

Abstract
The chemosensory pathway of bacterial chemotaxis has become a paradigm for the two-component superfamily of receptor-regulated phosphorylation pathways. This simple pathway illustrates many of the fundamental principles and unanswered questions in the field of signaling biology. A molecular description of pathway function has progressed rapidly because it is accessible to diverse structural, biochemical, and genetic approaches. As a result, structures are emerging for most of the pathway elements, biochemical studies are elucidating the mechanisms of key signaling events, and genetic methods are revealing the intermolecular interactions that transmit information between components. Recent advances include (a) the first molecular picture of a conformational transmembrane signal in a cell surface receptor, (b) four new structures of kinase domains and adaptation enzymes, and (c) significant new insights into the mechanisms of receptor-mediated kinase regulation, receptor adaptation, and the phospho-activation of signaling proteins. Overall, the chemosensory pathway and the propulsion system it regulates provide an ideal system in which to probe molecular principles underlying complex cellular signaling and behavior.
Keywords: transmembrane signaling, kinase regulation, histidine kinase, aspartate kinase, protein methylation
INTRODUCTION
Bacterial cells, like their eukaryotic counterparts, possess elegant signaling pathways that monitor critical parameters of the external environment and internal physiology. The resulting information is used to regulate cellular function as the environment changes and the cell matures. In all prokaryotes examined to date, intracellular signaling is dominated by an ancient pathway motif consisting of receptor, histidine kinase, and aspartate kinase elements (Figure 1) (Bourret et al 1991, Parkinson & Kofoid 1992, Hoch & Silhavy 1995). In Escherichia coli alone, dozens of these His-Asp kinase or two-component pathways monitor different external and internal cues and regulate diverse cellular processes. 1 More generally, bacterial two-component pathways control a dazzling array of functions including cell division, virulence, antibiotic resistance, metabolite fixation and utilization, response to environmental stress, sporulation, and taxis. Highly homologous pathways have recently been discovered in eukaryotic organisms including Saccharomyces cerevisiae, Arabidopsis, Neurospora, and Dictyostelium, where receptor-regulated histidine kinases and aspartate kinases are used to regulate development, respond to osmotic and oxidative stress, and detect hormones (Swanson et al 1994, Alex et al 1996, Appleby et al 1996, Chang 1996, Schuster et al 1996, Wang et al 1996). Eukaryotic systems appear to use two-component signaling for highly specialized applications: The complete genome of S. cerevisiae, for example, contains just one phosphorelay of this type (Posas et al 1996). Due to the abundance and greater importance of two-component pathways in prokaryotes, the conserved elements of these pathways represent promising targets for a new generation of broad-spectrum antibiotics.
Figure 1.

Information flow through a two-component signaling pathway. Shown are the standard prokaryotic and eukaryotic signaling components including (a) the sensor module, typically a transmembrane receptor with two putative membrane-spanning helices; (b) a transmitter histidine kinase that is regulated by the receptor and catalyzes autophosphorylation on histidine; and (c) a receiver or response regulator whose active site catalyzes phosphotransfer from the transmitter, thereby yielding autophosphorylation on aspartate. The response regulator can catalyze its own dephosphorylation, but some pathways require a separate phosphatase to generate more rapid dephosphorylation, or to provide additional pathway regulation. Different pathways display highly specialized assemblages of the modular elements; e.g. the sensor, transmitter, and response regulator modules can be separate proteins or can be fused together in various combinations (see text for references).
Currently, the best-characterized two-component pathway is the chemosensory system of E. coli, Salmonella typhimurium, and related enteric bacteria (Hazelbauer & Adler 1971). This pathway enables bacterial cells to sense and swim up or down gradients of specific chemical attractants and repellents, respectively, which include certain amino acids, sugars, and metal ions. Chemosensing may also play a role in the entry of S. typhimurium into eukaryotic host cells during infection (Jones et al 1992). Owing to the ready accessibility of the pathway components to genetic, biochemical, and physical approaches, this chemosensory system has become one of the leading models in which to analyze complex cellular behavior in terms of the molecular mechanisms of individual receptors and signaling proteins. The present review introduces bacterial chemotaxis, then focuses on the two-component chemosensory pathway of the chemotaxis system. Other recent reviews of the chemosensory pathway include those by Stock & Surette (1996), Blair (1995), and Stock & Mowbray (1995). The fascinating rotary swimming motor and its flagellar propulsion system have been reviewed by Macnab (1996) and Blair (1995).
THE CHEMOSENSORY PATHWAY
Function: Regulation of Cellular Taxis
The swimming of bacterial cells in solution can be fully described as a series of simple switching events between two modes of cellular movement. Chemosensory signaling can, in turn, transform this simple swimming into complex cellular behavior (Berg 1993, Blair 1995). In the absence of a chemical gradient, a swimming bacterial cell executes a three-dimensional random walk consisting of runs of swimming in a straight line punctuated by tumbles during which the cell briefly stops and randomly reorients before swimming off in a new, arbitrary direction. When a gradient of an attractant or repellent is imposed, the chemosensory pathway monitors how the local concentration of the chemical species changes with time as the cell swims through different regions of the gradient. If the cell is swimming up an attractant gradient, the chemosensory pathway detects an increasing attractant concentration with time, and sends a signal to the propulsion motor, which decreases the probability of a tumble event, thereby lengthening the average run up the gradient. Alternatively, if the swimming cell monitors a decrease in the attractant concentration or an increase in the repellent concentration with time, the tumbling probability is increased so that runs in this direction become shorter. The net effect of this temporal sensing and regulation is to change the random walk into a biased walk, in which the cell tends to migrate up an attractant gradient or down a repellent gradient as it swims. Such simple chemotaxis can give rise to strikingly complex patterns of cellular distribution when bacteria migrate under controlled environmental conditions (Figure 2), providing a model system for pattern formation in eukaryotic development (Budrene & Berg 1995, Woodward et al 1995). In certain natural environments, the taxis of E. coli or S. typhimurium on the surface of a wetted substrate, rather than migration through solution, is also important (Harshey & Matsuyama 1994). Although such surface swimming has not yet been fully characterized, it likely shares some of the same switching behavior observed for swimming in solution.
Figure 2.

Self-organized swarm pattern generated by chemotaxing E. coli. Shown is the negative image of a bacterial culture in which growth was initiated at the center of a semisolid medium of defined composition. The pattern was formed over a period of three days as the cells swarmed outward from the center, migrating in response to self-generated gradients of chemical attractants. Dark areas indicate regions of high cell density (Budrene & Berg 1995).
Circuit Diagram of the Chemosensory Pathway
The present review focuses on the highly homologous chemosensory pathways of E. coli and S. typhimurium. These pathways share nearly the same set of components, differing only in their complement of soluble and transmembrane receptors that define the species-specific set of recognized attractants and repellents. Most pathway components can be swapped between species without loss of function. Table 1 lists the pathway components and their properties, Figure 3 summarizes their modular domain structures, and Figure 4 displays the circuit diagram of their interactions. Recent reviews have described the pathway organization (Blair 1995, Stock & Surette 1996). The phosphosignaling branch of the pathway begins with a set of at least four water-soluble binding proteins, located in the periplasmic compartment, which recognize different chemoattractants. Upon activation by ligand binding, the ligand-occupied form of a given binding protein docks to one of five transmembrane receptors, thereby initiating a signaling event. Alternatively, certain transmembrane receptors, in particular the aspartate, serine, and citrate receptors, can be regulated directly by the binding of a small-molecule ligand without the assistance of a soluble protein. Each receptor serves as the organizational framework for a stable, super-molecular receptor-kinase signaling complex, formed by associations with the cytoplasmic histidine kinase CheA and other pathway components. The docking of a binding protein or small-molecule attractant to the periplasmic sensory domain of a receptor generates an intramolecular conformational change that is transmitted across the bilayer to the bound cytoplasmic histidine kinase. The kinase, which is up-regulated by either the repellent-occupied or empty (apo) receptor, but is down-regulated by the attractant-occupied receptor, phosphorylates itself on a specific histidine sidechain. This same phosphoryl group is then transferred from the histidine to an aspartate in the active site of a response regulator protein, CheY or CheB, each of which is an autocatalytic aspartate kinase. Phospho-CheY dissociates from the signaling complex and diffuses to the rotary motor where it docks and increases the probability of the clockwise motor rotation, thereby favoring the formation of the tumbling swimming state. The steady state level of phospho-CheY thus serves as a diffusible tumble signal that controls the overall frequency of tumbling. This tumble signal is modulated by two opposing reactions: creation of phospho-CheY by the receptor-kinase complex, and destruction of phospho-CheY by hydrolysis of its acyl phosphate. CheZ speeds the latter hydrolysis reaction, acting as a phosphatase. Repellents stimulate the histidine kinase activity and speed the production of phospho-CheY, whereas attractants inhibit the histidine kinase and slow phospho-CheY formation, thereby raising or lowering the steady state tumble signal, respectively.
Table 1.
Chemotaxis protein and their properties
| Protien | Gene | Location | Monomer mass (kDa) | Oligomer number | Monomers/cell; total concentration | Affinity (Kd) for signaling complex μM | References | |
|---|---|---|---|---|---|---|---|---|
| (N) | (μM)a | |||||||
| Maltose-binding proteinb | malE | periplasm | 41 | 1 | 45,000 | 400 | 250 | 1, 2, 11 |
| Galactose/glucose-binding protein | mglB | periplasm | 33 | 1 | 20,000 | 200 | ND | 1, 2 |
| Ribose-binding protein | rbsB | periplasm | 30 | 1 | 40,000 | 400 | ND | 1, 2 |
| Dipeptide-binding proteinb | dpp | periplasm | 57 | 1 | ND | ND | ND | 2 |
| Ni2+-binding proteinb | nikA | periplasm | 57 | 1 | 20,000 | 200 | ND | 3 |
| Aspartate receptor | tar | membrane | 60 | 2 | 1,500 | 2 | ND | 6, 13, 16, 17 |
| Serine receptor | tsr | membrane | 59 | (2) | 3,000 | 4 | ND | 13, 16, 17 |
| Ribose/galactose receptor | trg | membrane | 59 | 2 | 150 | 0.2 | ND | 5, 16, 17 |
| Dipeptide receptorb | tap | membrane | 58 | (2) | 150 | 0.2 | ND | 2, 4, 16, 17 |
| Citrate receptorc | tcp | membrane | 59 | (2) | ND | ND | ND | 7 |
| CheA histidine kinase | cheA | cytoplasm | 71 | 2 | 4,000 | 5 | 3 | 12, 16, 17 |
| CheAS histidine kinase (short form) | cheAd | cytoplasm | 60 | 2 | 4,000 | 5 | ND | 14,16,17 |
| CheW coupling protein | cheW | cytoplasm | 18 | 1 | 4,000 | 5 | 2 | 12, 16, 17 |
| CheY response regulator | cheY | cytoplasm | 14 | 1 | 8,000 | 10 | 2.0 | 13, 16, 17 |
| CheR methyl transferase | cheR | cytoplasm | 33 | 1 | 850 | 1 | 2.1 | 2, 4, 8, 16, 17 |
| CheB methyl esterase/amidase | cheB | cytoplasm | 37 | 1 | 2,000 | 2 | 3.2 | 2, 4, 13, 16, 17 |
| CheZ phosphatase | cheZ | cytoplasm | 24 | ≥2 | 20,000 | 20 | — | 2, 4, 15, 16, 17 |
| FliM motor switch | fliM | motor | 37 | 1 | 1,500 | 2 | — | 9, 10 |
Assuming a volume of 1.7 ×10−16 L for the periplasm or 1.41 ×10−15 L for the cytoplasm (Goodsell 1991; Kuo & Koshland 1987).
Only present, or only used, in E. coli chemotaxis.
Only present in S. typhimurium.
CheAS, the short form of CheA, is generated by an alternate start site within the cheA gene (Smith & Parkinson 1980).
References: (1) Koman et al 1979; (2) Macnab 1987; (3) DePina et al 1995; (4) Stock et al 1991; (5) Hazelbauer & Adler 1971; (6) Milligan & Koshland 1988; (7) Yamamoto & Imae 1993; (8) Simms and Subbaramaiah 1991; (9) Kihara et al 1989; (10) Tang & Blair 1995; (11) Manson et al 1985; (12) Li et al 1995; (13) Gegner et al 1992; (14) Sanatinia et al 1995; (15) Blat & Eisenbach 1996b; (16) Bray et al 1993; (17) Bray & Bourret 1995
Figure 3.

Domain organization of chemosensory pathway components. Confirmed, proteolytically sensitive interdomain linkers that release stable isolated domains are indicated as horizontal bars; structural or functional subdomains are separated by vertical bars. The aspartate receptor is composed of a sensory ligand-binding and transmembrane-signaling domain, coupled to a cytoplasmic kinase regulation domain (TM, transmembrane; MH, methylation). The transmitter histidine kinase CheA is composed of four functionally distinct domains involved in phosphotransfer (P1), response regulatory docking (P2), dimerization, and histidine autophosphorylation and receptor coupling. CheY and CheB share homologous aspartate kinase receiver domains; CheB also possesses a separate methylesterase domain. Residues shown in bold indicate phosphorylation sites on CheA, CheY, and CheB. Ongoing studies are mapping the domain structures of CheZ and FliM (see text for references).
Figure 4.

The chemosensory two-component pathway of E. coli and S. typhimurium. Arrows indicate the action of one component on another. Attractants and repellents in the periplasm bind to specific transmembrane receptors or to soluble binding proteins that in turn bind to transmembrane receptors. The transmembrane receptors are coupled by a scaffolding protein (CheW) to a cytoplasmic histidine kinase (CheA), which in turn regulates two response regulators (CheB and CheY). Phosphorylation of CheB modulates the adaptation system in which CheR methylates specific regulatory glutamate side chains on the cytoplasmic surface of the receptor, whereas phospho-CheB hydrolyzes these modifications. The steady state level of receptor methylation provided by the opposing CheR and CheB reactions enables the pathway to adapt to background stimuli and also provides a simple chemical memory. Phosphorylation of CheY modulates the rotary flagellar motor as phospho-CheY docks to the motor switch apparatus, thereby controlling the direction of motor rotation and the swimming behavior of the cell. Although CheY can catalyze its own dephosphorylation, the rate of phospho-signal inactivation is enhanced by a phosphatase activity (CheZ) (see text for references).
The adaptation branch of the pathway enables the cell to adapt to a constant background stimulus so that it can chemotax even in a concentration gradient superimposed on a large constant level of attractant or repellent. Adaptation is controlled by a feedback loop that covalently modifies multiple glutamate side chains on the cytoplasmic surface of each receptor. The carboxylates of these glutamates are methyl esterified by the methyltransferase CheR, which binds tightly to the C terminus of the receptor and is thus part of the signaling complex. The resulting methyl esters, which increase the kinase activation signal of the receptor, are ultimately hydrolyzed by the methylesterase CheB. The relative rates of the methylation and demethylation reactions define the steady state level of receptor methylation, and feedback control of this adaptation system is generated by the receptor-kinase complex, which phosphorylates and thereby activates the methylesterase enzyme. As a result, the receptor controls its own methylation level, which changes when, for example, a sudden increase in the attractant concentration turns off the histidine kinase responsible for activating the methylesterase CheB. As the level of activated phospho-CheB drops due to spontaneous hydrolytic dephosphorylation, the constant rate of CheR methyltransferase activity gradually increases the average number of methyl esters per receptor. Eventually the rising level of methyl esters will stimulate histidine kinase activity, thereby counteracting the effect of bound attractant and resetting the receptor signal to its basal level. It should be noted that the methyltransferase and methylesterase enzymes act globally on all the chemotaxis receptors in the cell. Thus when fully adapted, the net methylation level of the receptor population exactly balances the signals generated by multiple attractants and repellents.
The receptor methylation level also provides a simple chemical memory used to ascertain whether the current direction of swimming is favorable or unfavorable. The methylation level is high if the attractant concentration was high in the recent past; conversely, the methylation level is low if the attractant concentration was low. Repellent memories are stored in the same way, but with reverse polarity. As the cell swims, it compares the current chemical environment, as monitored by the ligand occupancy of the receptor population, to the chemical environment of the recent past as remembered by the methylation level. If the environment has significantly improved or deteriorated, the histidine kinase activity of the receptor-kinase complex is inhibited or stimulated, respectively, thereby altering the probability of tumble events in the appropriate manner. For a review of these and other aspects of pathway function, see Blair (1995).
Spatial Distribution of the Circuit Components
The spatial distribution of the chemosensory components is nonrandom, which has important functional implications. The receptor-kinase complexes form large clusters and are often located at one end of the cell termed the nose, which is oriented either toward or against the direction of swimming (Maddock & Shapiro 1993, Parkinson & Blair 1993, Berg & Turner 1995). Receptor clustering is unaffected by the presence of ligand but is lost when CheW or CheA are absent, suggesting that the dimeric histidine kinase may cross-bridge some fraction of the receptor dimers, thereby maintaining the cluster (Maddock & Shapiro 1993). Each cell also possesses several motors and their associated flagella, which are more randomly distributed around the cell surface. Due to the spatial separation between the receptors and the motor, a tumble stimulus is followed by a ≈0.1 s time lag before the motor response is detected. This lag period represents the time required for phospho-CheY to be generated and diffused from a signaling complex to the motor (Khan et al 1995). One function of receptor clustering is to facilitate the global receptor methylation reaction that occurs, at least in part, via an inter-receptor mechanism (Wu et al 1996). Other functions are possible but unproven; in principle, receptor clustering could also serve to (a) facilitate still more complex interactions between pathway components, (b) increase the ligand sensing efficiency of the receptor population, or (c) prevent cross talk with other pathways (assuming that they, too, are localized).
