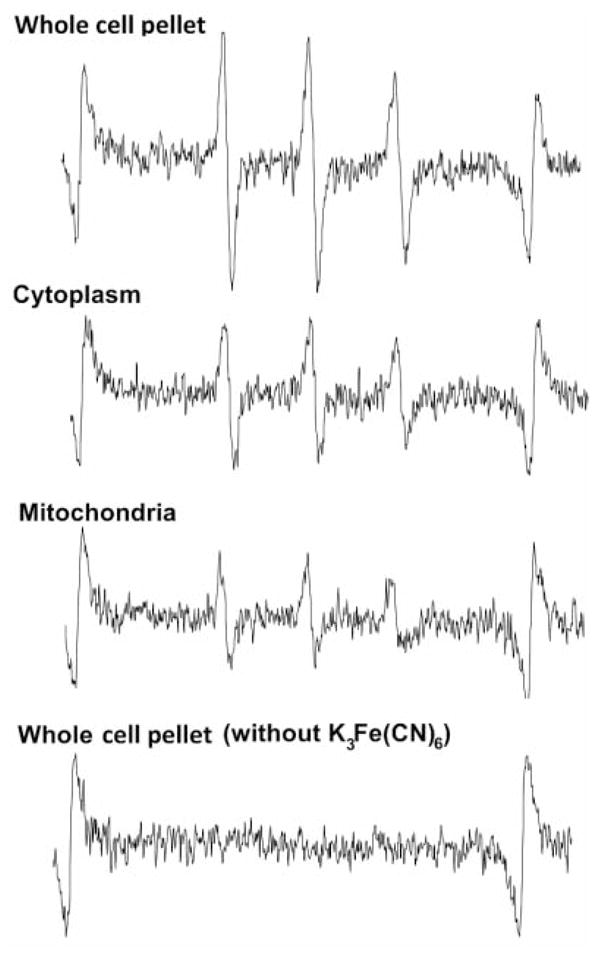
Representative EPR spectra demonstrating nitroxide signal in isolated subcellular fractions of JP4-039-treated cells. The top three spectra are from samples mixed with DMSO and addition of the oxidizing agent potassium ferricyanide (K3Fe(CN)6). The lower EPR spectrum is a negative control indicating no detectable EPR signal without addition of the oxidizing agent (n=3). No signal was detected in TEMPO-treated cells (n=3).
Figure 6.

EPR-based determination of nitroxide signal in subcellular fractions of 32D cl 3 cells. Cells were incubated for 24 h in exponential growth (flask) or pelleted form and were subsequently treated with 10 μM JP4-039. A) The percentage of JP4-039 in 32D cl 3 cells as a fraction of that in the whole cell pellet. B) The concentration of JP4-039 as calculated from EPR magnitude and isolated volumes. In pelleted cells, the mitochondrial concentration of JP4-039 was 10.9 μM and was significantly (*) higher than that in other subcellular compartments (p<0.001) and the mitochondrial concentration of JP4-039 in flasks (p=0.026).
In 32D cl 3 cells treated with JP4-039, there were significant differences in the level of nitroxide EPR signal detected from each of the subcellular fractions compared to cells treated with TEMPO (p<0.0001). Comparing the calculated amount of nitroxide in each subcellular fraction against that of the whole cell pellet, we determined the relative partitioning of the nitroxide inside cells (Figure 6). Calculated percentages summed to less than 100% due to loss during isolation and measurement procedures. In cells held in exponential growth, 5.0±0.3% of the cellular EPR signal was detected in mitochondria, 37.2±0.3% in nucleus and/or cellular debris and 19.4±1.6% in the cytoplasm. In pelleted cells, 3.9±0.2% of the nitroxide EPR signal in cells was localized to the mitochondria, 33.2±3.8% was located in the nuclei and/or cellular debris and 29.5±0.7% was detected in the cytoplasm. The concentration of JP4-039 was calculated in each of these subcellular compartments (Figure 7B). In pelleted cells, the mitochondrial concentration of JP4-039 was 10.90±0.48 μM and was significantly higher than the other subcellular fractions (p<0.001). In cells held in exponential growth, the mitochondrial concentration was 8.5±0.5 μM, but was lower than the concentration in the nuclei and cellular debris (10.1±0.1 μM) and the whole cell pellet (12.6±0.2 μM). The mitochondrial concentration of JP4-039 in pelleted cells was significantly higher than that of cells grown in flasks (p=0.026).
