

Abstract
The discovery and development of the asymmetric rhodium-catalyzed [2+2+2] cycloaddition of alkenyl isocyanates and exogenous alkynes to form indolizinone and quinolizinone scaffolds is described. This methodology has been expanded to include substituted alkenes and dienes, a variety of sterically and electronically diverse alkynes, and carbodiimides in place of the isocyanate. Through X-ray analysis of Rh(cod)/phosphoramidite complexes, additives that modify the enantio-determining step, and other experimental data, a mechanism has been proposed that explains lactam, vinylogous amide, and pyridone products and the factors governing their formation. Finally, we have applied this methodology to the synthesis of (+)-lasubine-II and (−)-209D.
INTRODUCTION
The rapid assembly of complex molecules is a long-standing goal in organic chemistry, and metal-catalyzed cycloadditions are one of the most efficient ways to achieve that goal. Previously, the formation of carbocyles has been the main focus, including [5+2] and [4+2] reactions [1]. Three component cycloadditions include [2+2+1] Pauson-Khand type reactions and [2+2+2] cyclotrimerizations of alkynes [2]. Utilizing three separate π components in [2+2+2] cycloadditions presents the potential for numerous substitutions and stereocenters to be set in a single step. At the time we entered the field, the formation of nitrogen-containing heterocycles was limited to [2+1] aziridinations, 1,3-dipolar cycloadditions, and [2+2+2] pyridone formation [3]. The area of three component cycloadditions to form nitrogen-containing heterocycles with a carbon stereocenter was underdeveloped. This review will provide an overview of our discovery and development of rhodium-catalyzed [n+2+2] asymmetric cycloaddition reactions utilizing nitrogen-containing π components.
In 1977, Yamazaki reported a metal-catalyzed [2+2+2] reaction that coupled isocyanates and alkynes to form pyridones [4]. In the 1980’s, Hoberg and coworkers greatly advanced the field of metal-promoted cycloadditions with nickel-mediated and catalyzed transformations utilizing a combination of isocyanates, carbon dioxide, alkenes, or alkynes to form various heterocycles and carbocycles [5].
Vollhardt [6] demonstrated the ability to control the regioselectivity of cycloadditions of tethered alkynes in the formation of pyridone products and found alkyne electronics affect this selectivity. Itoh [7] and Louie [8] independently showed that both nickel and ruthenium work effectively to synthesize bicyclic pyridones. Tanaka [9] established the first asymmetric synthesis of pyridones by exploiting a cationic rhodium/bidentate phosphine catalyst to generate atropisomers of 2-pyridones with good enantioselectivity.
 |
(1) |
 |
(2) |
 |
(3) |
We sought to increase the synthetic utility of these reactions by replacing one of the alkynes with an alkene; such a substitution would introduce a carbon stereocenter in the newly formed cycloadduct. The coupling of a nitrogen-containing heterocumulene, alkene, and alkyne in an asymmetric metal-catalyzed cycloaddition would provide an efficient route to numerous indolizidine and quinolizidine natural products (Fig. 1).
Fig. 1.

Indolizidine and quinolizidine natural products
Although synthetically useful, the introduction of an alkene π component in place of an alkyne is hardly trivial. Inherently, alkynes are more electronegative than alkenes and bind more tightly to transition metals due to Mdπ-Cπ* back bonding. To coax the alkene to participate in the cycloaddition in the presence of excess alkyne, we elected to tether the alkene to the isocyanate to give the alkene an entropic advantage over an exogenous alkyne.
DEVELOPMENT OF A RHODIUM-CATALYZED [2+2+2] CYCLOADDITION WITH ALKENYL ISOCYANATES
Reaction Discovery
Our investigation of three-component [2+2+2] metal-catalyzed cycloadditions began with alkene-tethered isocyanates and internal, symmetrical alkynes [10]. The complex generated from [Rh(C2H4)2Cl]2 and a triarylphosphine ligand was initially found to catalyze the desired cycloaddition. In the case of alkyl alkynes, the expected lactam product (3) was formed, but we were surprised to find a second cycloadduct product in addition to lactam 3 (eq 4). X-ray analysis of the second cycloadduct established its structure as vinylogous amide 4, which arises from fragmentation of the isocyanate.
With our first generation catalyst, we further demonstrated that the product selectivity between these two adducts is influenced by the sterics and electronics of the alkyne. While aryl or conjugated internal alkynes generate vinylogous amide 4, lactam 3 is predominantly formed with internal, alkyl alkynes. An initial substrate scope revealed that a large variety of internal alkynes are tolerated and electron-donating aryl groups afford better yields than their electron-withdrawing counterparts (Scheme 1).
 |
(4) |
Scheme 1.