Relationship to Other Two-Component Pathways
The organization of the chemosensory pathway is highly specialized for multi-stimulus chemotactic sensing. The histidine kinase CheA acts as the central processing unit of the chemosensory circuit, integrating the signals of five transmembrane sensor proteins and controlling two response regulators. In many other prokaryotic and eukaryotic two-component pathways only a single stimulus is sensed, so that only a single receptor is needed and is typically fused directly to the histidine kinase (Parkinson & Kofoid 1992, Swanson et al 1994). Despite such differences in modular organization, the mechanisms of corresponding two-component modules appear to be similar or identical. For example, the sensing and transmembrane signaling modules of different chemoreceptors can be swapped to generate functional chimeric receptors with the expected changes to ligand specificity (Krikos et al 1985). Module swapping has also been successful between chemoreceptors and the distantly related EnvZ osmosensor: The resulting chimeras possessing chemosensory domains confer chemical regulation onto the EnvZ histidine kinase and its signaling pathway in vivo (Rampersaud et al 1991, Baumgartner et al 1994). More generally, the predicted transmembrane topologies of most prokaryotic and eukaryotic receptor modules in two-component pathways are the same as those of the chemosensory receptors, and the active sites of CheA and CheY are highly homologous to those of other two-component histidine and aspartate kinase modules, respectively (Parkinson & Kofoid 1992, Swanson et al 1994, Stock & Surette 1996). Thus despite their specialization for different tasks, many prokaryotic and eukaryotic two-component pathways appear to share the same structural and mechanistic principles exhibited by the receptors and signaling proteins of the bacterial chemosensory pathway.
SOLUBLE RECEPTORS
For many chemoattractants, the signal transduction process begins with a set of four soluble binding proteins in the periplasmic compartment that act as primary receptors by engulfing their ligands, then docking to specific transmembrane receptors. These monomeric soluble receptors range in size from 30 to 51 kDa (Table 1) and include the maltose-, galactose/glucose-, ribose-, and dipeptide-binding proteins (Macnab 1987, Boos & Lucht 1996). Recently, a fifth binding protein has also been implicated in the chemosensing of Ni2+, a repellent (DePina et al 1995). Together, the sensory-binding proteins form a small subset of the large class of periplasmic binding proteins, most of which serve solely as transport pathway components and are not involved in chemosensing (Boos & Lucht 1996, Quiocho & Ledvina 1996). All binding proteins exhibit a conserved, two-domain architecture wherein each α/β domain consists of a β-sheet sandwiched between two layers of α-helices (Quiocho & Ledvina 1996). Ligand binds in the cleft between the two domains, where loops linking secondary structure elements provide the key recognition elements. The buried ligand recognition site provides intricate hydrogen-bonding networks that confer specificity, as well as hydrophobic contacts that help drive the binding equilibrium (Vyas et al 1988, Aqvist & Mowbray 1995, Quiocho & Ledvina 1996). Ligand-binding affinities, measured as dissociation constants (Kd), are typically on the order of 100 nM and the residence time of bound ligand approaches 1 s, enabling the diffusing protein to maintain its grasp on the activating ligand until it encounters and docks to its membrane receptor (Miller et al 1983).
Binding protein activation is triggered by ligand-induced closure of the interdomain cleft. 19F NMR studies have shown that the activation cleft of the galactose/glucose-binding protein can open at least transiently in solution, but closes when a sugar molecule binds (Luck & Falke 1991a). These results, together with crystallographic and small angle X-ray scattering analyses of binding proteins in their apo and ligand-occupied states (Sharff et al 1992, Shilton et al 1996), reveal that the cleft can open as much as 20 to 40° in the apo state, as illustrated by crystal structures of the maltose-binding protein in Figure 5. Cleft opening events are essential for ligand binding and release, whereas ligand-induced cleft closure appears to be required for successful docking to a transmembrane receptor. Consistent with this simple picture is the observation that covalent cleft closure by an interdomain disulfide generates a high intrinsic affinity for receptor docking (Zhang et al 1996). Cleft closure regulates a large receptor docking surface defined by genetic and structural studies of the maltose- and ribose-binding proteins. In each of these soluble receptors, the docking surface spans the interdomain cleft, such that residues on both domains are implicated in receptor recognition (Vyas et al 1991, Zhang et al 1992, Binnie et al 1992). Cleft closure brings together the distinct docking patches on the two protein domains, thereby generating a contiguous docking surface. In addition, 19F NMR studies of the galactose/glucose-binding protein reveal widespread conformational changes within both domains upon sugar binding to the inter-domain cleft, but not upon metal binding to a structural Ca2+-binding site (Luck & Falke 1991b,c, Danielson & Falke 1996). These findings raise the possibility that the global intradomain conformational rearrangments triggered specifically by the regulatory sugar ligand may contribute to the regulation of receptor docking surfaces, thereby augmenting the regulation provided by cleft closure.
Figure 5.

Ligand-induced cleft closure in a periplasmic binding protein. Shown are crystal structures of the maltose binding protein (MBP) in its sugar-occupied (upper panel) and apo (lower panel) states (Scharff et al 1992). The ligand-binding site lies in a deep cleft separating the two domains. Bound ligand stabilizes the closed conformation of the cleft, whereas the apo cleft can open by at least 35° and also exhibits an 8° hinge twist. The structural and dynamic differences between these two states regulates the docking of binding proteins to their specific transmembrane receptors (Careaga & Falke 1992). Dark α-carbon spheres denote the genetically defined receptor-docking surface (Zhang et al 1992).
Although ligand-induced structural changes are clearly important in binding protein activation, these proteins also illustrate the potential importance of induced dynamical changes. In principle, molecular on-off switches could be controlled by an entropic component arising from activation-induced changes to protein thermal dynamics. In the galactose/glucose-binding protein, disulfide trapping studies have revealed spontaneous, large amplitude thermal fluctuations of the protein backbone structure including 15 Å helix translations and 36° domain rotations. The frequencies of these fluctuations are approximately 10 s−1, which represents ten fluctuations during the average 1 s lifetime of bound ligand (Careaga & Falke 1992, Careaga et al 1995, Butler & Falke 1996). Notably, these fluctuations are greatly enhanced in the absence of ligand, indicating that they provide an entropic component to on-off switching. Specifically, in the apo binding protein, the enhanced thermal fluctuations will help prevent the accidental juxtaposition of receptor docking elements, thereby disfavoring unproductive membrane docking events (Careaga et al 1995). This type of entropic on-off switching could play an important role in the activation of many receptors and signaling proteins (Falke & Koshland 1987, Kim 1994, Seeley et al 1996), although studies to date have typically focused on the structural, rather than the dynamic, aspects of on-off switching.
THE RECEPTOR-KINASE SIGNALING COMPLEX
Molecular Organization
The supermolecular signaling complex is a widespread motif in signaling biology, and the bacterial chemosensory pathway is one of the best-described examples. Each of the membrane-spanning chemoreceptors is a stable homodimer of identical ≈60 kDa subunits (Milligan & Koshland 1988, Yeh et al 1993, Lee et al 1994, 1995, Chervitz et al 1995). The dimeric receptor, in turn, provides the structural framework for a multifunctional, receptor-kinase complex (Figure 6). The protein components of the complex are listed in Table 1, together with their estimated binding affinities for the receptor (Borkovich et al 1989, Ninfa et al 1991, Gegner et al 1992, Schuster et al 1993, Li et al 1995, Wu et al 1996). The receptor consists of a periplasmic sensory region, a transmembrane region, and a cytoplasmic region. The latter domain binds the cytoplasmic proteins CheW, CheA, and CheR, yielding a polypeptide stoichiometry of 2:2:2:2 within the signaling complex. The response regulators CheY and CheB also dock competitively to the complex where they await a phosphotransfer event. Thus at some point during pathway function, the signaling complex may contain nearly all of the pathway components, the only exceptions being the phosphatase CheZ and the motor components regulated by CheY. Assembly of the core receptor-CheW-CheA ternary complex occurs slowly, requiring up to an hour in vitro, suggesting that significant structural rearrangements of the isolated components are needed for complex formation (Borkovich & Simon 1991). However, once formed, the ternary complex remains intact for 20 minutes or more, and its stability is independent of receptor occupancy (Gegner et al 1992, Schuster et al 1993).
Figure 6.

A typical receptor-kinase signaling complex illustrated by the aspartate receptor. The transmembrane receptor provides the architectural framework of the super-molecular signaling complex (Borkovich et al 1989, Ninfa et al 1991, Gegner et al 1992, Schuster et al 1993, Wu et al 1996). Most of the chemosensory pathway components are associated with this complex, either stably or transiently. The kinetically stable core ternary complex is composed of the dimeric receptor (illustrated as a collection of helices), the coupling protein CheW, and the dimeric histidine kinase CheA. Other components are believed to be in rapid equilibrium between bound and soluble forms, including periplasmic binding proteins, the methyltransferase CheR, the methylesterase CheB, and the motor response regulator CheY.
The Receptor Sensory Domain
The transmembrane receptors possess a sensory domain that recognizes specific chemical signals and thereby initiates the events that alter the receptor signaling state. In most cases, the sensory domain is located in the periplasm where it recognizes a binding protein or small molecule ligand (Stock & Surette 1996). The five chemoreceptors of this type and their respective chemoattractants are (a) the aspartate receptor (aspartate, maltose-binding protein); (b) the serine receptor (serine); (c) the ribose and galactose receptor (ribose-binding protein, galactose/glucose-binding protein); (d) the dipeptide receptor (dipeptide-binding protein); and (e) the citrate receptor (citrate or citrate-binding protein). Recently, a sixth receptor involved in aerotaxis has been described (Bibikov et al 1997), which also appears to regulate the CheA/CheY two-component signaling pathway (Bespalov et al 1996). This FAD-containing receptor has a different domain organization that places its specialized sensory domain at its N terminus in the cytoplasm, which is a suitable location for sensing the redox potential of the electron transport chain or other components of the cell interior.
The periplasmic domain of the transmembrane aspartate receptor has been cloned as a water-soluble fragment and extensively characterized, providing a molecular view of ligand binding and negative cooperativity. This homodimeric fragment, consisting of two 19-kDa subunits, includes virtually the entire periplasmic region of the receptor and retains the native ligand-binding properties even though the transmembrane helices have been removed (Milligan & Koshland 1993, Danielson et al 1994). Crystallographic structures of the isolated domain reveal that each subunit is an antiparallel four-helix bundle in which the individual helices are labeled α1 through α4, yielding a roughly cylindrical dimeric domain approximately 20 Å in diameter and 70 Å in length (Figure 7) (Milburn et al 1991, Bowie et al 1995, Yeh et al 1996). Two symmetric aspartate-binding sites are located at the interface of the two subunits, near the extreme periplasmic end of the domain. Considerable evidence indicates that the other transmembrane chemoreceptors share the same overall architecture, although each periplasmic sensory domain is specialized for the binding of certain ligands (Jeffery & Koshland 1993, Lee et al 1994).
Figure 7.
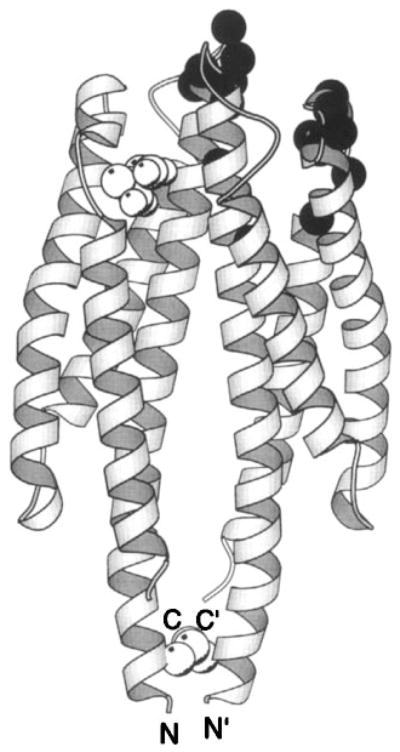
The periplasmic sensory domain of the transmembrane aspartate receptor. The crystal structure of this water-soluble, isolated domain reveals a homodimer of identical four-helix bundles (Milburn et al 1991, Yeh et al 1996). The engineered interdomain disulfide bond (CPK, open sphere, bottom) stabilizes native interactions present in the full-length receptor (Falke & Koshland 1987, Chervitz et al 1995), wherein the membrane-spanning helices would continue in a downward direction. Ligand binding occurs at the opposite, extreme periplasmic end of the domain. Shown is the single molecule of bound aspartate observed in the crystal structure (CPK, open sphere, upper), as well as the genetically defined docking surface for a single molecule of MBP comprised by residues on both receptor subunits (α-carbon, filled sphere; Gardina et al 1997).
The E. coli aspartate receptor is regulated by the maltose-binding protein (MBP) and aspartate, providing the unique opportunity to compare the molecular recognition of large-and small-molecule ligands. Genetic studies indicate that both domains of MBP dock to the extreme periplasmic end of the aspartate receptor, whereas an intersubunit complementation analysis suggests that the bound MBP molecule establishes simultaneous, asymmetric contacts on both receptor subunits (Zhang et al 1992, Gardina et al 1997). In particular, the receptor recognition elements appear to include loops at the ends of helices α1 and α2 in one subunit, and α4′ in the other subunit (prime denotes different subunits). These findings are consistent with a modeled structure in which MBP was computationally docked to the sensory domain fragment (Stoddard & Koshland 1992), yielding a ligand-receptor complex analogous to the crystallographically characterized complex between human growth hormone and its dimeric receptor (Wells 1994). The modeled MBP ligand blocks one of the two symmetric aspartate-binding sites but is proposed to leave the other intact, suggesting that the receptor can simultaneously bind only one MBP and one aspartate. Such a picture is consistent with the observed independent and additive effects of maltose and aspartate on receptor signaling (Mowbray & Koshland 1987, Stoddard & Koshland 1992).
The binding of aspartate to its receptor has been characterized in detail, revealing key thermodynamic, kinetic, and structural aspects of small-molecule recognition. Aspartate binding to the two symmetric sites exhibits strong negative cooperativity, as evidenced by direct binding measurements utilizing the membrane bound receptor, and by crystallographic and solution 19F NMR studies of the isolated sensory domain (Milburn et al 1991, Danielson et al 1994, Biemann & Koshland 1994, Yeh et al 1996). Owing to this negative cooperativity, the binding of the first aspartate molecule substantially reduces the affinity of the second aspartate-binding event. In the case of the S. typhimurium aspartate receptor, a second aspartate molecule may still bind weakly, whereas the E. coli receptor exhibits true half-of-sites binding in which the second aspartate is virtually excluded (Biemann & Koshland 1994). Similar half-of-sites binding has been observed for the E. coli serine chemoreceptor, and the binding proteins are also proposed to dock to the chemoreceptors in a half-of-sites fashion (Lin et al 1994, Stoddard & Koshland 1992, Gardina et al 1997). It follows that negative cooperativity is a general feature of attractant binding to the chemoreceptor family.
Solution 19F NMR has characterized the kinetics of ligand binding and release for the isolated sensory domain of the aspartate receptor. The first, high-affinity aspartate binds to the domain at a rate approaching the diffusion limit, indicating that aspartate binding and the conformational change it induces are extremely rapid (Danielson et al 1994). Thus the ligand-induced rearrangement likely involves a small-amplitude, low-energy structural transition.
The structural basis of aspartate recognition is defined by crystal structures for the apo and aspartate-occupied states of the S. typhimurium sensory domain (Yeh et al 1996). The ligand-occupied structures exhibit full aspartate occupancy of the first, high-affinity site, and at most partial occupancy of the second weaker site. The high-affinity aspartate binds to a site dominated by direct contacts to α4 (Tyr149, Gln152, Thr154) and α1 (Arg64) from one subunit, with supplemental contacts to α1′ (Arg69′, Arg73′) from the other subunit (Figure 8). Together these contacts provide an extensive array of protein-ligand and protein-solvent-ligand hydrogen bonds and salt bridges that are presumed to control ligand specificity by an undetermined mechanism.
Figure 8.

The aspartate-binding site of the transmembrane aspartate receptor. The dimeric receptor possesses two aspartate-binding sites that are symmetric in the apo dimer. The first molecule of aspartate binds with high affinity to one of these sites, which has been characterized crystallographically as shown (Milburn et al 1991, Yeh et al 1996). Highlighted are the protein residues and four water molecules that provide direct and indirect aspartate coordination, as well as the Ser68 residue implicated in negative cooperativity between the two sites (Kolodziej et al 1996). Owing to this negative cooperativity, the first aspartate binding event substantially weakens or completely prevents the second binding event (Biemann & Koshland 1994, Danielson et al 1994, Yeh et al 1996).
Recently, the Ser68 residue has been implicated as a critical component of the cooperative interactions between the two aspartate-binding pockets (Kolodziej et al 1996). The Ser68 side chain hydroxyl provides indirect coordination to the high-affinity aspartate via two bridging water molecules and also lies at the subunit interface in close proximity to Ser68′ of the other site, providing a plausible molecular pathway for intersite information transfer. Side chain substitutions at this position yield mutants possessing dramatically altered cooperativities, ranging from increased negative cooperativity to no cooperativity or even to positive cooperativity.
More generally, the aspartate-binding site represents a conserved small-molecule ligand-binding motif shared by several of the chemoreceptors. The serine-binding site likely possesses a very similar architecture (Jeffery & Koshland 1993, Yeh et al 1996, Wang et al 1997), whereas the citrate-binding site differs at one coordinating position (T154A) but retains the three conserved arginine residues utilized in aspartate and serine binding (Yamamoto & Imae 1993). For large-molecule ligands, the docking of MBP probably represents typical binding protein-receptor interactions (Vyas et al 1991, Gardina et al 1997). Both small- and large-molecule attractants break the symmetry of the receptor dimer via negative cooperativity and asymmetric binding to the two receptor subunits. It follows that receptor symmetry is likely to play an important role in the mechanism of attractant-triggered transmembrane signaling and kinase regulation. However, the mechanism of repellent binding and signaling remains uncharacterized. Another key unresolved issue is the mechanism of ligand specificity, both for small and large molecule ligands. Interestingly, the aspartate- and serine-binding sites are believed to use the same coordinating side chains to provide different ligand specificities, which are controlled by subtle features of the surrounding protein context (I Kawagishi, personal communication). Further comparative studies of the chemoreceptors will reveal the molecular principles used to adapt conserved binding motifs for the recognition of different small- and large-molecule ligands.