In cells treated with TEMPO under either condition, the nitroxide signal was below the detectable threshold. The TEMPO signal was detected in the cell media; however, no signal was detected in the whole cell pellet or any of the subcellular fractions.
These experiments establish that JP4-039 effectively partitioned into 32D cl 3 cells compared to TEMPO in cells incubated in either logarithmic growth in flasks or under pelleted conditions. Furthermore, under pelleted conditions, JP4-039 concentrated in the mitochondria more effectively than in other measured subcellular compartments and in mitochondria isolated from cells under exponential growth conditions. The increased concentration of JP4-039 in 24 h-pelleted cells was consistent with its superior radiation mitigation effect observed in the clonogenic radiation survival curves.
Discussion
The present studies demonstrate a simple, novel technique to achieve contact inhibition of hematopoietic cells and hold a large proportion of cells in the G1-phase, facilitating PLDR conditions. The current work establishes that JP4-039, a mitochondria-targeted nitroxide drug, enhances PLDR in 32D cl 3 cells as determined by clonogenic radiation survival curves. Cell cycle analysis revealed that cells held in pellets were in the G1-phase of the cell cycle and EPR spectroscopy data demonstrated an increased concentration of drug in pelleted cells. Thus, the higher concentration of JP4-039 in the mitochondria of pelleted cells may have had a role in the observed increased radiation-mitigation effect.
Nitroxides are efficient free radical scavengers that ameliorate the oxidative stress induced by ionizing radiation. However, the delivery of sufficient quantities of nitroxide to the mitochondria has been challenging (22). Novel nitroxide compounds such as JP4-039, which contains an alkene peptide isostere fragment of hemigramicidin S as a targeting unit, enable selective delivery of ROS-scavenging agents to the mitochondria, the purported site of origin of irradiation-induced apoptosis. A noteworthy structural feature of JP4-039 is its small size and enhanced lipophilicity which may help explain its greater ability for radiation mitigation in these studies as compared to TEMPO (22).
In these studies, JP4-039 concentrated in the mitochondria of pelleted cells more than in exponentially growing cells. Differing pharmacokinetic properties between quiescent and cycling cells has previously been documented. Busulfan, for example, is an alkylating agent that is particularly toxic for quiescent hematopoietic stem cells (27, 28). Whether the increased mitochondrial concentration of JP4-039 presented here is due to an active or passive process in the quiescent cell should be explored in future studies.
Various methods have been employed to induce conditions for PLDR. Post-irradiation liquid holding for a recovery period followed by delayed plating has been shown to increase cell survival compared to cells plated immediately after irradiation (29). Cycloheximide and deoxyadenosine, two inhibitors of DNA synthesis, were also shown to increase PLDR capacity and cell survival, perhaps by delaying the expression of damaged DNA and allowing for time for repair (1, 30). Balanced salt solution incubation (26) and hypothermia (4, 5) have also been described as mechanisms to enable PLDR. Importantly, these prior studies demonstrated the significance of holding cells in the G1-phase (31). Recent studies have suggested that simply arresting the cell cycle is not sufficient to induce the conditions for PLDR (32). In the present study, the majority of pelleted cells demonstrated cell cycle arrest in G1 after 24 h. Furthermore, after 24 h these pelleted cells were not hypoxic compared to exponentially growing cells. The data suggest that cell contact inhibition, nutrient deprivation and perhaps factors other than hypoxia may have a played a role in establishing the ideal microenvironment for PLDR.
The clinical relevance of PLDR in radiotherapy continues to be a topic of debate (2). In vitro techniques for PLDR including incubation in balanced salt solutions or hypothermia are difficult to translate to clinical paradigms. Conversely, studies of plateau phase cell cultures and pelleted cells, both of which have large proportions of nonproliferating G1-phase cells, have been shown to have clinical relevance (33). Some studies sought to demonstrate PLDR in normal tissue in vivo. A study of irradiated rat thyroid in vivo followed 24 h later by transplantation was found to increase cell survival compared to tissue transplanted immediately after irradiation (34). In an experiment conducted by Watanabe et al., six weeks after thyroid irradiation was the time for transplantation when the frequency of irradiation-induced chromosomal lesions was less. The authors found that the longer holding time reduced colony-forming efficiency by a factor of 2 (35). Our data are consistent with both these studies.