Initial reaction scope
Mechanistic Proposa
The current mechanistic proposal that rationalizes the formation of lactam, vinylogous amide, and pyridone products is illustrated in Fig. 2 [11]. Initial coordination of the isocyanate and alkyne to rhodium occurs orthogonal to the square plane (I) and both lactam and vinylogous amide products can be formed from this coordination complex by oxidative cyclization, which is believed to be irreversible. For the lactam product (pathway 3), oxidative cyclization results in C-C bond formation generating IIa that undergoes a subsequent 1,2-migratory insertion to form IIIa in what is the enantio-determining step. Reductive elimination of IIIa yields lactam 3. Alternatively, oxidative cyclization through pathway 4 forms a C-N bond (IIb). In this case, the alkene cannot immediately insert due to a strained transition state geometry. A CO migration occurs via IIIb en route to rhodacycle IVb, which can then undergo alkene insertion (Vb). Reductive elimination of Vb furnishes vinylogous amide 4. This mechanism also provides an explanation for the formation of 2- and 4-pyridone side products.
Fig. 2.

Proposed mechanistic pathway
Enantioselective Cycloadditions
After demonstrating that alkenes participate in the three component coupling under our conditions, we sought to render the reaction asymmetric and include the more ubiquitous terminal alkynes. Under the initially developed catalytic conditions, yields with terminal alkynes are low due to a competing alkyne dimerization [12]. Chiral phosphoramidites were found to prevent dimerization and to be the most efficient ligands at promoting the enantioselective rhodium-catalyzed [2+2+2] cycloaddition. Commercially available MonoPhos L4 works in poor yields, while Taddol-based phosphoramidites (L1–L3) provide the best yields and enantioselectivities (Scheme 2). Manipulation of the amine portion of the phosphoramidite allows further optimization of enantioselectivity; ultimately, L2 was found to promote the reaction with the highest yields and enantio- and product selectivities [13].
 |
(5) |
Scheme 2.
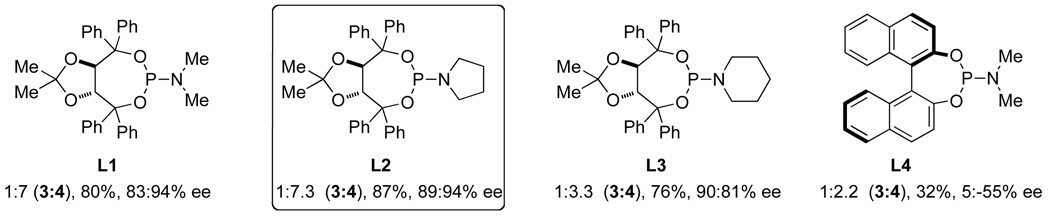
Phosphoramidite ligand screen
As the substrate scope was flushed out, we observed that the sterics and electronics of the alkyne had a marked influence on product selectivity. Sterically large terminal, aryl alkynes favor vinylogous amide 4. However, as aryl alkynes become more electron-deficient, selectivity erodes and lactam 3 is formed in increasing amounts. Alkyl alkynes, in contrast, provide lactam 3 ordinarily, but shift to vinylogous amide 4 as their steric bulk is increased.
 |
(6) |
We sought a more general strategy to control product selectivity that is dependent on the catalyst and not the nature of the alkyne. Such control would enable access to a variety of indolizidine natural products: cylindricine A, C-F and (−)-209D, derived from a vinylogous amide-selective reaction with alkyl alkynes (Fig. 1). With Taddol-based phosphoramidite ligands and alkyl alkynes, product selectivity favors lactam, and we sought to reverse this bias through modification of the ligand. Results with commercially available MonoPhos (L4) were poor (22%, 72% ee), but the product selectivity favors vinylogous amide (1:2.2, 3:4). This was a promising result, and we decided to explore modifiying the BINOL backbone to improve product selectivity. The 3,3’-disubstituted-trimethylsilyl BINOL (GuiPhos, L6) further improved product ratio (1:3.6, 3:4). Ultimately, a 3,3’-disubstituted tert-butylbiphenyl phosphoramidite (L7) was shown to give the highest ratio in favor of vinylogous amide (1:6.2, 3:4) and excellent enantioselectivity (91% ee) (Scheme 4) [14].
 |
(7) |
Scheme 4.