The Receptor-Mediated Transmembrane Signal
Considerable progress has been made in describing the molecular basis of transmembrane signaling by the chemoreceptors. Unlike the large class of receptors that signal by dimerization, the chemotaxis receptors are stable homodimers that signal via an intra-dimer conformational change. Conclusive evidence that a monomer-dimer equilibrium is not required for signaling is provided by engineered intersubunit disulfides, which yield covalent dimerization but have no effect on histidine kinase regulation in vitro, nor on the resulting chemotactic behavior in vivo (Chervitz et al 1995, Lee et al 1995).
STRUCTURE OF THE TRANSMEMBRANE SIGNALING DOMAIN
Extensive proteolysis of the aspartate receptor yields a distinct, proteolytically resistant structural domain (29 kDa per subunit) consisting of the periplasmic sensory domain and the membrane-spanning segments (Mowbray et al 1985), herein termed the transmembrane signaling domain. The well-characterized architecture of this domain includes four extremely long α-helices, each of which spans the entire length of the domain from the periplasmic ligand-binding site to the cytoplasm. The packing of these four massive helices has been defined both by the crystal structure of the periplasmic sensory domain (Milburn et al 1991, Yeh et al 1993) and by a combinatorial disulfide mapping analysis of the trans-bilayer region (Pakula & Simon 1992). The resulting packing model has been bolstered by the results of a random mutagenesis approach (Maruyama et al 1995) and by studies utilizing targeted disulfide bonds, especially those that have systematically scanned engineered disulfides down the entire length of the major inter- and intrasubunit helix contacts (Falke & Koshland 1987, Lee et al 1994, 1995, Chervitz & Falke 1995, Chervitz et al 1995). Together, these independent approaches have generated the medium-resolution structure of the sensory and transmembrane signaling domain shown in Figure 6, in which the first transbilayer helix (α1/TM1) spans ≈150 Å from its cytoplasmic N terminus to the extreme periplasmic end of the sensory domain. Throughout most of this distance, the helix packs against its symmetric partner from the other subunit (α1′/TM1′), thereby dominating the subunit interface in the periplasmic and transmembrane regions. The second transmembrane helix (α4/TM2) also extends at least 150 Å from the periplasmic sensory domain to the cytoplasmic domain, which binds and regulates the CheA histidine kinase.
IDENTIFICATION OF THE SIGNALING HELIX
Several independent methods have revealed that the transmembrane signal is carried by the second transmembrane helix (α4/TM2), which is displaced by ligand binding so that it moves relative to a static subunit interface. The initial phases of this ligand-induced signal have been probed using an isolated periplasmic fragment of the aspartate receptor (Milburn et al 1991) in which an engineered, intersubunit disulfide bond (Cys36-Cys36′) stabilizes the native subunit interactions of the full-length active receptor (Falke & Koshland 1987, Chervitz & Falke 1995). Solution 19F NMR studies of this periplasmic fragment have revealed asparate-induced chemical shift changes for a probe on helix α4/TM2, but no detectable effects for probes on helix α1/TM1 at the subunit interface (Danielson et al 1994, Danielson & Falke 1996). Similarly, the crystal structures of the apo and one-aspartate-occupied states of the periplasmic fragment (Milburn et al 1991) have been compared using the distance-difference method to identify ligand-induced distance changes (Chervitz & Falke 1996). This model-independent method shows that the binding of a single molecule of aspartate induces an asymmetric displacement of helices α3 and α4/TM2 within only one of the two subunits (Chervitz & Falke 1996). Because α3 does not span the bilayer, the crystallographic results implicate one of the two α4/TM2 helices as a lone transmembrane signaling helix. By contrast, although a subtle intersubunit displacement is observed (Yeh et al 1996), especially in the vicinity of the aspartate-binding pocket where cooperative interactions between the subunits occur (Chervitz & Falke 1996, Kolodziej et al 1996), the crystallographic data indicate that the most of the periplasmic subunit interface formed by the two α1/TM1 helices of the dimer is unperturbed by aspartate binding (Milburn et al 1991, Chervitz & Falke 1996).
Engineered disulfide studies of full-length receptors in the native receptor-kinase complex also indicate that the second transmembrane helix carries the signal across the bilayer (Chervitz et al 1995, Chervitz & Falke 1995). Disulfide bonds covalently linking the signaling helix (α4/TM2) of the aspartate receptor to the adjacent helix in the same subunit (α1/TM1) invariably inhibit transmembrane kinase regulation in vitro, confirming that transmembrane signaling requires movement of the second transmembrane helix relative to the static subunit interface (Chervitz & Falke 1995). Two of these disulfides covalently trap a state resembling the normal on state, yielding high levels of histidine kinase activity and reduced aspartate affinity. Two other disulfides trap an off state in which the kinase is locked in an inactive mode, while aspartate affinity is increased significantly. The observed effects on lock-on and -off disulfides on ligand affinity suggest that these disulfides trap states resembling the normal on and off states, which correspond to the apo and asparate-occupied states, respectively. More generally, the lock-on and lock-off disulfides demonstrate that it is possible to engineer reversible, chemical constraints that trap the signaling states of a transmembrane receptor (Chervitz & Falke 1995).
In contrast to helix-helix disulfides involving α4/TM2, engineered disulfide bonds that link the α1/TM1 and α1′/TM1′ helices at the subunit interface often have little or no effect on transmembrane signaling and kinase regulation. This observation further confirms the static nature of the subunit interface (Chervitz et al 1995). Even removal of the small cytoplasmic ends of the interfacial transmembrane helices fails to block signaling; these helices do not transmit information to the cytoplasmic domain (Chen & Koshland 1995).
Engineered disulfide studies of the homologous ribose/galactose receptor have shown that its second membrane-spanning helix (α4/TM2) also carries the transmembrane signal and that its subunit interface is static. As in the aspartate receptor, disulfides between α1/TM1 and α1′/TM1′ at the subunit interface retain the signal, whereas disulfides that constrain the α4/TM2 helix block receptor function (Lee et al 1995). These studies of the ribose/galactose receptor have used a novel approach to generate periplasmic disulfides in vivo, enabling direct analysis of their effect on receptor function in cellular chemotaxis assays (Lee et al 1995).
MOLECULAR VIEW OF THE TRANSMEMBRANE SIGNAL
Recent studies of the chemo receptors have provided a molecular picture of the ligand-induced displacement of the signaling helix, which represents the first structural description of a conformational transmembrane signal (Chervitz & Falke 1996, Hughson & Hazelbauer 1996). The distance difference analysis of the aspartate receptor revealed that one subunit of the sensory domain is essentially unaltered by ligand binding: This static subunit has the same backbone conformation in the apo and aspartate-occupied crystal structures of the periplasmic fragment (Milburn et al 1991, Chervitz & Falke 1996). The static subunit has been used to guide the superimposition of the two crystal structures, thereby providing a molecular view of the ligand-induced displacement of signaling helix α4/TM2 in the nonstatic subunit (Figure 9) (Chervitz & Falke 1996). By contrast, the other three transmembrane helices within the dimer (α1/TM1, α1′/TM1′, α4′/TM2′), including the bulk of the subunit interface formed by two of these helices (Chervitz & Falke 1996), are not detectably perturbed (Figure 9).
Figure 9.

The aspartate-induced displacement of the transmembrane signaling helix. Shown are the periplasmic regions of the four membrane-spanning helices, two provided by each subunit. When the apo and aspartate-occupied crystal structures (Milburn et al 1991) are superimposed using their static B subunits as a guide, aspartate is observed to displace only the α4/TM2 transmembrane helix in subunit A, termed the signaling helix (Chervitz & Falke 1996). This displacement consists of a vertical, 1.6 Å piston component directed down toward the cytoplasm as well as a subtle 5° helix tilt (difficult to visualize in this perspective). The kink or notch near the upper N-terminal end of the signaling helix is generated by conserved Pro153. This proline creates an indentation in the signaling helix complementary to the shape of the bound ligand, thereby controlling the vertical position the helix. Gray and black helices represent the apo and aspartate-occupied structures, respectively; cross-sectional shapes specify helices from subunits A (elliptical) and B (square), also denoted by primes.
The observed displacement of the signaling helix likely begins within the aspartate-binding site, where conserved Pro153 creates a striking indentation in the signaling helix that surrounds the bound ligand and acts as a positioning “notch.” When aspartate binds to the three conserved Arg residues at the subunit interface, the notch must be moved to accommodate the shape of the bound attractant, thereby generating a signaling helix displacement that can be deconvoluted into a “piston” component and a “tilting” component. The piston component is a 1.6 Å displacement of the signaling helix toward the cytoplasm (Figure 9) (Chervitz & Falke 1996). This piston displacement is not an artifact of the isolated periplasmic fragment or of crystal packing, because essentially the same displacement, indistinguishable in magnitude and direction, is revealed by the engineered lock-on and lock-off disulfides that trap receptor signaling states in the active receptor-kinase complex (see Figure 10) (Chervitz & Falke 1995, 1996). Thus both crystallographic studies of the isolated periplasmic sensory domain and engineered lock-on and lock-off disulfides in the native, receptor-kinase complex reveal the same aspartate-induced piston displacement of the signaling helix, which demonstrates that the aspartate-triggered piston displacement of the signaling helix toward the cytoplasm is a bona fide feature of the native transmembrane signal.
Figure 10.

Engineered cysteine pairs that yield lock-on and lock-off disulfide bonds in the full-length, aspartate receptor-kinase complex. Shown are the periplasmic ends of the four transmembrane helices in the dimer (Milburn et al 1991), two of which have been extended by modeling into the bilayer region (Chervitz & Falke 1996). A disulfide formed between cysteines Cys25/Cys197 or between Cys39/Cys183 locks the kinase on and decreases aspartate affinity. At the other extreme, a disulfide linkage between cysteines Cys176/Cys43 or between Cys179/Cys39 locks kinase activity off and increases the aspartate affinity (Chervitz & Falke 1996). These properties mirror those expected for the native on and off states of the receptor-kinase complex, respectively, in which aspartate binding causes kinase inactivation. Lock-on disulfides trap upward vertical displacements of the signaling helix; lock-off disulfides (analogous to aspartate binding) trap downward displacements toward the cytoplasm.
In the ribose/galactose receptor, disulfide formation rates between pairs of engineered cysteines on different helices have been used to analyze ligand-induced displacements of the transmembrane helices in vivo. Addition of ribose in the presence of the ribose-binding protein has a negligible effect on disulfide formation rates at the subunit interface, but disulfide formation rates between the α4/TM2 signaling helix and the adjacent transmembrane helix of the same subunit are reproducibly changed by receptor occupancy (Hughson & Hazelbauer 1996). The observed pattern of ligand-induced rate changes indicates that the docking of ribose-binding protein triggers a piston-type movement of the signaling helix toward the cytoplasm while maintaining a static subunit interface, exactly as observed for attractant signaling in the aspartate receptor. It follows that the piston mechanism is triggered by small- and large-molecule attractants and is a general feature of the chemosensory receptors (Chervitz & Falke 1996, Hughson & Hazelbauer 1996). Overall, the piston mechanism is analogous (though opposite in direction) to the piston model of transmembrane signaling originally hypothesized by Lynch & Koshland (1992).
Besides the piston displacement, the crystallographic analysis of the sensory domain also reveals an intriguing, ligand-induced 5° tilt of the signaling helix (Chervitz & Falke 1996). This 5° tilt could be an artifact of the periplasmic fragment or an important component of the transmembrane signal. Most likely, the piston component would transmit the signal across the bilayer more effectively than the bending component because the stiffness of the helix toward compression and stretching is considerably greater than its stiffness toward bending. On the other hand, if the helix is rigid and does not bend, the observed 5° tilt would displace the cytoplasmic end of the signaling helix by as much as 6 Å, thus yielding a larger diplacement than the piston component (Chervitz & Falke 1996). Although the significance of the piston component to signaling is established, it remains to be determined whether the tilting component is also required for function. Other types of signaling helix rotations, such as rotations about its long axis, appear to be ruled out because they are not observed by any of the three independent methods used to analyze the signaling helix displacement (Chervitz & Falke 1995, 1996, Hughson & Hazelbauer 1996).
Two features of the transmembrane signal warrant further discussion: the asymmetry of the signal and its small magnitude. The signal asymmetry arises directly from the preferential binding of aspartate to one of the two binding pockets. The observation that ligand binding displaces only one of the two signaling helices raises the possibility that the transmembrane signal is generated completely within one subunit (Chervitz & Falke 1996). Recent genetic studies have confirmed this prediction and have shown, just as observed in the crystallographic analysis (Figure 9), that the signal is transmitted largely or completely within the subunit in which the signaling helix contacts the bound aspartate. In fact, the cytoplasmic domain of the other subunit can be almost completely removed without blocking the signal (Tatsuno et al 1996, Gardina & Manson 1996). These findings provide additional evidence that the asymmetric nature of the piston displacement observed in the crystal structure is a real feature of the native receptor-kinase complex.
The small magnitude of the transmembrane signal is also striking. The 1.6 Å displacement of the signaling helix observed in the crystal structure lies within the range of helix displacements (>2 Å) that can maintain side chain contacts between adjacent helices (Chothia & Lesk 1985, Gerstein et al 1994). For example, when aspartate binds, the signaling helix retains the majority of its specific side chain contacts to adjacent helices including multiple inter-helix salt bridges, hydrogen bonds, and hydrophobic side chain contacts (Milburn et al 1991, Chervitz & Falke 1996). Moreover, side chains at the membrane-water interface can shift to accommodate a displacement of this magnitude without being dragged into a different solvent phase. In short, the small magnitude of the signaling helix displacement ensures that its energetic cost will be relatively minor and easily triggered by aspartate binding.
The Receptor Cytoplasmic Domain
GENERAL FEATURES
The transmembrane signaling helix (α4/TM2) ultimately enters the cytoplasm, where it is contiguous with the homodimeric C-terminal domain of the receptor. It is this cytoplasmic domain (31 kDa per subunit) that contains the covalent modification sites responsible for receptor adaptation, as well as the docking site that regulates the histidine kinase. Because each of the chemoreceptors binds the same CheW and CheA proteins, their cytoplasmic domains are highly conserved and exhibit pairwise sequence identities as high as 85% (LeMoual & Koshland 1996). Moreover, these domains belong to a superfamily of at least 56 prokaryotic cytoplasmic domains exhibiting pairwise sequence identities over 22% (LeMoual & Koshland 1996, Danielson 1997). Thus the cytoplasmic domain represents a conserved structural motif that is widespread in prokaryotic receptors. Much remains to be learned about the structure and function of the cytoplasmic domain, which holds the key to understanding the mechanisms of receptor adaptation and kinase regulation.
The dynamic and heterogeneous nature of the isolated cytoplasmic domain has hindered high-resolution structural studies, although it is clearly an assemblage of α-helices. In solution NMR experiments, the isolated, water-soluble domain exhibits chemical shift and hydrogen exchange parameters similar to those observed for molten globule proteins, indicating motional flexibility or extensive solvent exposure (Seeley et al 1996). Circular dichroism measurements indicate that the domain is predominantly α-helical (Mowbray et al 1985, Wu et al 1995), whereas its hydrodynamic parameters reveal a highly elongated shape (Long & Weis 1992), accounting for at least part of the extensive solvent exposure. Sequence alignments of homologous domains from over 56 prokaryotic receptors reveal five regions of high α-helix propensity (α5-α9) (Figure 11), separated by linkers identified by insertions, deletions, phase shifts, Gly, or Pro residues (LeMoual & Koshland 1996, Danielson 1997). Most regions of the putative helices exhibit a repeating heptad pattern (a-b-c-d-e-f-g) characteristic of coiled coils and four-helix bundles, in which the first and fourth residues (a, d) are typically hydrophobic (Paliakasis & Kokkinidis 1992, Kohn et al 1997). Polar hydrogen-bonding side chains, sometimes found at these a and d positions, help stabilize a specific register and oligomeric state of the helical aggregate, thereby minimizing structural heterogeneity (Lumb & Kim 1995, Gonzalez et al 1996a). Under certain conditions, the isolated domain can exhibit a complex equilibrium between its monomeric state and two oligomeric states, where the latter states are reminiscent of the dimers and trimers formed by mutant leucine zippers (Gonzalez et al 1996b). Thus the observation of oligomeric heterogeneity suggests that there are multiple ways the amphiphilic helices of the cytoplasmic domain can associate in pairs or bundles, some of which may represent nonnative structures. Current structural models propose that the ten putative helices of the dimeric cytoplasmic domain form three distinct functional regions: the linker, the methylation region, and the kinase-signaling domain (Stock et al 1991, LeMoual & Koshland 1996, Danielson 1997). This organization is summarized in a schematic working model of cytoplasmic domain architecture (Figure 6).
Figure 11.

Model for the cytoplasmic domain of the transmembrane receptors. A secondary-structure analysis of aligned sequences from over 56 related receptors suggests that each subunit of the homodimeric domain contains five amphiphilic helices (α5 to α9) and a short region of β-strand (β1) (LeMoual & Koshland 1996, Danielson 1997). Functionally, the domain is divided into the linker region, which provides the interface to the transmembrane signaling helix; the methylation region, which contains the sites of adaptive methylation (large black circles); and the signaling domain, which promotes CheW and CheA binding (see text for references). Also shown are the locations of lock-on and lock-off mutations in the serine receptor (white and black small circles, respectively), as well as second site suppressors of the inhibitory A19K mutation in the first transmembrane helix of the aspartate receptor (white small squares) (Ames et al 1988, Oosawa & Simon 1986). Both sets of mutations identify critical regulatory regions.