PLDR has been demonstrated utilizing an in vitro colony assay of murine lung irradiated in vivo. An increase in the fraction of surviving cells was detected 6 h after irradiation, and there was no further change between 6 and 24 h (36). Parenchymal hepatocytes were also studied using an in vivo clonogenic assay system after exposure to irradiation. Hepatocytes which were allowed to remain in situ for 24 h before the assay for survival were found to be more radioresistant than hepatocytes assayed after 30 min, as evidenced by an increase in Ñ but not D0. The authors claimed that in these normal parenchymal hepatocytes in quiescence, PLDR was responsible for the increased survival (37). Collectively, in accordance with this, the data presented here show that PLDR was most effective at 24 h.
In the present study, no statistically significant radiation mitigation was detected 48 or 72 h after irradiation with either JP4-039 or TEMPO. This finding correlated with the increased percentage of apoptotic cells measured by flow cytometry at these later time points compared to a 24 h. PLDR conditions at these longer incubation time points may have been overwhelmed by apoptosis. Several studies have suggested that PLDR is nearly complete 6 h after irradiation (26). Our findings indicate that the effects of PLDR persist for up to 24 h after irradiation.
One future clinical application of the present findings may involve hematopoietic stem cells (HSCs). HSCs are characterized by their ability to differentiate to all blood cell types while retaining the capacity for self-renewal (38). Prior studies demonstrated that long-term, reconstituting HSCs are not mitotically active and that only 15–20% of these cells are cycling (cell phase S/G2/M) (39, 40). The quiescent state of HSCs, in conjunction with other factors in the hematopoietic microenvironment, may be critical to the relative radioresistance of this population of cells (41, 42). Quiescence of the HSCs resembles the state of the pelleted 32D cl 3 cells presented here. Future studies will be required to demonstrate PLDR in HSCs in vivo.
Acknowledgments
This project was supported by award number U19AI068021 from the National Institute of Allergy and Infectious Diseases and T32AG21885 from the National Institutes of Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Allergy and Infectious Diseases or the National Institutes of Health.
References
- 1.Phillips RA, Tolmach LJ. Repair of potentially lethal damage in x-irradiated HeLa cells. Radiat Res. 1966;29:413–432. [PubMed] [Google Scholar]
- 2.Hall EJ, Giaccia AJ. Radiobiology for the Radiologist. 6. Philadelphia: Lippincott Williams & Wilkins; 2006. pp. 60–84. [Google Scholar]
- 3.Weiss BG, Tolmach LJ. Modification of x-ray-induced killing of HeLa s3 cells by inhibitors of DNA synthesis. Biophys J. 1967;7:779–795. doi: 10.1016/S0006-3495(67)86622-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Whitmore GF, Gulyas S. Studies on recovery processes in mouse L cells. Natl Cancer Inst Monogr. 1967;24:141–156. [PubMed] [Google Scholar]
- 5.Winans LF, Dewey WC, Dettor CM. Repair of sublethal and potentially lethal x-ray damage in synchronous Chinese hamster cells. Radiat Res. 1972;52:333–351. [PubMed] [Google Scholar]
- 6.Belli JA, Shelton M. Potentially lethal radiation damage: repair by mammalian cells in culture. Science. 1969;165:490–492. doi: 10.1126/science.165.3892.490. [DOI] [PubMed] [Google Scholar]
- 7.Little JB. Repair of sub-lethal and potentially lethal radiation damage in plateau phase cultures of human cells. Nature. 1969;224:804–806. doi: 10.1038/224804a0. [DOI] [PubMed] [Google Scholar]