Product selectivity shift
The scope of terminal alkyl alkynes with L7 shows tolerance of a variety of functional groups and select examples are shown in Scheme 5. Sterically hindered alkynes and reactive functional groups such as alkyl chlorides, Weinreb amides, and tethered diynes proceed smoothly.
 |
(8) |
Scheme 5.

Scope with L7
Model for Product Selectivity
As part of our effort to control product selectivity, we sought a better understanding of how the rhodium-catalyzed [2+2+2] cycloaddition takes place with terminal alkynes. X-ray crystal analysis of Rh(cod)/phosphoramidite complexes (Fig. 3) in cooperation with experimental data allowed us to propose the following explanation for product selectivity [11].
Fig. 3.

X-ray crystal structures of Rh(cod)Cl/phosphoramidite
Examination of the Rh(cod)Cl/phosphoramidite complexes led us to hypothesize that the steric environment of the phosphoramidite controls the coordination of the isocyanate and alkyne to the rhodium center. Both Taddol and BINOL-based phosphoramidites hinder one face of the rhodium square plane and this causes the π components to coordinate with the larger substituents opposite the hindered rhodium face (Fig. 3). From this orientation both the lactam and vinylogous amide products can be accessed depending on the direction that the π components rotate during oxidative cyclization (Fig. 4, TSI and TSII). If the two small components tilt away from the rhodium center during cyclization, C-C bond formation occurs as shown in TSI to form IIa en route to lactam 3, whereas if they tilt towards the rhodium center, C-N bond formation takes place as in TSII to form IIb leading to vinylogous amide 4. The direction of cyclization and thus product selectivity of the reaction are governed by both steric and electronic factors.
Fig. 4.

Explanation of product selectivity
Lactam products are favored with small alkyl alkynes, which have little steric interaction with the ligand in TSI. Large alkyl and aromatic alkynes have a disfavored steric interaction with the ligand (TSI) en route to lactam and thus prefer to cyclize through TSII where steric interactions are minimized, favoring the vinylogous amide pathway. Taddol phosphoramidites lead to lactam products due to longer Rh-P bond lengths, whereas BINOL/Biaryl phosphoramidites have shorter Rh-P bonds that exacerbate the alkyne-ligand interactions in TSI and lead to increased amounts of vinylogous amide. According to Stockis and Hoffman’s calculations of metallacyclopentadienes [15], there is an electronic preference for the largest LUMO coefficient to be beta to the metal. Lactam formation, in which the isocyanate LUMO controls selectivity, is either reinforced or disfavored by the LUMO coefficient of the alkyne. Thus, electron-deficient aryl and small alkyl alkynes favor lactam formation while large or electron rich alkynes favor vinylogous amide.
Application to Total Synthesis
The ability to control and predict product selectivity with terminal alkynes in a highly enantioselective fashion allows for quick access to a variety of natural products. The rapid syntheses of (+)-lasubine II [12] and (−)-209D [14] were achieved with our rhodium-catalyzed [2+2+2] cycloaddition as the key step. The cycloadditions proceed with moderate to good yields and excellent enantioselectivities. For (+)-lasubine-II, hydrogenation of quinolizinone 6 reduces both the vinylogous amide and the carbonyl to provide quinolizidine 7. A Mitsunobu reaction completes the synthesis, yielding (+)-lasubine II in four steps and 18% overall yield (Scheme 6). For indolizidine (−)-209D, hydrogenation of cycloadduct 4 yields alcohol 8, which was reduced with a Barton-McCombie deoxygenation to furnish (−)-209D in 5 linear steps from commercially available starting material in 24% overall yield.
Scheme 6.