THE LINKER REGION
The small, ≈30 residue linker region couples the transmembrane signaling helix α2/TM2 to the cytoplasmic domain, thereby providing a critical interface for the communication of periplasmic information to the cytoplasm. The conservation of many linker residues throughout the superfamily of cytoplasmic domain structures (LeMoual & Koshland 1996, Danielson 1997) indicates that the linker has extensive conserved packing interactions. This region appears to be important for signaling because genetic studies have identified lock-on and -off mutations in the linker (Ames et al 1988). In addition, receptor dimers lacking one cytoplasmic domain are able to retain signaling function only when both subunits, including the truncated subunit, possess the linker region (Tatsuno et al 1996, Gardina & Manson 1996). In the native receptor-kinase signaling complex, it is plausible to propose that the linker region has a stable structure suitable for transmitting signals from the second transmembrane helix to the cytoplasmic domain, even though the isolated domain is dynamic.
The predictions of sequence analysis remain largely untested but have enabled the development of a working model for certain features of linker structure (Danielson 1997). Among the first ≈15 positions of the linker, the conserved hydrophobic residues lack a defined periodicity, but the second ≈15 positions exhibit the hydrophobic periodicity expected for an amphiphilic α-helix (LeMoual & Koshland 1996), termed putative helix α5 (Danielson 1997). The end of the linker is defined by a proteolytic hot spot (Arg259 in the aspartate receptor) followed closely by a phase shift in the putative α-helical periodicity (LeMoual & Koshland 1996, Danielson 1997), suggesting the presence of a bend or loop between putative helices α5 and α6 of the methylation region.
THE METHYLATION REGION
Following the linker region is the first of two methylation segments containing the regulatory methylation sites targeted by the adaptation pathway. The first methylation segment is especially critical to kinase regulation, as indicated by several lines of evidence: (a) This segment is directly coupled via the linker to the transmembrane signaling helix and thus lies on the path of information transfer from the periplasmic ligand binding site to the kinase. (b) This segment contains the majority of the consensus methylation sites (Terwilliger et al 1986, Stock & Surette 1996). (c) Genetic studies have revealed a number of lock-on and lock-off point mutations in this segment, as well as most of the second site revertants that restore signaling to an inactive receptor possessing a mutant lysine in its first transmembrane helix (Ames et al 1988, Oosawa & Simon 1986). Overall, it appears that the methylation region gathers, integrates, and interprets the multiple input signals sent by the transmembrane signaling domain and the methylated side chains. Concomitantly, an output signal is transmitted to the kinase regulation machinery.
The methylation sites are glutamate side chains, in some cases generated by the CheB-catalyzed deamidation of glutamine. The most rapidly methylated sites conform to the consensus recognition sequence Glu-UGlu-X-X-Ala-Ser/Thr, where the methylated residue is underlined (Terwilliger et al 1986). The methylation sites and the recognition side chains within both methylation segments exhibit the pattern expected for their display on the same face of an α-helix, suggesting that both methylation segments are helical (Terwilliger & Koshland 1984, Stock & Surette 1996). Moreover, both segments lie within regions of the primary structure predicted to be α-helical by sequence analysis (α6 and α9 in Figure 11) (Stock et al 1991, LeMoual & Koshland 1996, Danielson 1997). The first methylation segment on putative helix α6 contains three methylation sites, two of which are typically consensus sites, whereas the second methylation segment on putative helix α9 contains either one or two nonconsensus methylation sites depending on the type of chemoreceptor (Kehry & Dahlquist 1982, Kehry et al 1983, Terwilliger & Koshland 1984, Nowlin et al 1987, Rice & Dahlquist 1991). Both putative helices are strongly amphiphilic, exhibiting a hydrophobic face and a separate charged face containing the methylation sites and recognition side chains. Recent cysteine and disulfide scanning studies have confirmed that putative helix α6 is indeed helical and have mapped its buried hydrophobic face and its solvent-exposed methylation face (Danielson et al 1997). It is clear that the hydrophobic face of helix α6 packs against the corresponding face of its symmetric helix α6′ in the other subunit because certain engineered disulfide bonds cross-linking this interface retain full kinase regulation (Danielson et al 1997). A working model for the packing arrangement postulates that the first methylation helices (α6, α6′) form a parallel, coiled-coil helix pair at the subunit interface, whereas the remaining two symmetric methylation helices (α9, α9′) each dock in an antiparallel orientation relative to the central pair, yielding an antiparallel four-helix bundle (see Figure 6) (Stock et al 1991, Danielson et al 1997). Many features of this putative packing arrangement remain to be tested.
A simple model for the regulation of the methylation region proposes that the subunit interface of this module serves as a variable dimmer switch controlling kinase activity. The maximum kinase activity is provided by an optimal configuration of the interface, while kinase activity is down-regulated when the interface is perturbed by a displacement of the signaling helix or by an altered methylation state. The methylation sites bestow a broad, relatively continuous tuning range onto the regulatory machinery because both the number and location of methyl groups can be varied; in the extreme, the fully demethylated, anionic carboxylates down-regulate histidine kinase activity as much as 50-fold relative to the fully methylated receptor by an unknown mechanism (Ninfa et al 1991, Borkovich et al 1992). A key interaction defining the signaling state of the methylation region is the extensive contact between the α6 and α6′ helices, which form the subunit interface and possess the majority of the methylation sites. This interface can be driven toward its tightly associated, symmetric kinase-activating state by (a) certain intersubunit disulfide bonds that covalently cross-link the α6-α6′ interface (Danielson et al 1997); (b) a leucine zipper dimerization motif fused to the N-terminal end of the α6 helix in the isolated cytoplasmic domain (Cochran & Kim 1996, Surette & Stock 1996); or (c) high-level expression of a fragment containing just the α6 helix and the signaling module, which is believed to yield dimers stabilized by the α6-α6′ interaction (Surette & Stock 1996).
One discrepancy is that this simple model for interfacial regulation of the methylation module requires interactions between two subunits of the cytoplasmic domain. In contrast, genetic studies have shown that receptor dimers lacking one cytoplasmic subunit are functional (Tatsuno et al 1996, Gardina & Manson 1996). This apparent contradiction has been rationalized by proposing that when necessary, the cytoplasmic domain subunits of different receptor dimers can interact to bind and regulate the histidine kinase. Such interactions are not unreasonable, since a growing body of evidence indicates that receptor dimers are clustered at one end of the cell and, at least in certain cases, receptor methylation is a result of dimer-dimer interactions (Maddock & Shapiro 1993, Wu et al 1996). Clearly, many structural and mechanistic features of the methylation module remain to be elucidated, including the molecular mechanism of methylation-based receptor adaptation.
THE METHYLTRANSFERASE CheR
The CheR methyltransferase, a 33-kDa soluble enzyme, covalently modifies the receptor cytoplasmic domain. A pool of free CheR exists in the cytoplasm (Clarke et al 1980) where the aspartate, serine, and citrate receptors display a specific CheR-docking motif (Wu et al 1996). Thus in certain receptors the methyltransferase is an integral component of the receptor-kinase signaling complex. The motif used to bind CheR consists of the five-residue sequence Asn-Trp-Glu-Thr/Ser-Phe-C, which is located at the extreme C-terminal end of the receptor cytoplasmic domain (Wu et al 1996). The isolated five-residue peptide displays the same CheR affinity as the full-length receptor (Kd = 2 μM), indicating that most of the binding energy is provided by this simple docking site. Because the ribose/galactose and dipeptide receptors lack the consensus docking sequence and are methylated poorly in the absence of the aspartate or serine receptors, it has been proposed that methyltransferase bound to one dimer can methylate other dimers (Wu et al 1996; I Kawagishi, personal communication). Intra-dimer methylation may also occur because the receptor-methyltransferase complex can be solubilized from the membrane with retention of considerable methylation activity (Bogonez & Koshland 1985). The receptors that bind CheR possess an unconserved sequence of variable length lying just N-terminal to the CheR recognition motif. This unconserved stretch may act as a flexible tether, enabling the bound CheR to collide with the methylation sites on the same dimer or on nearby dimers.
A recent crystal structure of the CheR methyltransferase significantly extends the molecular description of the adaptation machinery (Djordjevic & Stock 1997). The CheR enzyme, which utilizes S-adenosyl-methionine as the methyl donor, consists of two domains (Figure 12). The smaller domain contains four helices arranged in a perpendicular fashion, whereas the larger domain exhibits an α/β folding motif in which a central seven-stranded β-sheet is sandwiched between two layers of α-helices (Djordjevic & Stock 1997). The active site cleft lies between the two domains where, in the crystal structure, it is occupied by the product inhibitor S-adenosyl-homocysteine. The smaller domain, by analogy with other methyltransferases of known structure, is proposed to dock to the substrate, which in this case is a methylation helix. Support for this hypothesis is provided by the discovery of a positively charged surface on the small domain suitable for docking to the anionic face of a methylation helix (Djordjevic & Stock 1997). It is not yet known where the C-terminal tail of the receptors binds to the enzyme, but this issue may soon be resolved by new crystals of the co-complex between the methyltransferase and the five-residue receptor peptide (S Djordjevic & AM Stock, personal communication).
Figure 12.
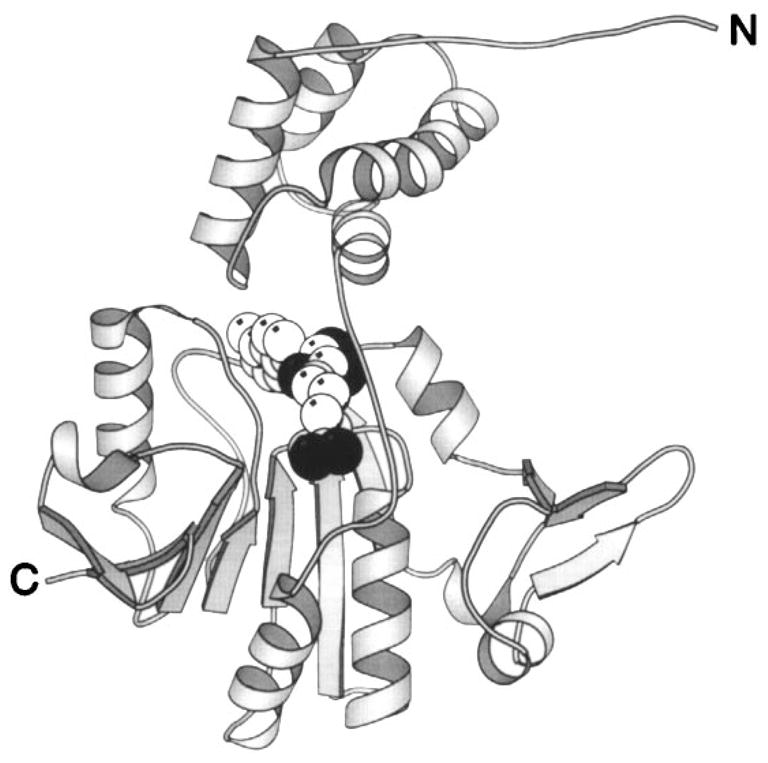
Structure of the CheR methyltransferase enzyme. The CheR protein uses S-adenosyl-methionine as a substrate for methyl transfer to the adaptation sites of transmembrane chemosensory receptors. This crystal structure reveals two distinct domains connected by a long, single-strand hinge (Djordjevic & Stock 1997). The N-terminal domain is an assembly of perpendicular helices; the C-terminal domain exhibits the α/β folding motif. The bound S-adenosyl-homocysteine molecule (CPK, sphere), a product of the methylation reaction, identifies the location of the active site region between the two domains. (Black spheres indicate oxygen atoms.)
THE SIGNALING DOMAIN
The final region of the cytoplasmic domain forms the ternary complex with CheW and CheA and is responsible for regulating the bound histidine kinase activity. This region, corresponding to a 16-kDa stretch of primary structure between the two methylation helices, is proposed to contain two helices per subunit (α7, α8 in Figure 11) and is the most highly conserved region of the large superfamily of prokaryotic cytoplasmic domains (LeMoual & Koshland 1996, Danielson 1997). The importance of the signaling region to kinase regulation is emphasized by the observation that several randomly generated lock-on mutations have been found here; in fact, the prevalence of such mutations is higher in this domain than in other regions of the receptor (Ames et al 1988). In the serine receptor, the signaling region is an independent folding domain because liberation and expression of the isolated domain in cells containing CheW and CheA lead to substantial histidine kinase regulation in vivo and in vitro (Ames & Parkinson 1994, Ames et al 1996). The corresponding fragment of the aspartate receptor exhibits greater kinase modulation activity when it is forced to dimerize by the inclusion of the first methylation helix α6 or a leucine zipper dimerization motif (Surette & Stock 1996). Together these findings indicate that the signaling module is an independent functional unit containing the minimal receptor sequences required for ternary complex formation and kinase activation (Ames et al 1996) and that dimerization of the module enhances kinase activation (Surette & Stock 1996). In vitro binding and activity measurements have confirmed that, as predicted by earlier genetic studies, the isolated signaling domain directly associates with CheW (Liu & Parkinson 1991, Ames & Parkinson 1994, Surette & Stock 1996).
Structurally, the signaling module remains enigmatic, although a simple arrangement of the four putative helices (α7, α8, α7′, α8′) would result in an antiparallel four-helix bundle (see Figure 6). Such an arrangement requires a hairpin turn between the two helices in the same subunit (α7, α8), and sequence analysis reveals a cluster of Gly residues at the appropriate location, as well as a short putative β-strand that could stabilize the subunit interaction by forming an intersubunit β-sheet (LeMoual & Koshland 1996). These putative structural features remain to be tested.
THE COUPLING PROTEIN CheW
The 18-kDa CheW protein possesses no known regulatory or catalytic function. Instead, it is a simple scaffold protein that couples CheA to the signaling module of the receptor and is strictly required for receptor-mediated activation of histidine kinase activity (Borkovich et al 1989, Ninfa et al 1991, Ames & Parkinson 1994). Although E. coli CheW is prone to aggregation and has been inaccessible to structural methods, a related CheW isolated from the hyperthermophile Thermatoga maritima is more well-behaved and its NMR solution structure is nearly complete (Swanson et al 1996; FW Dahlquist, personal communication). The preliminary results define the secondary structure, which is dominated by β-sheet. A class of CheW mutants previously shown to perturb the receptor interaction should help define the specific face of CheW involved in receptor docking (Liu & Parkinson 1991); the residues involved in CheA docking have not yet been identified. Due to its dual receptor and kinase docking sites, sufficiently high concentrations of free CheW can actually dissociate the ternary complex by driving the formation of separate CheW-receptor and CheW-CheA complexes (Liu & Parkinson 1989, 1991).
The Histidine Kinase CheA
Ultimately, the signaling output of the receptor is used to regulate the activity of the bound histidine kinase CheA. The isolated kinase is a dimer of identical 71-kDa subunits containing two symmetric active sites, each of which utilizes Mg2+-ATP to drive phosphorylation of His48 (Hess et al 1987, Wylie et al 1988, Swanson et al 1993a; review by Surette & Stock 1996). When bound to a receptor, the histidine kinase activity is modulated by the receptor occupancy and methylation state over at least a 102-fold range (Borkovich et al 1989, 1992, Ninfa et al 1991). The isolated CheA dimer exhibits a low rate of intrinsic autophosphorylation activity and dissociates to become completely inactive at low concentrations, suggesting that phosphorylation proceeds only in trans between subunits, or that the active site requires a native subunit interface (Surette et al 1996). The architecture of CheA is highly modular: each subunit can be divided into four functional regions (Figure 3) (Parkinson & Kofoid 1992, Stock & Surette 1996). At least three of these regions have been shown to be distinct folding domains.
THE P1 PHOSPHOTRANSFER DOMAIN
The N-terminal P1 domain, which folds independently in its isolated form, possesses the reactive His48 residue that serves as the site of autophosphorylation (Hess et al 1988, Swanson et al 1993b, Morrison & Parkinson 1994). The isolated domain has no intrinsic enzymatic activity, but it can be phosphorylated by a separate CheA molecule, and following purification, the resulting phospho-P1 domain is fully functional as a phosphotransfer substrate for CheY or CheB even in the absence of the other CheA domains (Swanson et al 1993b). Thus the P1 domain possesses the minimal recognition elements required for roles as a phospho-acceptor substrate of the CheA active site and as a phospho-donor substrate for the CheY and CheB active sites (Swanson et al 1993b; note that a different domain of CheA provides the high-affinity docking site for CheY and CheB, as discussed below).
The solution NMR structure of the isolated P1 domain displayed in Figure 13 A reveals a monomeric, 18-kDa antiparallel bundle of five helices (Zhou et al 1995, Zhou & Dahlquist 1997). The fifth, C-terminal helix is more weakly associated and can be proteolytically removed with retention of the domain activity (Morrison & Parkinson 1994, Zhou et al 1995). The His48 residue is located on helix α2, where the imidazole ring is exposed to solvent (Zhou et al 1995, Zhou & Dahlquist 1997). NMR pH titrations have shown that the environment of the unphosphorylated imidazole ring stabilizes the normally unfavored tautomer, in which hydrogen is bonded to the Nδ1 nitrogen (Zhou & Dahlquist 1997). Significantly, this tautomeric state leaves the Nε2 nitrogen of the ring available for phosphorylation. NMR analysis has confirmed that this nitrogen is indeed the phosphorylation site and that the phosphorylation-induced structural change is highly localized to the vicinity of the phosphorylated residue (Zhou & Dahlquist 1997). Together, these findings suggest that the P1 domain is a relatively inert structural unit that simply accepts the phosphoryl moiety from the catalytic module and allows it to be transferred to a response regulator. Three highly conserved residues in the vicinity of the reactive histidine (Gly52, His67, Glu70) play an important functional role (Stock & Surette 1996), perhaps stabilizing the appropriate imidazole tautomer and optimizing its orientation for the phosphorylation and phosphotransfer reactions (Zhou & Dahlquist 1997).