- 8.Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- 9.Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. 2004;116:205–219. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- 10.Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- 11.Wang ZB, Liu YQ, Cui YF. Pathways to caspase activation. Cell Biol Int. 2005;29:489–496. doi: 10.1016/j.cellbi.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 12.Soule BP, Hyodo F, Matsumoto K, Simone NL, Cook JA, Krishna MC, Mitchell JB. The chemistry and biology of nitroxide compounds. Free Radic Biol Med. 2007;42:1632–1650. doi: 10.1016/j.freeradbiomed.2007.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mitchell JB, DeGraff W, Kaufman D, Krishna MC, Samuni A, Finkelstein E, Ahn MS, Hahn SM, Gamson J, Russo A. Inhibition of oxygen-dependent radiation-induced damage by the nitroxide superoxide dismutase mimic, tempol. Arch Biochem Biophys. 1991;289:62–70. doi: 10.1016/0003-9861(91)90442-l. [DOI] [PubMed] [Google Scholar]
- 14.Hahn SM, Tochner Z, Krishna CM, Glass J, Wilson L, Samuni A, Sprague M, Venzon D, Glatstein E, Mitchell JB, et al. Tempol, a stable free radical, is a novel murine radiation protector. Cancer Res. 1992;52:1750–1753. [PubMed] [Google Scholar]
- 15.Jiang J, Belikova NA, Hoye AT, Zhao Q, Epperly MW, Greenberger JS, Wipf P, Kagan VE. A mitochondria-targeted nitroxide/hemigramicidin S conjugate protects mouse embryonic cells against gamma irradiation. Int J Radiat Oncol Biol Phys. 2008;70:816–825. doi: 10.1016/j.ijrobp.2007.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hoye AT, Davoren JE, Wipf P, Fink MP, Kagan VE. Targeting mitochondria. Acc Chem Res. 2008;41:87–97. doi: 10.1021/ar700135m. [DOI] [PubMed] [Google Scholar]
- 17.Fink MP, Macias CA, Xiao J, Tyurina YY, Jiang J, Belikova N, Delude RL, Greenberger JS, Kagan VE, Wipf P. Hemigramicidin-TEMPO conjugates: novel mitochondria-targeted anti-oxidants. Biochem Pharmacol. 2007;74:801–809. doi: 10.1016/j.bcp.2007.05.019. [DOI] [PubMed] [Google Scholar]
- 18.Prenner EJ, Lewis RN, Neuman KC, Gruner SM, Kondejewski LH, Hodges RS, McElhaney RN. Nonlamellar phases induced by the interaction of gramicidin S with lipid bilayers. A possible relationship to membrane-disrupting activity. Biochemistry. 1997;36:7906–7916. doi: 10.1021/bi962785k. [DOI] [PubMed] [Google Scholar]
- 19.Epperly MW, Pierce JG, Dixon T, Franicola D, Wipf P, Greenberger JS. The mitochondrial targeted GS-nitroxide JP4-039 is radioprotective in vitro and in vivo. Int J Radiat Oncol Biol Phys. 2008;72:S82–S82. [Google Scholar]
- 20.Epperly MW, Gretton JE, Sikora CA, Jefferson M, Bernarding M, Nie S, Greenberger JS. Mitochondrial localization of superoxide dismutase is required for decreasing radiation-induced cellular damage. Radiat Res. 2003;160:568–578. doi: 10.1667/rr3081. [DOI] [PubMed] [Google Scholar]
- 21.Epperly MW, Bray JA, Carlos TM, Prochownik E, Greenberger JS. Biology of marrow stromal cell lines derived from long-term bone marrow cultures of Trp53-deficient mice. Radiat Res. 1999;152:29–40. [PubMed] [Google Scholar]
- 22.Wipf P, Xiao J, Jiang J, Belikova NA, Tyurin VA, Fink MP, Kagan VE. Mitochondrial targeting of selective electron scavengers: synthesis and biological analysis of hemigramicidin-TEMPO conjugates. J Am Chem Soc. 2005;127:12460–12461. doi: 10.1021/ja053679l. [DOI] [PubMed] [Google Scholar]
- 23.Behringer W, Safar P, Kentner R, Wu X, Kagan VE, Radovsky A, Clark RS, Kochanek PM, Subramanian M, Tyurin VA, Tyurina YY, Tisherman SA. Antioxidant Tempol enhances hypothermic cerebral preservation during prolonged cardiac arrest in dogs. J Cereb Blood Flow Metab. 2002;22:105–117. doi: 10.1097/00004647-200201000-00013. [DOI] [PubMed] [Google Scholar]
- 24.Kanai AJ, Pearce LL, Clemens PR, Birder LA, VanBibber MM, Choi SY, de Groat WC, Peterson J. Identification of a neuronal nitric oxide synthase in isolated cardiac mitochondria using electrochemical detection. Proc Natl Acad Sci USA. 2001;98:14126–14131. doi: 10.1073/pnas.241380298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Epperly M, Bray J, Kraeger S, Zwacka R, Engelhardt J, Travis E, Greenberger J. Prevention of late effects of irradiation lung damage by manganese superoxide dismutase gene therapy. Gene Ther. 1998;5:196–208. doi: 10.1038/sj.gt.3300580. [DOI] [PubMed] [Google Scholar]