Synthesis of (+)-lasubine II and (−)-209D.
Alkene substitution
We sought to explore substitution on the tethered alkene as substituted olefins could be used to create tetrasubstituted stereocenters found in natural products, such as cylindricines A, C-F and FR901483. When a variety of 1,1-disubstituted olefins are used, the desired cycloadducts are afforded in good yield and selectivity [16]. The scope of the substituted olefins is shown below in Scheme 7 and both alkyl and aryl alkynes are tolerated in the reaction. Ligand L8 was found to give the best product and enantioselectivity for lactam 3 with alkyl alkynes. A number of different alkyl olefin substitutions are tolerated; however, sterically bulky substituents disfavor migratory insertion of the alkene resulting in lower yields and increased 2-pyridone formation. Cyclohexyl substitution yields 19% of the desired cycloadduct while methyl and butenyl substituted olefins give upwards of 75% yield. Nevertheless, the size of the substituent does not appear to diminish enantioselectivities, which remain greater than 87%.
 |
(9) |
Scheme 7.

Scope of 1,1-disubstituted alkenes
Internal, unsymmetrical alkynes
A variety of symmetrical, internal alkynes have been tested in this methodology, but we had not yet explored how unsymmetrical, internal alkynes would behave. Fortunately, unsymmetrical alkynes participate in a highly regioselective and predictable manner [17]. A number of alkynes were explored (Scheme 8) and we found that electron-withdrawing substituents end up alpha to the carbonyl and electron-donating groups are beta in the products. The vinylogous amide (4) was almost exclusively the major product with unsymmetrical, internal alkynes. Substitution on the olefin is well tolerated, even with sterically bulky groups. The 1,1-disubstituted alkenes also show a significant increase in enantioselectivity. We suggest that regioselectivity arises from stabilization of the forming partial positive charge (Fig. 4, TSII) by the more electron-donating group. Mayr’s scale of nucleophilicity [18] provides a convenient method for predicting the alkyne insertion.
 |
(10) |
Scheme 8.

Unsymmetrical alkyne incorporation
Carbodiimides
One of the challenges to improving the substrate scope of the reaction was the formation of lactam 3 using aryl alkynes. We addressed this issue through the use of a carbodiimide in place of the isocyanate [19]. Exchange of the isocyanate for the carbodiimide biases oxidative cyclization towards metallacycle IIa, allowing for selective lactam formation even with vinylogous amide-favoring aryl alkynes (Scheme 9). Electron-rich alkynes are able to partially override this preference. These amidine products can be modified by reduction of the imine to the amine or hydrolysis to give the lactam, illustrating their synthetic utility.
 |
(11) |
Scheme 9.

Carbodiimide scope
Additive effect on enantioselectivity
The advent of GuiPhos allowed us to examine asymmetric cycloadditions of diarylalkynes (tolanes). In an initial survey, we found that enantioselectivities varied as a function of alkyne electronics in a non-linear fashion. We hypothesized that in the presence of excess alkyne, the alkyne can coordinate to the octahedral rhodium(III) metallacycle IVb and alter the enantioselectivity of the alkene insertion. To test this hypothesis, we investigated a number of weakly coordinating additives that would not participate in the reaction to standardize enantioselectivities [20]. Methyl nicotinate was found to be the best additive and numerous examples showed a leveling of enantioselectivities as seen in Scheme 10. We believe the exogenous ligand binds to the octahedral rhodium(III) species and favors IVb2 over the other diastereomeric transition states for the olefin insertion. The composition of the alkyne no longer plays a role in the enantioselective step of the catalytic cycle due to the additive.
 |
(12) |
Scheme 10.