Figure 13.

Two domains of the CheA histidine kinase. (A) The N-terminal phosphotransfer domain, termed P1, provides the phospho-histidine used as a substrate during phosphotransfer from CheA to response regulators. The NMR solution structure of the phosphotransfer domain reveals a bundle of five helices (Zhou et al 1995, Zhou & Dahlquist 1997). The site of phosphorylation is the Nε2 nitrogen atom (black) of the His48 imidazole ring (CPK, sphere), located on the surface of helix α2. Phosphorylation yields a local structural change limited to the immediate environment of the phospho-histidine. (B) The solution structure of the response regulator docking domain, designated P2, displays an open-faced β-sandwich folding motif (McEvoy et al 1995, 1996). The residues implicated in CheY binding (α-carbon, black sphere) are clustered to a distinct docking surface.
The phosphoramidate linkage of phospho-His is the most unstable of the known phospho-amino acids (His, Asp, Tyr, Ser, Thr); in fact, the standard free energy of phosphotransfer from ATP to histidine is positive (Stock & Surette 1996). Thus it is not surprising that the CheA autophosphorylation reaction is reversible, such that phospho-CheA can phosphorylate ADP in vitro (Tawa & Stewart 1994, Surette et al 1996). As a result, CheA efficiently catalyzes the exchange of phosphoryl groups between ATP and ADP. Under cellular conditions, the high ratio of ATP to ADP and rapid phosphotransfer to response regulators help speed CheA autophosphorylation relative to the backward reaction, thereby making CheA an effective kinase.
THE P2 RESPONSE-REGULATOR DOCKING DOMAIN
The P2 domain, which provides the docking site for CheY and CheB, lies C-terminal to the P1 domain and can also be liberated as a stable, well-folded protein fragment (Parkinson & Kofoid 1992, Swanson et al 1993b, Morrison & Parkinson 1994). The solution NMR structure of the 14-kDa monomeric domain is an open-faced β-sandwich in which two antiparallel helices lie on one surface of an antiparallel, four-stranded β-sheet (Figure 13B) (McEvoy et al 1995, 1996). All the residues implicated in response regulator docking lie near one end of the helices and their contiguous loops. Many of these solvent-exposed docking residues are hydrophobic, and together they define a clear docking surface that binds a single CheY molecule. Although the P1 domain must also interact transiently with the bound response regulator during the phosphotransfer event, removal of P1 from CheA does not affect the affinity of CheY binding (Swanson et al 1993b). Similarly, removal of the catalytic domain has little effect on CheY binding to the P1–P2 region (Li et al 1995). It follows that the primary CheY binding elements reside entirely on P2. The CheB protein competes with CheY for binding to the P1–P2 region and presumably uses the same docking site on the P2 domain (Li et al 1995).
Remarkably flexible linkers connect the P2 domain to the rest of the CheA molecule. These linkers are sufficiently dynamic to be proteolytic hot spots (Parkinson & Kofoid 1992, Morrison & Parkinson 1994), allowing the P2 domain to tumble rapidly relative to the rest of the protein. As a result, the NMR spectrum of intact CheA clearly reveals the resonances of the P2 domain even though other regions of the molecule tumble too slowly to be detected (McEvoy et al 1997). Many histidine kinases encode specificity within other domains and lack the P2 domain entirely, suggesting that CheA and its homologues have evolved this domain for a specific purpose other than phosphotransfer specificity (Parkinson & Kofoid 1992). The remarkable mobility of the P2 domain and its bound response regulator could serve to facilitate inter-dimer phosphotransfer reactions between adjacent signaling complexes, thereby amplifying the phosphorylation signal.
THE CATALYTIC DOMAIN
The most highly conserved region of CheA is its catalytic domain (Figure 3): Homologous domains are found in all prokaryotic and eukaryotic two-component signaling pathways (Parkinson & Kofoid 1992, Stock & Surette 1996). No structure yet exists for any of these catalytic modules. The CheA catalytic domain folds independently, and the isolated domain can efficiently phosphorylate the isolated P1 domain (Swanson et al 1993b). It believed that the catalytic domain contains all of the motifs needed to bind Mg2+-ATP, recognize the P1 domain, and catalyze its phosphorylation. Several highly conserved clusters of residues are proposed to form the ATP-binding site and the active site, although the catalytic residues have not yet been directly identified (Stock & Surette 1996). Isolated CheA exhibits a slow autophosphorylation rate estimated at ≈10 turnovers min−1 (Tawa & Stewart 1994, Surette et al 1996), while CheA bound to attractant-saturated receptors exhibits an even lower activity. The apo state of the receptor-kinase complex stimulates the autophosphorylation rate over 102-fold relative to the isolated dimer, but the mechanism of this activation remains unknown (Borkovich & Simon 1990, Ninfa et al 1991).
One possible mechanism of receptor-mediated regulation is suggested by the dimeric structure of CheA. The enzyme is active only when it is a dimer; when diluted, it dissociates to form inactive monomers (Surette et al 1996). The dimerization motif lies primarily within the catalytic domain, since the isolated domain retains its functional, dimeric structure (Swanson et al 1993b, Surette et al 1996). Thus receptor-mediated signals could serve to modulate the CheA subunit interface, thereby controlling kinase activity either by directly altering the kinase active site or by changing its interaction with the substrate His48 on the P1 domain of the other subunit.
THE RECEPTOR DOCKING REGION
The C-terminal region of CheA is essential for receptor-mediated regulation, although the structure of the module and its precise role in receptor-kinase coupling have not yet been determined. Truncation of this module from CheA does not inhibit the intrinsic kinase activity, but does eliminate all receptor-mediated stimulation of kinase activity, both in vitro and in vivo (Bourret et al 1993a). The observed loss of receptor stimulation could stem from a failure to dock to CheW or to the receptor; alternatively, the truncation could block signal transmission from the receptor to the bound kinase. Genetic studies suggest that distinct CheW- and receptor-binding sites exist within this region of CheA (Parkinson & Kofoid 1992).
CheAS—A SHORT FORM OF THE HISTIDINE KINASE
Chemotaxing bacteria express CheA both as the full-length molecule and a short form termed CheAS, the latter generated by an alternative translational start site (Figure 3) (Kofoid & Parkinson 1991). Both forms share the same C terminus, but CheAS lacks the P1 domain and cannot be phosphorylated. Although CheAS is not strictly required for chemotaxis (Sanatinia et al 1995), a 1:1 mole ratio of CheA to CheAS provides optimal cellular motility (Wang & Matsumura 1997). A recent explanation for this finding came from the observation that the P1 domain serves two opposing functions. In addition to its role as the histidine kinase substrate, a given P1 domain also inhibits the kinase activity of its own subunit, perhaps by simple steric hindrance (Levit et al 1996, Garzon & Parkinson 1996). As a result, the isolated catalytic domain phosphorylates the free P1 domain more rapidly than does CheA itself. Similarly, a mixed dimer containing the catalytic domain and full-length CheA exhibits an approximately fivefold higher autophosphorylation rate than the native CheA dimer (Levit et al 1996). The latter mixed dimer is analogous to the heterodimers formed between CheA and CheAS under cellular conditions, which explains, in part, why cells express the short form. A second, unrelated function of the CheAS molecule as a component of the CheZ phosphatase complex is discussed below.
RESPONSE REGULATORS AND PHOSPHATASE
The Aspartate Kinase Domain of CheY and CheB
In the modular design of two-component signaling pathways, the histidine kinase element serves as an information transmitter, and an aspartate kinase element acts as a receiver (Parkinson & Kofoid 1992, Parkinson 1993). Considerable progress has been made in determining the structure and mechanism of the asparatate kinase receiver element, of which CheY is the prototypical example.
CheY: THE PROTOTYPICAL RECEIVER DOMAIN
All response regulators contain an autocatalytic aspartate kinase domain homologous to the CheY protein; in most cases this CheY-like domain is coupled to a second output domain that carries out a specific phosphorylation-regulated function, such as the methylesterase domain of CheB (Figure 3) or a DNA-binding domain (Parkinson & Kofoid 1992). In the chemosensory pathway, the CheY protein needs no output domain. Prior to phosphorylation, CheY binds to the P2 domain of CheA until the reversible phosphotransfer event generates phospho-CheY, which is quickly released because of its lower affinity for CheA (Swanson et al 1993b, Morrison & Parkinson 1994, Li et al 1995, Stewart 1997). Phospho-CheY subsequently dissociates and diffuses to the flagellar motor, where it binds to the FliM protein in the switch apparatus that controls the direction of motor rotation (Welch et al 1994). Docking of phospho-CheY to the switch element increases the probability of a tumbling event, thereby controlling cellular swimming behavior (Barak & Eisenbach 1992a).
The structure of the unphosphorylated 14-kDa CheY protein has been determined by crystallography or NMR under different conditions in several laboratories (Stock et al 1989, 1993, Volz & Matsumura 1991, Bellsolell et al 1994, Moy et al 1994, Lowry et al 1994, Santoro et al 1995, Ganguli et al 1995). Together, these structures yield a molecular picture of the receiver domain tertiary fold, active site geometry, and protein docking surfaces. The structures are in good agreement although some subtle differences have been debated (Stock & Surette 1996).
CheY exhibits an α/β folding motif (Figure 14A) in which a central five-stranded parallel β-sheet is sandwiched between five α-helices. The phosphorylation site is Asp57 in the kinase active site (Sanders et al 1989), where the active site residues are provided by loops at one end of the β-sheet. Phosphorylation of Asp57 yields a global conformational change, detected by 19F NMR and mapped out in detail by multidimensional heteronuclear NMR (Drake et al 1993, Lowry et al 1994) (Figure 14B). The spatial range of this global conformational change is much greater than that caused by constitutively activating or inactivating CheY point mutations (Bourret et al 1993b) and also dwarfs the localized structural changes triggered by phosphorylation of the relatively static P1 domain of CheA (Zhou & Dahlquist 1997). These findings indicate that the CheY molecule is poised to undergo widespread structural rearrangements upon phosphorylation. Relative to its unphosphorylated state, phospho-CheY exhibits a decreased affinity for CheA but a significantly increased affinity for the motor protein FliM and the phosphatase CheZ (Welch et al 1994, Li et al 1995, Blat & Eisenbach 1996a,b,c).
Figure 14.

Structure of the response regulator CheY, illustrating the phospho-induced conformational regulation of three docking surfaces. (A) The CheY molecule serves as a receiver of signals from CheA and as the response regulator for motor switching. This crystal structure, which includes a bound catalytic Mg2+ ion, displays the α/β folding motif of unphosphorylated CheY (Stock et al 1993). The site of phosphorylation is Asp57 located at the upper edge of the parallel β-sheet (CPK, side chain). Other crystallographic and NMR structures have yielded the same overall backbone fold (see references in text). (B) View of CheY from the same perspective showing residues (α-carbon, sphere) perturbed by the phosphorylation-induced global conformational change, as revealed by aromatic side chain (Drake et al 1993) or backbone (Lowry et al 1994) NMR frequency changes. The large, phospho-regulated surface is seen to cover most of the protein. In addition, smaller backbone frequency changes are observed throughout the molecule, indicating that the conformational change is global (Lowry et al 1994). (C) Same perspective, illustrating a CheA docking surface defined by NMR (α-carbon, black sphere, McEvoy et al 1995, 1996) and surfaces implicated by genetic studies as important to motor switch docking (α-carbon, white sphere; Roman et al 1992, Sockett et al 1992) or CheZ interactions (α-carbon, gray sphere; Sanna et al 1995). The three regions are largely distinct, and none of the interfaces directly overlaps the phosphorylation site (Asp57 is indicated as a ball-and-stick side chain). Some overlap exists between the CheA and motor docking surfaces (Shukla & Matsumura 1995). Phospho-activation of CheY generates a global conformational change that alters the conformation of these docking regions (Drake et al 1993, Lowry et al 1994).
The CheY docking surfaces that interact with each of its three effector proteins have been identified by NMR (CheA P2 domain) or implicated by genetic studies (P2, FliM, CheZ) (Parkinson et al 1983, Yamaguchi et al 1986, Magariyama et al 1990, Roman et al 1992, Sockett et al 1992, Sanna et al 1995, Shukla & Matsumura 1995, Swanson et al 1995). None of these distinct docking surfaces appears to directly overlap the phosphorylation site (Figure 14C). Instead, the available evidence indicates that the phosphorylation-induced conformational change, rather than the presence of acyl phosphate on the protein surface, is responsible for regulating these docking regions. For example, some CheY mutants are phosphorylated but fail to promote tumbling, whereas others are not phosphorylated but do stimulate tumbling (Bourret et al 1990, 1993b, Lukat et al 1991, Welch et al 1994), suggesting that tumbling is modulated by a specific conformational change at the the motor-docking surface. Moreover, the P2-docking surface on CheY is distinct from the Asp57 phosphorylation site, implying conformational regulation (Li et al 1995). Together, such findings indicate that the phosphorylation-induced global conformational change within CheY directly controls distinct docking regions covering as much as half of its surface. Comparison of the surface maps for phospho-activation and inter-protein contacts plainly illustrates this conformational control of docking interactions (compare Figure 14B,C).
THE ASPARTATE KINASE ACTIVE SITE
The active site of CheY, which is highly homologous to those of other aspartate kinases, is illustrated in Figure 15. The conserved catalytic residues include the Asp57 side chain that forms the acyl phosphate, as well as Asp12 and Asp13, which coordinate the essential catalytic Mg2+ (Lukat et al 1991, Stock et al 1993, reviewed by Stock & Surette 1996). The Thr87 residue likely serves as a general acid-base catalyst, whereas Lys109 provides acid-base catalysis or electrostatic stabilization. The Lys109 side chain could also serve as a molecular switch by forming a salt bridge to Asp57 or phospho-Asp57 (Lukat et al 1991, Stock et al 1993, Bourret et al 1993, Stock & Surette 1996). This active site can catalyze autophosphorylation of Asp57 using one of several substrates, such as the isolated phospho-P1 domain of CheA, or the small-molecule phospho-donors phosphoramidate, acetyl phosphate, or carbamoyl phosphate (Lukat et al 1991, Swanson et al 1993b).
Figure 15.
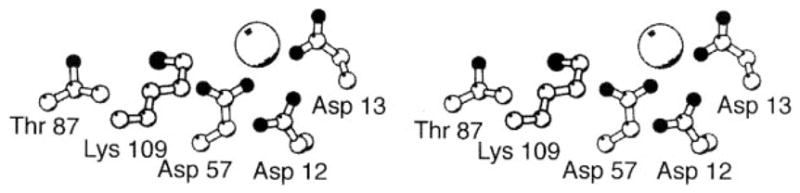
The aspartate kinase active site of CheY. Shown is the Mg2+-occupied structure of the unphosphorylated active site (Stock et al 1993), illustrating the highly conserved catalytic residues. Asp57 serves as the site of phosphorylation, and the aspartate triad (Asp12, Asp13, Asp57) provides both direct and indirect Mg2+ coordination, the latter via solvent. Lys109 and Thr87 act as acid-base catalysts. The Mg2+ ion serves as an essential cofactor in both the autocatalytic phosphorylation and dephosphorylation reactions. (For additional references, see text.)
Plausible chemical mechanisms for the aspartate kinase phosphorylation and dephosphorylation reactions have been suggested based on the crystal structure of Mg2+-occupied CheY (Stock et al 1993; reviewed by Stock & Surette 1996). The phosphorylation reaction is proposed to form a pentavalent phosphate transition state with trigonal bipyramidal geometry, in which the side chain carboxylate oxygen of Asp57 is positioned by its environment to perform a nucleophilic in-line attack on the incoming phospho-donor. The additional negative charge on the transitional pentavalent phosphate is stabilized by the bound Mg2+, whereas the acid-base catalyst activates the leaving group by donating a proton. The dephosphorylation reaction follows a similar reverse pathway, in which a solvent oxygen performs an in-line attack on the acyl phosphate. This hydrolysis uses the acid-base catalyst as a proton acceptor to activate the attacking solvent, yielding a reactive hydroxide ion; the Mg2+ ion again stabilizes the pentavalent phosphate transition state. Under physiological conditions, the autocatalytic dephosphorylation of phospho-CheY is quite rapid, exhibiting a half time under 10 s (Hess et al 1988, Stock et al 1988, Lukat et al 1991). This rapid autodephosphorylation is characteristic of a fast signaling pathway because response regulators from slower two-component pathways can remain phosphorylated for up to an hour or longer (Stock & Surette 1996).
THE RECEIVER DOMAIN OF CheB
The N-terminal receiver domain of CheB is structurally homologous to CheY and also serves as a phospho-activation module. The aspartate kinase activity of CheB catalyzes phosphotransfer from phospho-His48 of CheA to its own active site, yielding phosphotransfer kinetics similar to those of CheY (Hess et al 1988, Lupas & Stock 1989). Once formed, however, phospho-CheB undergoes autocatalytic dephosphorylation much faster than phospho-CheY (Stewart 1993). CheB requires this ultrafast auto-hydrolysis reaction because, unlike phospho-CheY, the phospho-CheB molecule is not dephosphorylated by CheZ (Hess et al 1988). The short lifetime of phospho-CheB is also correlated with the smaller distance it needs to travel to reach its target—generally a receptor within the same cluster of signaling complexes. CheY, by contrast, must diffuse from the receptor cluster to a distant motor.
Genetic studies of CheB have suggested that the aspartate kinase active site contains the same complement of side chains observed in CheY. The corresponding residues in CheB are Asp10, Asp11, Asp56 (thought to be the site of phosphorylation), and Lys107 (Stewart 1993, Stock & Surette 1996). Mutations replacing the key active site residues have similar effects on the CheY and CheB kinases, including analogous lock-on mutations that cause CheY-mediated tumble behavior in vivo and enhanced CheB esterase activity in vitro (D13K and D11K, respectively) (Bourret et al 1990, Stewart 1993). These findings suggest that the activation-induced conformational change in the CheB receiver domain is similar to that observed for CheY.