- 26.Iliakis G. Radiation-induced potentially lethal damage: DNA lesions susceptible to fixation. Int J Radiat Biol Relat Stud Phys Chem Med. 1988;53:541–584. doi: 10.1080/09553008814550901. [DOI] [PubMed] [Google Scholar]
- 27.Westerhof GR, Down JD, Blokland I, Wood M, Boudewijn A, Watson AJ, McGown AT, Ploemacher RE, Margison GP. O6-Benzylguanine potentiates BCNU but not busulfan toxicity in hematopoietic stem cells. Exp Hematol. 2001;29:633–638. doi: 10.1016/s0301-472x(01)00631-2. [DOI] [PubMed] [Google Scholar]
- 28.Greenberger JS. Gene therapy approaches for stem cell protection. Gene Ther. 2008;15:100–108. doi: 10.1038/sj.gt.3303004. [DOI] [PubMed] [Google Scholar]
- 29.Frankenberg-Schwager M, Frankenberg D, Harbich R. Potentially lethal damage repair is due to the difference of DNA double-strand break repair under immediate and delayed plating conditions. Radiat Res. 1987;111:192–200. [PubMed] [Google Scholar]
- 30.Terasima T, Tolmach LJ. X-ray sensitivity and DNA synthesis in synchronous populations of HeLa cells. Science. 1963;140:490–492. doi: 10.1126/science.140.3566.490. [DOI] [PubMed] [Google Scholar]
- 31.Little JB. Factors influencing the repair of potentially lethal radiation damage in growth-inhibited human cells. Radiat Res. 1973;56:320–333. [PubMed] [Google Scholar]
- 32.van Bree C, Rodermond HM, ten Cate R, de Vos J, Stalpers LJ, Haveman J, Medema JP, Franken NA. G0 cell cycle arrest alone is insufficient for enabling the repair of ionizing radiation-induced potentially lethal damage. Radiat Res. 2008;170:184–191. doi: 10.1667/RR0845.1. [DOI] [PubMed] [Google Scholar]
- 33.Barendsen GW, Van Bree C, Franken NA. Importance of cell proliferative state and potentially lethal damage repair on radiation effectiveness: implications for combined tumor treatments (review) Int J Oncol. 2001;19:247–256. doi: 10.3892/ijo.19.2.247. [DOI] [PubMed] [Google Scholar]
- 34.Mulcahy RT, Gould MN, Clifton KH. The survival of thyroid cells: in vivo irradiation and in situ repair. Radiat Res. 1980;84:523–528. [PubMed] [Google Scholar]
- 35.Watanabe H, Hendry JH. Clonogen number and radiosensitivity in rat thyroid follicles. Radiat Res. 1991;128:222–224. [PubMed] [Google Scholar]
- 36.Deschavanne PJ, Guichard M, Malaise EP. Repair of sublethal and potentially lethal damage in lung cells using an in vitro colony method. Br J Radiol. 1981;54:973–977. doi: 10.1259/0007-1285-54-647-973. [DOI] [PubMed] [Google Scholar]
- 37.Jirtle RL, Michalopoulos G, Strom SC, DeLuca PM, Gould MN. The survival of parenchymal hepatocytes irradiated with low and high LET radiation. Br J Cancer Suppl. 1984;6:197–201. [PMC free article] [PubMed] [Google Scholar]
- 38.Adams GB, Scadden DT. The hematopoietic stem cell in its place. Nat Immunol. 2006;7:333–337. doi: 10.1038/ni1331. [DOI] [PubMed] [Google Scholar]
- 39.Morrison SJ, Weissman IL. The long-term repopulating subset of hematopoietic stem cells is deterministic and isolatable by phenotype. Immunity. 1994;1:661–673. doi: 10.1016/1074-7613(94)90037-x. [DOI] [PubMed] [Google Scholar]
- 40.Fleming WH, Alpern EJ, Uchida N, Ikuta K, Spangrude GJ, Weissman IL. Functional heterogeneity is associated with the cell cycle status of murine hematopoietic stem cells. J Cell Biol. 1993;122:897–902. doi: 10.1083/jcb.122.4.897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Arai F, Hirao A, Ohmura M, Sato H, Matsuoka S, Takubo K, Ito K, Koh GY, Suda T. Tie2/angiopoietin-1 signaling regulates hematopoietic stem cell quiescence in the bone marrow niche. Cell. 2004;118:149–161. doi: 10.1016/j.cell.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 42.Calvi LM, Adams GB, Weibrecht KW, Weber JM, Olson DP, Knight MC, Martin RP, Schipani E, Divieti P, Bringhurst FR, Milner LA, Kronenberg HM, Scadden DT. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature. 2003;425:841–846. doi: 10.1038/nature02040. [DOI] [PubMed] [Google Scholar]