Methyl nicotinate additive effect
[4+2+2] Cycloaddition
As part of our effort to expand this methodology, we changed the alkene to a diene and found that a [4+2+2] azocene cycloadduct formed as the major product (Scheme 11) [21]. Yields vary depending on alkyne electronics. In general, conditions that favor lactam formation also favor the [4+2+2] reaction with dienes; alkyl alkynes proceed in good yields, while the aryl alkynes result in lower yields. The enantioselectivity is both excellent and invariant. Because lactam favoring conditions give the highest yields of the [4+2+2] product, we initially hypothesized that the first step is oxidative cyclization of the alkyne and isocyanate to form rhodacycle IIa. However, further mechanistic exploration suggests that the diene is involved in the first irreversible step, which we believe is formation of rhodacycle IIc. This would also be the enantio-determining step and explain the invariant ee’s (Scheme 11).
 |
(13) |
Scheme 11.

[4+2+2] cycloaddition and proposed mechanism
In conclusion, we have detailed our exploration of the rhodium-catalyzed, enantio-, and regioselective cycloaddition of alkenes, alkynes, isocyanates, and carbodiimides [22]. We have proposed a mechanism that accounts for regio- and product selectivity and this methodology has been expanded to include 1,1-disubstituted alkenes to generate tetrasubstituted carbinolamine stereocenters. We have explored the regioselective incorporation of internal, unsymmetrical alkynes and succeeded in predicting selectivity based on sterics and electronics. Through the use of non-participating, exogenous ligands, we have modified the enantio-determining step of alkene insertion using tolanes. Exchange of an isocyanate for a carbodiimide reverses product selectivity to favor lactam. The replacement of the tethered olefin with a diene generates azocenes. Finally, we have shown that [2+2+2] cycloadditions are an efficient way to assemble indolizidine and quinolizidine natural products.
Scheme 3.