The Output Domain of CheB
In its role as the methylesterase of the adaptation system, the 37-kDa phospho-CheB monomer either dissociates from the P2 domain of receptor-bound CheA or utilizes the intrinsic mobility of the P2 domain to collide with nearby receptor methylation helices. During such a collision, the C-terminal methylesterase domain of CheB catalyzes the hydrolysis of regulatory methyl esters, yielding free methanol (Stewart & Dahlquist 1988). In some cases, receptor methylation sites are expressed as glutamines, and the CheB methylesterase can hydrolyze these to yield ammonia and a bare glutamate side chain that is then available for methylation (Sherris & Parkinson 1981, Terwilliger & Koshland 1984). Phosphorylation of the CheB receiver domain increases the methylesterase activity at least tenfold, representing the major regulatory event that controls the steady state level of receptor methylation (Lupas & Stock 1989). A similar level of methylesterase activation is achieved when the unconserved, flexible linker between the receiver and output domains is cleaved by proteolysis (Lupas & Stock 1989, Stock & Surette 1996). It follows that the receiver domain of intact CheB autoinhibits the methyltransferase domain and that this autoinhibition is released when the receiver domain is phosphorylated.
The crystal structure of the isolated 21-kDa methylesterase domain reveals an α/β folding motif, in which the central seven-stranded parallel β-sheet is sandwiched between six α-helices and a β-hairpin (Figure 16) (West et al 1995). The active site lies on one edge of the β-sheet, where loops provide the catalytic residues. Figure 17 shows the novel arrangement of three side chains, Ser164, His190 and Asp286, that form a catalytic triad analogous in function, although different in structure, to the catalytic triad of serine proteases (West et al 1995). The Ser164 side chain is essential for activity and probably carries out nucleophilic attack on the methyl ester (Krueger et al 1992). The location of the Ser164 hydroxyl is suitable to allow base catalysis by the His190-Asp286 pair, thereby activating the hydroxyl oxygen by proton removal (West et al 1995). A putative oxyanion hole containing amide protons available for transition state stabilization is found at the appropriate location, and all the active site residues are located in a linear indentation of size and shape appropriate for the docking of a methylation helix. The mechanism by which the receiver domain autoinhibits the activity of the methyltransferase domain remains undetermined, as does the nature of the molecular recognition used by the methylesterase domain to correctly identify a substrate helix. Some of these intriguing questions may be answered by the nearly completed crystal structure of the intact, two-domain protein (A West & AM Stock, personal communication).
Figure 16.
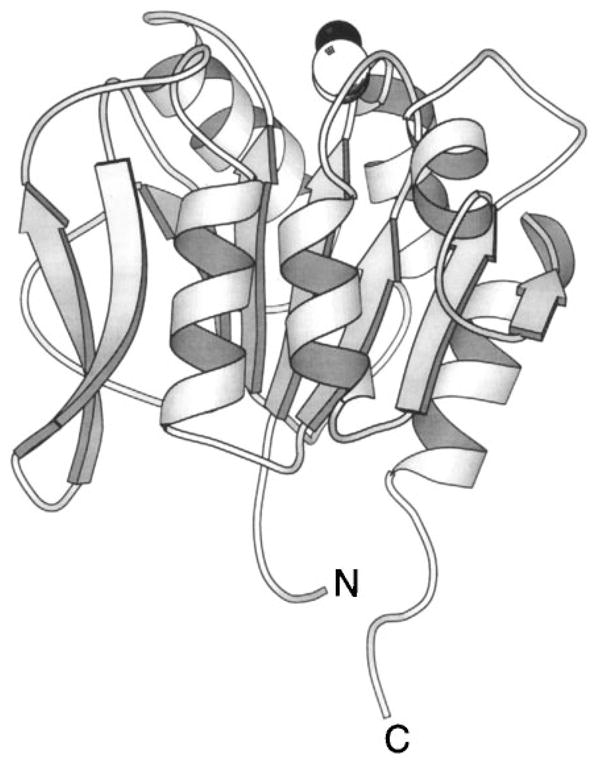
The methylesterase domain of CheB. The C-terminal domain of CheB is a methylesterase that hydrolyzes the regulatory methyl esters and amides of the receptor adaptation sites. In the full-length protein this activity is regulated by phosphorylation of the N-terminal receiver domain (not shown). The crystal structure of the isolated methylesterase domain (West et al 1995) displays an α/β folding motif coupled to a β-hairpin (extreme left). The highlighted Ser164 side chain (CPK, sphere), located on one edge of the β-sheet, acts as the nucleophile in ester and amide hydrolysis.
Figure 17.
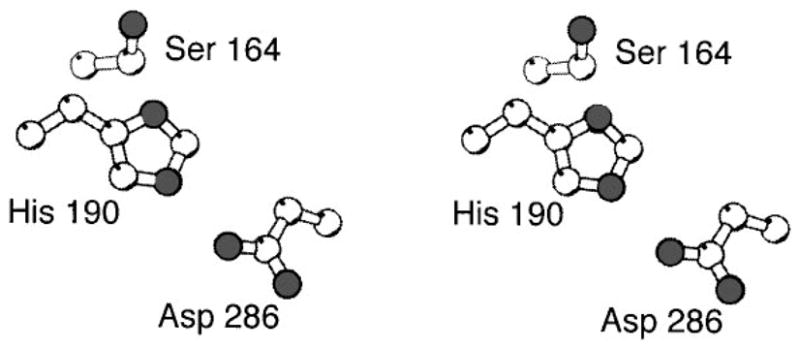
The methyltransferase active site of CheB. Shown are the catalytic residues, including the Ser164 residue essential for catalytic activity and proposed to act as the nucleophile in the methyl ester and amide hydrolysis (West et al 1995). Together the Ser164, His190 and Asp286 side chains form a novel catalytic triad that is functionally, but not structurally, analogous to the catalytic triads of serine proteases.
The Phosphatase CheZ
Of all the pathway components, the CheZ protein remains the least understood. CheZ serves to inactivate the tumble signal of the chemosensory pathway by speeding the hydrolysis of phospho-CheY, either directly using a CheZ active site residue to carry out a nucleophilic attack, or indirectly via a conformational change that stimulates the intrinsic auto-phosphatase activity of the CheY active site (Hess et al 1988). In the latter case, CheZ would be more accurately described as an allosteric effector rather than a true phosphatase. The phosphatase activity of CheZ is essential for the rapid response time of a chemotaxing bacterial cell, because the swimming cell must detect stimuli and modulate the phospho-CheY tumble signal on a ≈1 s timescale, while the intrinsic half-life of phospho-CheY is ≈10 s.
Little is known about the structure of CheZ. It is isolated as a dimer of 24-kDa subunits but assembles into higher-order oligomers in vitro and in vivo (Stock & Stock 1987, Blat & Eisenbach 1996a,b). The phospho-CheY protein, but not unphosphorylated CheY, drives the formation of a large, higher-order oligomer containing a 2:1 mole ratio of CheZ to phospho-CheY, and the resulting oligomer is thought to bind and increase the rate of free phospho-CheY hydrolysis (Blat & Eisenbach 1994, 1996b,c, Bren et al 1996). CheZ also forms mixed oligomers with CheAS that enhance CheY phosphatase activity (Wang & Matsumura 1996, 1997). Other than the stimulation of CheZ oligomerization by its own target, phospho-CheY, no regulation of CheZ has yet been demonstrated.
Recently, a deletion map and random mutagenesis has revealed evidence for distinct functional domains within CheZ (Figure 3) (Blat & Eisenbach 1996c, Sanna & Simon 1996a,b). The C-terminal 19 residues form part of the CheY-docking site because the corresponding peptide fragment specifically binds phospho-CheY in vitro, albeit with low affinity; moreover, suppressors of a mutant CheY are found in this region (Blat & Eisenbach 1996, Sanna & Simon 1996a). The N-terminal region of CheZ may also contribute to the CheY-docking surface (Sanna & Simon 1996b). Much remains to be learned about the CheZ protein, including its structure, the mechanism by which it stimulates phospho-CheY hydrolysis, the nature of phospho-CheY docking, and its regulatory inputs (if any).
THE MOTOR ROTATIONAL SWITCH
Phospho-CheY acts as a tumble signal to alter swimming behavior by specifically binding to the FliM protein, which is a component of the switch apparatus that controls the rotational state of the flagellar motor (Blair 1995, Macnab 1996). FliM is located in the C-ring, a labile structure that projects out from the motor into the cytoplasm (Francis et al 1994). It is estimated that the C-ring contains about 40 copies of FliM, which is also found in the cytoplasm as a soluble protein (Zhao et al 1996). The multiplicity of the FliM assembly suggests that phospho-CheY binding may be highly cooperative, although this aspect of switch activation has not yet been characterized.
Purified FliM is a 37-kDa monomer that binds phospho-CheY more tightly than unphosphorylated CheY (Welch et al 1993, 1994). Certain phospho-CheY mutants are not bound by FliM (Welch et al 1994), further indicating that the switch protein recognizes the conformation of CheY rather than its phosphoryl group (Bourret et al 1990, 1993b, Lukat et al 1991; see above). Recent deletion and mutational studies of FliM suggest that the primary structure contains at least four overlapping functional regions and that the N-terminal region is essential for phospho-CheY binding (Toker et al 1996; D Blair, personal communication). Figure 3 illustrates the proposed functional domains. It is not known how FliM regulates the rotational switching of the motor. For recent reviews of motor structure and function see Macnab (1996) and Blair (1995).
CONCLUSIONS AND FUTURE DIRECTIONS
Considerable headway has been made in defining the structures of chemosensory pathway components, making this an excellent system in which to probe the molecular mechanisms of signal transduction and cellular behavior. For those components and domains that have been resistant to structural determination thus far, homologous thermophilic proteins are being isolated and have significant advantages for structural studies (Swanson et al 1996, Lee & Stock 1996). Alternatively, engineered disulfides can be incorporated to stabilize dynamic proteins for structural analysis.
A number of key chemosensing events have now been described, and mechanistic studies have provided molecular information concerning the activation of soluble and transmembrane receptors by their ligands, the generation of a transmembrane signal, and the activation of a response regulator by phosphorylation. In the membrane-embedded chemoreceptors, the observed piston or tilting piston displacement of the signaling helix represents the first molecular picture of a transmembrane conformational signal, whereas structural studies of CheY phosphoactivation reveal a global conformational change that regulates multiple docking sites. Other critical molecular features of the pathway and its components remaining to be determined include (a) the mechanism of repellent binding and signaling; (b) the structure of the receptor cytoplasmic domain and its mechanism of regulation by methylation and receptor occupancy; (c) the structure, chemical mechanism, and activation mechanism of the CheA histidine kinase domain; (d ) the principles of molecular recognition underlying the specific interactions between pathway components, particularly in the signaling complex; (e) the mechanism by which phospho-CheY regulates the motor switch apparatus; and (f ) the role of conformational dynamics in receptor and signaling protein activation.
Additional important unresolved questions remain concerning the function of the fully integrated chemotaxis system in vivo. (a) The sensory system is able to adapt and chemotax at attractant concentrations at least two orders of magnitude higher than the KD for receptor binding (Mesibov et al 1973, Clarke & Koshland 1979), yet the mechanism of this impressive adaptibility remains unknown. (b) The chemosensory pathway is extraordinarily sensitive: As little as a single attractant molecule can trigger a detectable motor response (Block et al 1983, Segall et al 1986), suggesting that the pathway possesses an unknown amplification mechanism. (c) The clustering of signaling complexes at one pole of the cell must have an important function (Maddock & Shapiro 1993). At one extreme, it could simply facilitate inter-receptor methylation reactions (Wu et al 1996); alternatively, it could serve as an intricate receptor-kinase signaling network that serves as a central processing unit. (d ) The roles of alternate signaling inputs into the pathway via Ca2+ fluxes, acetyl phosphate, fumarate, or cross talk with the PTS sugar-sensing system need to be determined (Tisa & Adler 1992, Barak & Eisenbach 1992b, McCleary & Stock 1994, Lux et al 1995). (e) Finally, initial attempts have demonstrated the promise of quantitative descriptions of pathway function (Bray & Bourret 1997, Bray et al 1993). Ultimately, these approaches will computationally reproduce the working pathway in terms of the microscopic equilibrium and kinetic constants of its individual components.
Acknowledgments
We are indebted to two former group members, C Careaga and L Luck, who made important contributions to our research in this area. In addition, we gratefully acknowledge the following colleagues for coordinates, preprints, helpful discussions, or critical reading of the manuscript: D Blair, R Dahlquist, S Djordjevic, G Hazelbauer, I Kawagishi, S-H Kim, D Koshland, R Macnab, M Manson, M McEvoy, JS Parkinson, R Stewart, A Stock, J Stock, L Thompson, and R Weis. Funding was provided by National Institutes of Health grant GM40731 (to JJF).
Footnotes
A stringent search of the recently completed E. coli genome reveals at least 16 CheA and 34 CheY homologues, respectively (M Danielson & J Falke, unpublished data).
Literature Cited
- Alex LA, Borkovich KA, Simon MI. Hyphal development in Neurospora crassa. Involvement of a 2-component histidine kinase. Proc Natl Acad Sci USA. 1996;93:3416–21. doi: 10.1073/pnas.93.8.3416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ames P, Chen J, Wolff A, Parkinson JS. Structure-function studies of bacterial chemosensors. Cold Spring Harbor Symp Quant Biol. 1988;53:59–66. doi: 10.1101/sqb.1988.053.01.010. [DOI] [PubMed] [Google Scholar]
- Ames P, Parkinson JS. Constitutively signaling fragments of Tsr, the Escherichia coli serine chemoreceptor. J Bacteriol. 1994;176:6340–48. doi: 10.1128/jb.176.20.6340-6348.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ames P, Yu AL, Parkinson JS. Methylation segments are not required for chemotactic signaling by cytoplasmic fragments of Tsr, the methyl-accepting serine chemoreceptor of Escherichia coli. Mol Microbiol. 1996;19:737–46. doi: 10.1046/j.1365-2958.1996.408930.x. [DOI] [PubMed] [Google Scholar]
- Appleby JL, Parkinson JS, Bourret RB. Signal transduction via the multistep phosphorelay. Not necessarily a road less traveled. Cell. 1996;86:845–48. doi: 10.1016/s0092-8674(00)80158-0. [DOI] [PubMed] [Google Scholar]
- Aqvist J, Mowbray SL. Sugar recognition by a glucose/galactose receptor. Evaluation of binding energetics from molecular dynamics stimulations. J Biol Chem. 1995;270:9978–81. [PubMed] [Google Scholar]
- Barak R, Eisenbach M. Correlation between phosphorylation of the chemotaxis protein CheY and its activity at the flagellar motor. Biochemistry. 1992a;31(6):1821–26. doi: 10.1021/bi00121a034. Erratum. 1992. Biochemistry 31:(19):4736. [DOI] [PubMed] [Google Scholar]
- Barak R, Eisenbach M. Fumarate and fumarate metabolite restores switching ability to rotating flagella of bacterial envelopes. J Bacteriol. 1992b;174:643–45. doi: 10.1128/jb.174.2.643-645.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgartner JM, Kim C, Brissette RE, Inouye M, Park C, Hazelbauer GL. Transmembrane signaling by a hybrid protein-communication from the domain of chemoreceptor Trg that recognizes sugar binding proteins to the kinase/phosphatase domain of osmosensor EnvZ. J Bacteriol. 1994;176:1157–63. doi: 10.1128/jb.176.4.1157-1163.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellsolell L, Prieto J, Serrano L, Coll M. Magnesium binding to the bacterial chemotaxis protein CheY results in large conformational changes involving its functional surface. J Mol Biol. 1994;238:489–95. doi: 10.1006/jmbi.1994.1308. [DOI] [PubMed] [Google Scholar]
- Berg HC. Random Walks in Biology. 2 Princeton: Princeton Univ. Press; 1993. [Google Scholar]
- Berg HC, Turner L. Cells of Escherichia coli swim either end forward. Proc Natl Acad Sci USA. 1995;92:477–79. doi: 10.1073/pnas.92.2.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bespalov VA, Zhulin B, Taylor BL. Behavioral responses of E. coli to changes in redox potential. Proc Natl Acad Sci USA. 1996;93:10084–89. doi: 10.1073/pnas.93.19.10084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibikov SI, Biran R, Rudd KE, Parkinson JS. A signal transducer for aerotaxis in E. coli. J Bacteriol. 1997 doi: 10.1128/jb.179.12.4075-4079.1997. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biemann H-D, Koshland DE. Aspartate receptors of Escherichia coli and Salmonella typhimurium bind ligand with negative and half-of-the-sites cooperativity. Biochemistry. 1994;33:629–34. doi: 10.1021/bi00169a002. [DOI] [PubMed] [Google Scholar]
- Binnie RA, Zhang H, Mowbray SL, Hermodson MA. Mutations of the ribose-binding protein of Escherichia coli which affect chemotaxis and transport. Protein Sci. 1992;1:1642–51. doi: 10.1002/pro.5560011212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blair DF. How bacteria sense and swim. Annu Rev Microbiol. 1995;49:489–522. doi: 10.1146/annurev.mi.49.100195.002421. [DOI] [PubMed] [Google Scholar]
- Blat Y, Eisenbach M. Phosphorylation-dependent binding of the chemotaxis signal molecule CheY to its phosphatase, CheZ. Biochemistry. 1994;33:902–6. doi: 10.1021/bi00170a008. [DOI] [PubMed] [Google Scholar]
- Blat Y, Eisenbach M. Mutants with defective phosphatase activity show no phosphorylation-dependent oligomerization of CheZ. The phosphatase of bacterial chemotaxis. J Biol Chem. 1996a;271:1232–36. doi: 10.1074/jbc.271.2.1232. [DOI] [PubMed] [Google Scholar]
- Blat Y, Eisenbach M. Oligomerization of the phosphatase CheZ upon interaction with the phosphorylated form of CheY. The signal protein of bacterial chemotaxis. J Biol Chem. 1996b;271:1226–31. doi: 10.1074/jbc.271.2.1226. [DOI] [PubMed] [Google Scholar]
- Blat Y, Eisenbach M. Conserved C-terminus of the phosphatase CheZ is a binding domain for the chemotactic response regulator CheY. Biochemistry. 1996c;35:5679–83. doi: 10.1021/bi9530447. [DOI] [PubMed] [Google Scholar]
- Block SM, Segall JE, Berg HC. Adaptation kinetics in bacterial chemotaxis. J Bacteriol. 1983;154:312–23. doi: 10.1128/jb.154.1.312-323.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogonez E, Koshland DE. Solubilization of a vectorial transmembrane receptor in functional form. Aspartate receptor of chemotaxis. Proc Natl Acad Sci USA. 1985;82:4891–95. doi: 10.1073/pnas.82.15.4891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boos W, Lucht JM. Periplasmic binding protein-dependent ABC transporters. In: Neidhardt FC, editor. Escherichia coli and Salmonella: Cellular and Molecular Biology. 2 Washington, DC: ASM Press; 1996. pp. 1175–1209. [Google Scholar]
- Borkovich KA, Alex LA, Simon MI. Attenuation of sensory signaling by covalent modification. Proc Natl Acad Sci USA. 1992;89:6756–60. doi: 10.1073/pnas.89.15.6756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borkovich KA, Kaplan N, Hess JF, Simon MI. Transmembrane signal transduction in bacterial chemotaxis involves ligand-dependent activation of phosphate group transfer. Proc Natl Acad Sci USA. 1989;86:1208–12. doi: 10.1073/pnas.86.4.1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borkovich KA, Simon MI. The dynamics of protein phosphorylation in bacterial chemotaxis. Cell. 1990;63:1339–48. doi: 10.1016/0092-8674(90)90429-i. [DOI] [PubMed] [Google Scholar]
- Borkovich KA, Simon MI. Coupling of receptor function to phosphate transfer reactions in bacterial chemotaxis. Methods Enzymol. 1991;200:205–14. doi: 10.1016/0076-6879(91)00140-r. [DOI] [PubMed] [Google Scholar]
- Bourret RB, Borkovich KA, Simon MI. Signal transduction pathways involving protein phosphorylation in prokaryotes. Annu Rev Biochem. 1991;60:401–41. doi: 10.1146/annurev.bi.60.070191.002153. [DOI] [PubMed] [Google Scholar]
- Bourret RB, Davagnino J, Simon MI. The carboxyterminal portion of the CheA kinase mediates regulation of autophosphorylation by transducer and CheW. J Bacteriol. 1993a;175:2097–101. doi: 10.1128/jb.175.7.2097-2101.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourret RB, Drake SK, Chervitz SA, Simon MI, Falke JJ. Activation of the phospho-signaling protein CheY. Analysis of activated mutants by 19F NMR and protein engineering. J Biol Chem. 1993b;268:13089–96. [PMC free article] [PubMed] [Google Scholar]
- Bourret RB, Hess JF, Simon MI. Conserved aspartate residues and phosphorylation in signal transduction by the chemotaxis protein CheY. Proc Natl Acad Sci USA. 1990;87:41–45. doi: 10.1073/pnas.87.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowie JU, Pakula AA, Simon MI. The 3-dimensional structure of the aspartate receptor from Escherichia coli. Acta Cryst. 1995;51:145–54. doi: 10.1107/S0907444994010498. [DOI] [PubMed] [Google Scholar]
- Bray D, Bourret RB. Computer analysis of the binding reactions leading to a transmembrane receptor-linked multiprotein complex involved in bacterial chemotaxis. Mol Biol Cell. 1997;6:1367–80. doi: 10.1091/mbc.6.10.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray D, Bourret RB, Simon MI. Computer simulation of the phosphorylation cascade controlling bacterial chemotaxis. Mol Biol Cell. 1993;4:469–82. doi: 10.1091/mbc.4.5.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bren A, Welch M, Blat Y, Eisenbach M. Signal termination in bacterial chemotaxis. CheZ mediates dephosphorylation of free rather than switch-bound CheY. Proc Natl Acad Sci USA. 1996;93:10090–93. doi: 10.1073/pnas.93.19.10090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budrene EO, Berg HC. Dynamics of formation of symmetrical patterns by chemotactic bacteria. Nature. 1995;376:49–53. doi: 10.1038/376049a0. [DOI] [PubMed] [Google Scholar]
- Butler SL, Falke JJ. Effects of protein stabilizing agents on thermal backbone motions: a disulfide trapping study. Biochemistry. 1996;35:10595–600. doi: 10.1021/bi961107v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Careaga CL, Falke JJ. Thermal motions of surface alpha-helices in the D-galactose chemosensory receptor. Detection by disulfide trapping. J Mol Biol. 1992;226:1219–35. doi: 10.1016/0022-2836(92)91063-u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Careaga CL, Sutherland J, Sabeti J, Falke JJ. Large-amplitude twisting motions of an interdomain hinge. A disulfide trapping study of the galactose-glucose binding protein. Biochemistry. 1995;34:3048–55. doi: 10.1021/bi00009a036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C. The ethylene signal transduction pathway in Arabidopsis: an emerging paradigm. Trends Biochem Sci. 1996;21:129–33. [PubMed] [Google Scholar]
- Chen XM, Koshland DE. The N-terminal cytoplasmic tail of the aspartate receptor is not essential in signal transduction of bacterial chemotaxis. J Biol Chem. 1995;270:24038–42. doi: 10.1074/jbc.270.41.24038. [DOI] [PubMed] [Google Scholar]
- Chervitz SA, Falke JJ. Lock-on -off disulfides identify the transmembrane signaling helix of the aspartate receptor. J Biol Chem. 1995;270:24043–53. doi: 10.1074/jbc.270.41.24043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chervitz SA, Falke JJ. Molecular mechanism of transmembrane signaling by the aspartate receptor: a model. Proc Natl Acad Sci USA. 1996;93:2545–50. doi: 10.1073/pnas.93.6.2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chervitz SA, Lin CM, Falke JJ. Transmembrane signaling by the aspartate receptor: engineered disulfides reveal static regions of the subunit interface. Biochemistry. 1995;34:9722–33. doi: 10.1021/bi00030a010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chothia C, Lesk AM. Helix movements in proteins. Trends Biochem Sci. 1985;10:116–18. [Google Scholar]
- Clarke S, Koshland DE. Membrane receptors for aspartate and serine in bacterial chemotaxis. J Biol Chem. 1979;254:9695–702. [PubMed] [Google Scholar]
- Clarke S, Sparrow K, Panasenko S, Koshland DE. In vitro methylation of bacterial chemotaxis proteins. Characterization of protein methyltransferase activity in crude extracts of Salmonella typhimurium. J Supramol Struct. 1980;13:315–28. doi: 10.1002/jss.400130305. [DOI] [PubMed] [Google Scholar]
- Cochran AG, Kim PS. Imitation of Escherichia coli aspartate receptor signaling in engineered dimers of the cytoplasmic domain. Science. 1996;271:1113–16. doi: 10.1126/science.271.5252.1113. [DOI] [PubMed] [Google Scholar]
- Danielson MA. PhD thesis. Univ. Colo; Boulder: 1997. The molecular mechanism of transmembrane signaling and kinase regulation by the aspartate receptor of bacterial chemotaxis. [Google Scholar]
- Danielson MA, Bass RB, Kim T, Falke JJ. The first adaptive methylation segment of the chemotactic aspartate receptor is helical and lies at a subunit interface critical for kinase regulation. J Biol Chem. 1997 Submitted. [Google Scholar]
- Danielson MA, Biemann H-P, Koshland DE, Falke JJ. Attractant- and disulfide-induced conformational changes in the ligand-binding domain of the chemotaxis aspartate receptor: a F-19 NMR study. Biochemistry. 1994;33:6100–9. doi: 10.1021/bi00186a009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danielson MA, Falke JJ. Use of F-19 NMR to probe protein structure and conformational changes. Annu Rev Biophys Biomol Struct. 1996;25:163–95. doi: 10.1146/annurev.bb.25.060196.001115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePina K, Navarro C, McWalter L, Boxer DH, Price NC, et al. Purification and characterization of the periplasmic nickel-binding protein NikA of Escherichia coli K12. Eur J Biochem. 1995;227:857–65. doi: 10.1111/j.1432-1033.1995.tb20211.x. [DOI] [PubMed] [Google Scholar]
- Djordjevic S, Stock AM. Crystal structure of the chemotaxis receptor methyltransferase CheR. Structure. 1997;5:545–48. doi: 10.1016/s0969-2126(97)00210-4. [DOI] [PubMed] [Google Scholar]
- Drake SK, Bourret RB, Luck LA, Simon MI, Falke JJ. Activation of the phosphosignaling protein CheY. J Biol Chem. 1993;268:13081–88. [PMC free article] [PubMed] [Google Scholar]
- Falke JJ, Koshland DE. Global flexibility in a sensory receptor: a site directed cross-linking approach. Science. 1987;237:1596–600. doi: 10.1126/science.2820061. [DOI] [PubMed] [Google Scholar]
- Francis NR, Sosinsky GE, Thomas D, DeRosier DJ. Isolation, characterization, and structure of bacterial flagellar motors containing the switch complex. J Mol Biol. 1994;235:1261–70. doi: 10.1006/jmbi.1994.1079. [DOI] [PubMed] [Google Scholar]
- Ganguli S, Wang H, Matsumura P, Volz K. Uncoupled phosphorylation and activation in bacterial chemotaxis. The 2.1 angstrom structure structure of a threonine to isoleucine mutant at position 87 of CheY. J Biol Chem. 1995;270:17386–93. [PubMed] [Google Scholar]
- Gardina PJ, Bormans AF, Hawkins MA, Meeker JW, Manson MD. Maltose-binding protein interacts simultaneously and asymmetrically with both subunits of the Tar chemoreceptor. Mol Microbiol. 1997;23:1181–91. doi: 10.1046/j.1365-2958.1997.3001661.x. [DOI] [PubMed] [Google Scholar]
- Gardina PJ, Manson MD. Attractant signaling by an aspartate chemoreceptor dimer with a single cytoplasmic domain. Science. 1996;274:425–26. doi: 10.1126/science.274.5286.425. [DOI] [PubMed] [Google Scholar]
- Garzon A, Parkinson JS. Chemotactic signaling by the P1 phosphorylation domain liberated from the CheA histidine kinase of Escherichia coli. J Bacteriol. 1996;178:6752–58. doi: 10.1128/jb.178.23.6752-6758.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gegner JA, Graham DR, Roth AF, Dahlquist FW. Assembly of an MCP receptor, CheW, and kinase CheA complex in the bacterial chemotaxis signal transduction pathway. Cell. 1992;18:975–82. doi: 10.1016/0092-8674(92)90247-a. [DOI] [PubMed] [Google Scholar]
- Gerstein M, Lesk AM, Chothia C. Structural mechanisms for domain movements in proteins. Biochemistry. 1994;33:6739–49. doi: 10.1021/bi00188a001. [DOI] [PubMed] [Google Scholar]
- Gonzalez L, Jr, Brown RA, Richardson D, Alber T. Crystal structures of a single coiled-coil peptide in two oligomeric states reveal the basis for structural polymorphism. Nat Struct Biol. 1996a;3:1002. doi: 10.1038/nsb1296-1002. [DOI] [PubMed] [Google Scholar]
- Gonzalez L, Jr, Woolfson DN, Alber T. Buried polar residues and structural specificity in the GCN4 leucine zipper. Nat Struct Biol. 1996b;3:1011. doi: 10.1038/nsb1296-1011. [DOI] [PubMed] [Google Scholar]
- Goodsell DS. Inside a living cell. Trends Biochem Sci. 1991;16:203–6. doi: 10.1016/0968-0004(91)90083-8. [DOI] [PubMed] [Google Scholar]
- Harshey RM, Matsuyama T. Dimorphic transition in Escherichia coli and Salmonella typhimurium: surface-induced differentiation into hyperflagellate swarmer cells. Proc Natl Acad Sci USA. 1994;91(18):8631–35. doi: 10.1073/pnas.91.18.8631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazelbauer GL, Adler J. Role of the galactose binding protein in chemotaxis of Escherichia coli toward galactose. Nat New Biol. 1971;230:101–4. doi: 10.1038/newbio230101a0. [DOI] [PubMed] [Google Scholar]
- Hazelbauer GL, Engstrom P. Multiple forms of methyl-accepting chemotaxis proteins distinguished by a factor in addition to multiple methylation. J Bacteriol. 1981;145:35–42. doi: 10.1128/jb.145.1.35-42.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess JF, Oosawa K, Kaplan N, Simon MI. Phosphorylation of three proteins in the signaling pathway of bacterial chemotaxis. Cell. 1988;53:79–87. doi: 10.1016/0092-8674(88)90489-8. [DOI] [PubMed] [Google Scholar]
- Hess JF, Oosawa K, Matsumura P, Simon MI. Protein phosphorylation is involved in bacterial chemotaxis. Proc Natl Acad Sci USA. 1987;84:7609–13. doi: 10.1073/pnas.84.21.7609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoch JA, Silhavy TJ. Two-Component Signal Transduction. Washington, DC: Am. Soc. Microbiol; 1995. [Google Scholar]
- Hughson AG, Hazelbauer GL. Detecting the conformational change of transmembrane signaling in a bacterial chemoreceptor by measuring effects on disulfide cross-linking in vivo. Proc Natl Acad Sci USA. 1996;93:11546–51. doi: 10.1073/pnas.93.21.11546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffery CJ, Koshland DE. Three-dimensional structural model of the serine receptor ligand binding domain. Protein Sci. 1993;2:559–66. doi: 10.1002/pro.5560020407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones BD, Lee CA, Falkow S. Invasion by Salmonella typhimurium is affected by the direction of flagellar rotation. Infect Immun. 1992;60:2475–80. doi: 10.1128/iai.60.6.2475-2480.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehry MR, Dahlquist FW. The methyl-accepting chemotaxis proteins of Escherichia coli. Identification of the multiple methylation sites on methyl-accepting chemotaxis protein. J Biol Chem. 1982;257:10378–86. [PubMed] [Google Scholar]
- Kehry MR, Engstrom FW, Dahlquist FW, Hazelbauer GL. Multiple covalent modifications of Trg, a sensory transducer of Escherichia coli. J Biol Chem. 1983;258:5050–55. [PubMed] [Google Scholar]
- Khan S, Spudich JL, McCray JA, Trentham DR. Chemotactic signal integration in bacteria. Proc Natl Acad Sci USA. 1995;92:9757–61. doi: 10.1073/pnas.92.21.9757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kihara M, Homma M, Kutsukake K, Macnab RM. Flagellar switch of Salmonella typhimurium: gene sequences and deduced protein sequences. J Bacteriol. 1989;171:3247–57. doi: 10.1128/jb.171.6.3247-3257.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S-H. “Frozen” dynamic dimer model for transmembrane signaling in bacterial chemotaxis receptors. Protein Sci. 1994;3:159–65. doi: 10.1002/pro.5560030201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofoid EC, Parkinson JS. Tandem translation starts in the cheA locus of Escherichia coli. J Bacteriol. 1991;173:2116–19. doi: 10.1128/jb.173.6.2116-2119.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohn WD, Mant CT, Hodges RS. Alpha-helical protein assembly motifs. J Biol Chem. 1997;272:2583–86. doi: 10.1074/jbc.272.5.2583. [DOI] [PubMed] [Google Scholar]