Select examples of terminal alkynes
Acknowledgments
We thank our many coworkers without whose numerous intellectual and experimental contributions this project would not have been possible. Special thanks go to Robert Yu who started this entire project and provided many of its early key findings. Generous support for this work has been provided by NIGMS (GM80442). DMD thanks NSF-LSAMP Bridge to the Doctorate Program for support. KMO and DMD thank Oren Anderson and Susie Miller (CSU) for support and guidance. TR thanks the Monfort Family Foundation for a Monfort Professorship, and Amgen for support. We thank Johnson Matthey for a loan of rhodium salts.
References
- 1.(a) Robinson JE. Rhodium(I)-Catalyzed [4+2] and [4+2+2[ Carbocyclizations. In: Evans PA, editor. Modern Rhodium-Catalyzed Organic Reactions. Weinheim: Wiley-VCH; 2005. [Google Scholar]; (b) Wender PA, Gamber GG, Williams TJ. Rhodium(I)-Catalyzed [5+2], [6+2] and [5+2+1] Cycloadditions: New Reactions for Organic Synthesis. In: Evans PA, editor. Modern Rhodium-Catalyzed Organic Reactions. Weinheim: Wiley-VCH; 2005. [Google Scholar]
- 2. Strbing D, Beller M. Pauson-Khand Reactions. In: Beller M, Bolm C, editors. Transition Metals for Organic Synthesis. Weinheim: Wiley-VCH; 2004. Brummond KM, Kent JL. Tetrahedron. 2000;56:3263. Galan BR, Rovis T. Angew. Chem. Int. Ed. 2009;48:2830. doi: 10.1002/anie.200804651. and references therein.
- 3. Mobner C, Bolm C. Catalyzed Asymmetric Aziridinations. In: Beller M, Bolm C, editors. Transition Metals for Organic Synthesis. Weinheim: Wiley-VCH; 2004. Savizky RM, Austin DJ. Rhodium(II)-Catalyzed 1,3-Dipolar Cycloaddition Reactions. In: Evans PA, editor. Modern Rhodium-Catalyzed Organic Reactions. Weinheim: Wiley-VCH; 2005. Nakamura I, Yamamoto Y. Chem. Rev. 2004;104:2830. doi: 10.1021/cr020095i. Oberg KM, Lee EE, Rovis T. Tetrahedron. 2009;65:5056. doi: 10.1016/j.tet.2009.02.021. and references therein.
- 4.(a) Hong P, Yamazaki H. Synthesis. 1977;50 [Google Scholar]; (b) Hong P, Yamazaki H. Tetrahedron Lett. 1977;15:1333. [Google Scholar]
- 5.(a) Hoberg H, Oster BW. Synthesis. 1982:324. [Google Scholar]; (b) Hoberg H, Oster BW. J. Organomet. Chem. 1982;234:C35. [Google Scholar]; (c) Hoberg H, Oster BW. J. Organomet. Chem. 1983;252:359. [Google Scholar]
- 6.(a) Earl RA, Vollhardt KPC. J. Am. Chem. Soc. 1983;105:6991. [Google Scholar]; (b) Earl RA, Vollhardt KPC. J. Org. Chem. 1984;49:4786. [Google Scholar]
- 7. Yamamoto Y, Kinpara K, Saigoku T, Takagishi H, Okuda S, Nishiyama H, Itoh K. J. Am. Chem. Soc. 2005;124:5059. doi: 10.1021/ja045694g. and references therein.
- 8.Duong HA, Cross MJ, Louie J. J. Am. Chem. Soc. 2004;126:11438. doi: 10.1021/ja046477i. [DOI] [PubMed] [Google Scholar]
- 9.(a) Tanaka K, Takahashi Y, Suda T, Hirano M. Synlett. 2008:1724. [Google Scholar]; (b) Tanaka K, Wada A, Nogushi K. Org. Lett. 2005;7:4737. doi: 10.1021/ol052041b. [DOI] [PubMed] [Google Scholar]
- 10.Yu RT, Rovis T. J. Am. Chem. Soc. 2006;128:2782. doi: 10.1021/ja057803c. [DOI] [PubMed] [Google Scholar]
- 11.Dalton DM, Oberg KM, Yu RT, Lee EE, Perreault S, Oinen ME, Pease ML, Malik G, Rovis T. J. Am. Chem. Soc. 2009;131:15717. doi: 10.1021/ja905065j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu RT, Rovis T. J. Am. Chem. Soc. 2006;128:12370. doi: 10.1021/ja064868m. [DOI] [PubMed] [Google Scholar]
- 13.(a) Albano P, Aresta M. J. Organomet. Chem. 1980;190:243. [Google Scholar]; (b) Schafer H, Marcy R, Ruping T, Singer H. J. Organomet. Chem. 1982;240:17. [Google Scholar]; (c) Ohshita J, Furumori K, Matsuguchi A, Ishikawa M. J. Org. Chem. 1990;55:3277. [Google Scholar]
- 14.Yu RT, Lee EE, Malik G, Rovis T. Angew. Chem. Int. Ed. 2009;48:2379. doi: 10.1002/anie.200805455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stockis A, Hoffmann RJ. J. Am. Chem. Soc. 1980;102:2952. [Google Scholar]
- 16.Lee EE, Rovis T. Org. Lett. 2008;10:1231. doi: 10.1021/ol800086s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Keller Friedman R, Rovis T. J. Am. Chem. Soc. 2009;131:10775. doi: 10.1021/ja903899c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.(a) Mayr H, Ofial AR. J. Phys. Org. Chem. 2008;21:584. [Google Scholar]; (b) Mayr H, Kempf B, Ofial AR. Acc. Chem. Res. 2003;36:66. doi: 10.1021/ar020094c. [DOI] [PubMed] [Google Scholar]
- 19.Yu RT, Rovis T. J. Am. Chem. Soc. 2008;130:3262. doi: 10.1021/ja710065h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Oinen ME, Yu RT, Rovis T. Org. Lett. 2009;11:4943. doi: 10.1021/ol9020805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu RT, Keller Friedman R, Rovis T. J. Am. Chem. Soc. 2009;131:13250. doi: 10.1021/ja906641d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Perreault S, Rovis T. Chem. Soc. Rev. 2009;38:3149. doi: 10.1039/b816702h. [DOI] [PMC free article] [PubMed] [Google Scholar]