- Kolodziej AF, Tan T, Koshland DE. Producing positive, negative, and no cooperativity by mutations at a single residue located at the subunit interface in the aspartate receptor of Salmonella typhimurium. Biochemistry. 1996;35:14782–92. doi: 10.1021/bi961481v. [DOI] [PubMed] [Google Scholar]
- Koman A, Harayama S, Hazelbauer GL. Relation of chemotactic response to the amount of receptor: evidence for different efficiencies of signal transduction. J Bacteriol. 1979;138:739–47. doi: 10.1128/jb.138.3.739-747.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krikos A, Conley MP, Boyd A, Berg HC, Simon MI. Chimeric chemosensory transducers of Escherichia coli. Proc Natl Acad Sci USA. 1985;82:1326–30. doi: 10.1073/pnas.82.5.1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger JK, Stock J, Schutt CE. Evidence that the methylesterase of bacterial chemotaxis may be a serine hydrolase. Biochim Biophys Acta. 1992;1119:322–26. doi: 10.1016/0167-4838(92)90220-8. [DOI] [PubMed] [Google Scholar]
- Kuo SC, Koshland DE. Roles of cheY and cheZ gene products in controlling flagellar rotation in bacterial chemotaxis of Escherichia coli. J Bacteriol. 1987;169:1307–14. doi: 10.1128/jb.169.3.1307-1314.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo SC, Koshland DE. Multiple kinetic states for the flagellar motor switch. J Bacteriol. 1989;171:6279–87. doi: 10.1128/jb.171.11.6279-6287.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee GF, Burrows GG, Lebert MR, Dutton DP, Hazelbauer GL. Deducing the organization of a transmembrane domain by disulfide cross-linking. The bacterial chemoreceptor Trg. J Biol Chem. 1994;269:29920–27. [PubMed] [Google Scholar]
- Lee GF, Lebert MR, Lilly AA, Hazelbauer GL. Transmembrane signaling characterized in bacterial chemoreceptors by using sulfhydryl cross-linking in vivo. Proc Natl Acad Sci USA. 1995;92:3391–95. doi: 10.1073/pnas.92.8.3391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee PJ, Stock AM. Characterization of the genes and proteins of a 2-component system from the hyperthermophylic bacterium Thermotoga maritima. J Bacteriol. 1996;178:5579–85. doi: 10.1128/jb.178.19.5579-5585.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeMoual H, Koshland DE. Molecular evolution of the C-terminal cytoplasmic domain of a superfamily of bacterial receptors involved in taxis. J Mol Biol. 1996;261:568–85. doi: 10.1006/jmbi.1996.0483. [DOI] [PubMed] [Google Scholar]
- Levit M, Liu Y, Surette M, Stock J. Active-site interference and asymmetric activation in the chemotaxis protein histidine kinase CheA. J Biol Chem. 1996;271:32057–63. doi: 10.1074/jbc.271.50.32057. [DOI] [PubMed] [Google Scholar]
- Li JY, Swanson RV, Simon MI, Weis RM. The response regulators CheB and CheY exhibit competitive binding to the kinase CheA. Biochemistry. 1995;34:14626–36. doi: 10.1021/bi00045a003. [DOI] [PubMed] [Google Scholar]
- Lin LN, Li JY, Brandts JF, Weis RM. The serine receptor of bacterial chemotaxis exhibits half-site saturation for serine binding. Biochemistry. 1994;33:6564–70. doi: 10.1021/bi00187a025. [DOI] [PubMed] [Google Scholar]
- Liu JD, Parkinson JS. Genetic evidence for interaction between the CheW and Tsr proteins during chemoreceptor signaling by Escherichia coli. J Bacteriol. 1991;173:4941–51. doi: 10.1128/jb.173.16.4941-4951.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JD, Parkinson JS. Role of CheW protein in coupling membrane receptors to the intracellular signaling system of bacterial chemotaxis. Proc Natl Acad Sci USA. 1989;86:8703–7. doi: 10.1073/pnas.86.22.8703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long DG, Weis RM. Oligomerization of the cytoplasmic fragment from the aspartate receptor of Escherichia coli. Biochemistry. 1992;31:9904–11. doi: 10.1021/bi00156a007. [DOI] [PubMed] [Google Scholar]
- Lowry DF, Roth AF, Rupert PB, Dahlquist FW, Moy FJ, et al. Signal transduction in chemotaxis: a propagating conformation change upon phosphorylation of CheY. J Biol Chem. 1994;269:8348–54. [PubMed] [Google Scholar]
- Luck LA, Falke JJ. Open conformation of a substrate-binding cleft. F-19 NMR-studies of cleft angle in the D-galactose chemosesory receptor. Biochemistry. 1991a;30:6484–90. doi: 10.1021/bi00240a019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luck LA, Falke JJ. F-19 NMR studies of the D-galactose chemosensory receptor 1. Sugar binding yields a global structural change. Biochemistry. 1991b;30:4248–56. doi: 10.1021/bi00231a021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luck LA, Falke JJ. F-19 NMR studies of the D-galactose chemosensory receptor 2. Ca(II) binding yields a local structural change. Biochemistry. 1991c;30:4257–61. doi: 10.1021/bi00231a022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukat GS, Lee BH, Mottonen JM, Stock AM, Stock JB. Roles of the highly conserved aspartate and lysine residues in the response regulator of bacterial chemotaxis. J Biol Chem. 1991;266:8348–54. [PubMed] [Google Scholar]
- Lumb KJ, Kim PS. A buried polar interaction imparts structural uniqueness in a designed heterodimeric coiled-coil. Biochemistry. 1995;34:8642–48. doi: 10.1021/bi00027a013. [DOI] [PubMed] [Google Scholar]
- Lupas A, Stock JB. Phosphorylation of an N-terminal regulatory domain activates the CheB methylesterase in bacterial chemotaxis. J Biol Chem. 1989;264:17337–42. [PubMed] [Google Scholar]
- Lux R, Jahreis K, Bettenbrock K, Parkinson JS, Lengeler JW. Coupling the phosphotransferase system and the methyl-accepting chemotaxis protein-dependent chemotaxis signaling pathways of Escherichia coli. Proc Natl Acad Sci USA. 1995;92:11583–87. doi: 10.1073/pnas.92.25.11583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch BA, Koshland DE. Structural similarities between the aspartate receptor of bacterial chemotaxis and the trp repressor of E. coli. FEBS Lett. 1992;307:3–9. doi: 10.1016/0014-5793(92)80891-j. [DOI] [PubMed] [Google Scholar]
- Macnab RM. Flagella. In: Neidhardt FC, editor. Escherichia coli and Salmonella: Cellular and Molecular Biology. Washington, DC: ASM Press; 1987. pp. 70–83. [Google Scholar]
- Macnab RM. Flagella and motility. In: Neidhardt FC, editor. Escherichia coli and Salmonella: Cellular and Molecular Biology. 2 Washington, DC: ASM Press; 1996. pp. 123–45. [Google Scholar]
- Maddock JR, Shapiro L. Polar location of the chemoreceptor complex in the Escherichia coli cell. Science. 1993;259:1717–23. doi: 10.1126/science.8456299. [DOI] [PubMed] [Google Scholar]
- Magariyama Y, Yamaguchi S, Aizawa S-I. Genetic and behavioral analysis of flagellar switch mutants of Salmonella typhimurium. J Bacteriol. 1990;172:4359–69. doi: 10.1128/jb.172.8.4359-4369.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manson MD, Boos W, Bassford PJ, Rasmussen BA. Dependence of maltose transport and chemotaxis on the amount of maltose binding protein. J Biol Chem. 1985;260:9727–33. [PubMed] [Google Scholar]
- Maruyama IN, Mikawa YG, Maruyama HI. A model for transmembrane signaling by the aspartate receptor based a random cassette mutagenesis and site-directed disulfides. J Mol Biol. 1995;253:530–46. doi: 10.1006/jmbi.1995.0571. [DOI] [PubMed] [Google Scholar]
- McCleary WR, Stock JB. Acetyl phosphate and the activation of two-component response regulators. J Biol Chem. 1994;269:31567–72. [PubMed] [Google Scholar]
- McEvoy MM, Delacruz AFA, Dahlquist FW. Large modular proteins by NMR. Nat Struct Biol. 1997;4:9. doi: 10.1038/nsb0197-9. [DOI] [PubMed] [Google Scholar]
- McEvoy MM, Muhandiram DR, Kay LE, Dahlquist FW. Structure and dynamics of a CheY-binding domain of the chemotaxis kinase CheA determined by nuclear magnetic resonance spectroscopy. Biochemistry. 1996;35:5633–40. doi: 10.1021/bi952707h. [DOI] [PubMed] [Google Scholar]
- McEvoy MM, Zhou HJ, Roth AF, Lowry DF, Morrison TB, et al. Nuclear magnetic resonance assignments and global fold of a CheY-binding domain in CheA, the chemotaxis specific kinase of Escherichia coli. Biochemistry. 1995;34:13871–80. doi: 10.1021/bi00042a019. [DOI] [PubMed] [Google Scholar]
- Mesibov R, Ordal GW, Adler J. The range of attractant concentrations for bacterial chemotaxis and the threshold and size of response over this range. J Gen Physiol. 1973;62:203–23. doi: 10.1085/jgp.62.2.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milburn MV, Prive GG, Milligan DL, Scott WG, Yeh J, et al. Three-dimensional structures of the ligand-binding domain of the bacterial aspartate receptor with and without a ligand. Science. 1991;254:1342–47. doi: 10.1126/science.1660187. [DOI] [PubMed] [Google Scholar]
- Miller DM, III, Olson JS, Pflügrath JW, Quiocho FA. Rates of ligand binding to periplasmic proteins involved in bacterial transport and chemotaxis. J Biol Chem. 1983;258:13665–72. [PubMed] [Google Scholar]
- Milligan DL, Koshland DE. Site-directed cross-linking. Establishment of the dimeric structure of the aspartate receptor of bacterial chemotaxis. J Biol Chem. 1988;263:6268–75. [PubMed] [Google Scholar]
- Milligan DL, Koshland DE. Purification and characterization of the periplasmic domain of the aspartate receptor. J Biol Chem. 1993;268:19991–97. [PubMed] [Google Scholar]
- Morrison TB, Parkinson JS. Liberation of an interaction domain from the phosphotransfer region of CheA, a signaling kinase of Escherichia coli. Proc Natl Acad Sci USA. 1994;91:5485–89. doi: 10.1073/pnas.91.12.5485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mowbray SL, Foster DL, Koshland DE. Proteolytic fragments identified with domains of the aspartate receptor. J Biol Chem. 1985;260:11711–18. [PubMed] [Google Scholar]
- Mowbray SL, Koshland DE. Additive and independent responses in a single receptor: aspartate and maltose stimuli on the Tar protein. Cell. 1987;50:171–80. doi: 10.1016/0092-8674(87)90213-3. [DOI] [PubMed] [Google Scholar]
- Moy FJ, Lowry DF, Matsumura P, Dahlquist FW, Krywko JE, Domaille PJ. Assignments, secondary structure, global fold, and dynamics of chemotaxis Y protein using three- and four-dimensional heteronu-clear (13C, 15N) NMR spectroscopy. Biochemistry. 1994;33:10731–42. doi: 10.1021/bi00201a022. [DOI] [PubMed] [Google Scholar]
- Ninfa EG, Stock A, Mowbray SL, Stock J. Reconstitution of the bacterial chemotaxis signal transduction system from purified components. J Biol Chem. 1991;266:9764–70. [PubMed] [Google Scholar]
- Nowlin DM, Bollinger J, Hazelbauer GL. Sites of covalent modification in Trg, a sensory transducer of Escherichia coli. J Biol Chem. 1987;262:6039–45. [PubMed] [Google Scholar]
- Oosawa K, Simon MI. Analysis of mutations in the transmembrane region of the aspartate chemoreceptor in Escherichia coli. Proc Natl Acad Sci USA. 1986;83:6930–34. doi: 10.1073/pnas.83.18.6930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pakula AA, Simon MI. Determination of transmembrane protein structure by disulfide cross-linking: the Escherichia coli Tar receptor. Proc Natl Acad Sci USA. 1992;89:4144–48. doi: 10.1073/pnas.89.9.4144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paliakasis CD, Kokkinidis M. Relationships between sequence and structure for the 4 alpha helix bundle tertiary motif in proteins. Prot Eng. 1992;5:739–48. doi: 10.1093/protein/5.8.739. [DOI] [PubMed] [Google Scholar]
- Parkinson JS. Signal transduction schemes of bacteria. Cell. 1993;73:857–71. doi: 10.1016/0092-8674(93)90267-t. [DOI] [PubMed] [Google Scholar]
- Parkinson JS, Blair DF. Does E. coli have a nose? Science. 1993;259:1701–2. doi: 10.1126/science.8456297. [DOI] [PubMed] [Google Scholar]
- Parkinson JS, Kofoid EC. Communication modules in bacterial signaling proteins. Annu Rev Genet. 1992;26:71–112. doi: 10.1146/annurev.ge.26.120192.000443. [DOI] [PubMed] [Google Scholar]
- Parkinson JS, Parker SR, Talbert PB, Houts SE. Interactions between chemotaxis genes and flagellar genes in Escherichia coli. J Bacteriol. 1983;155:265–74. doi: 10.1128/jb.155.1.265-274.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posas F, Wurgler-Murphy SM, Maeda T, Witten EA, Thai TC, Saito TC. Yeast HOG1 MAP kinase cascade is regulated by a multistep phosphorelay mechanism in the SLN1-YPD1-SSK1 2-component osmosensor. Cell. 1996;86:865–75. doi: 10.1016/s0092-8674(00)80162-2. [DOI] [PubMed] [Google Scholar]
- Quiocho FA, Ledvina PS. Atomic structure and specificity of bacterial periplasmic receptors for active transport and chemotaxis. Variation of common themes. Mol Microbiol. 1996;20:17–25. doi: 10.1111/j.1365-2958.1996.tb02484.x. [DOI] [PubMed] [Google Scholar]
- Rampersaud A, Utsumi R, Delgado J, Forst SA, Inouye M. Ca2+-enhanced phosphorylation of a chimeric protein kinase involved with bacterial signal transduction. J Biol Chem. 1991;266:7633–37. [PubMed] [Google Scholar]
- Rice MS, Dahlquist FW. Sites of deamidation and methylation in Tsr, a bacterial chemotaxis sensory transducer. J Biol Chem. 1991;266:9746–53. [PubMed] [Google Scholar]
- Roman SJ, Meyers M, Volz K, Matsumura P. A chemotactic signaling surface on CheY defined by supressors of flagellar switch mutations. J Bacteriol. 1992;174:6247–55. doi: 10.1128/jb.174.19.6247-6255.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanatinia H, Kofoid EC, Morrison TB, Parkinson JS. The smaller of two overlapping CheA gene-products is not essential for chemotaxis in Escherichia coli. J Bacteriol. 1995;177:2713–20. doi: 10.1128/jb.177.10.2713-2720.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders DA, Gillece-Castro BL, Stock AM, Burlingame AL, Koshland DE. Identification of the site of phosphorylation of the chemotaxis response regulator, CheY. J Biol Chem. 1989;264:21770–78. [PubMed] [Google Scholar]
- Sanna MG, Simon MI. Isolation and in vitro characterization of CheZ suppressors for the Escherichia coli chemotactic response regulator mutant CheYN23D. J Biol Chem. 1996a;271:7357–61. doi: 10.1074/jbc.271.13.7357. [DOI] [PubMed] [Google Scholar]
- Sanna MG, Simon MI. Protein CheZ gain-of-function and loss-of-function mutants. J Bacteriol. 1996b;178:6275–80. doi: 10.1128/jb.178.21.6275-6280.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanna MG, Swanson RV, Bourret RB, Simon MI. Mutations in the chemotactic response regulator, CheY, that confer resistance to the phosphatase activity of CheZ. Mol Microbiol. 1995;15:1069–79. doi: 10.1111/j.1365-2958.1995.tb02282.x. [DOI] [PubMed] [Google Scholar]
- Santoro J, Bruix M, Pascual J, Lopez E, Serrano L, Rico M. Three-dimensional structure of chemotactic CheY protein in aqueous solution by nuclear magnetic resonance methods. J Mol Biol. 1995;247:717–25. doi: 10.1006/jmbi.1995.0175. [DOI] [PubMed] [Google Scholar]
- Schuster SC, Noegel AA, Oeme F, Gerish G, Simon MI. The hybrid histidine kinase DokA is part of the osmotic response system of Dictyostelium. EMBO J. 1996;15:3880–89. [PMC free article] [PubMed] [Google Scholar]
- Schuster SC, Swanson RV, Alex LA, Bourret RB, Simon MI. Assembly and function of a quaternary signal transduction complex monitered by surface plasmon resonance. Nature. 1993;365:343–47. doi: 10.1038/365343a0. [DOI] [PubMed] [Google Scholar]
- Seeley SK, Weis RM, Thompson LK. The cytoplasmic fragment of the aspartate receptor displays globally dynamic behavior. Biochemistry. 1996;35:5199–206. doi: 10.1021/bi9524979. [DOI] [PubMed] [Google Scholar]
- Segall JE, Block SM, Berg HC. Temporal comparisons in bacterial chemotaxis. Proc Natl Acad Sci USA. 1986;83:8987–91. doi: 10.1073/pnas.83.23.8987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharff AJ, Rodseth LE, Spurlino JC, Quiocho FA. Crystallographic evidence of a large ligand induced hinge-twist motion between the two domains of the maltodextrin binding protein involved in active transport and chemotaxis. Biochemistry. 1992;31:10657–63. doi: 10.1021/bi00159a003. [DOI] [PubMed] [Google Scholar]
- Sherris D, Parkinson JS. Post-translational processing of methyl-accepting chemotaxis proteins in Escherichia coli. Proc Natl Acad Sci USA. 1981;78:6051–55. doi: 10.1073/pnas.78.10.6051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shilton BH, Flocco MM, Nilsson M, Mowbray SL. Conformational changes of three periplasmic receptors for bacterial chemotaxis and transport. The maltose-binding, glucose/galactose-binding and ribose-binding proteins. J Mol Biol. 1996;264:350–63. doi: 10.1006/jmbi.1996.0645. [DOI] [PubMed] [Google Scholar]
- Shukla D, Matsumura P. Mutations leading to altered CheA binding cluster on a face of CheY. J Biol Chem. 1995;270:24414–19. doi: 10.1074/jbc.270.41.24414. [DOI] [PubMed] [Google Scholar]
- Simms SA, Subbaramaiah K. The kinetic mechanism of S-adenosyl-L-methionine: glutamylmethyltransferase from Salmonella typhimurium. J Biol Chem. 1991;266:12741–46. [PubMed] [Google Scholar]
- Smith RA, Parkinson JS. Overlapping genes at the cheA locus of Escherichia coli. Proc Nat Acad Sci USA. 1980;77:5370–74. doi: 10.1073/pnas.77.9.5370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sockett H, Yamaguchi S, Kihara M, Irikura VM, Mcnab RM. Molecular analysis of of the flagellar switch protein FliM of Salmonella typhimurium. J Bacteriol. 1992;174:793–806. doi: 10.1128/jb.174.3.793-806.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart RC. Activating and inhibitor mutations in the regulatory domain of CheB, the methylesterase in bacterial chemotaxis. J Biol Chem. 1993;268:1921–30. [PubMed] [Google Scholar]
- Stewart RC. Kinetic characterization of phosphotransfer between CheA and CheY in the bacterial chemotaxis signal transduction pathway. Biochemistry. 1997;36:2030–40. doi: 10.1021/bi962261k. [DOI] [PubMed] [Google Scholar]
- Stewart RC, Dahlquist FW. N-terminal half of CheB is involved in methylesterase response to negative chemotactic stimuli in Escherichia coli. J Bacteriol. 1988;170:5728–38. doi: 10.1128/jb.170.12.5728-5738.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stock AM, Martinez-Hackert E, Rasmussen BF, West AH, Stock JB, et al. Structure of the Mg2+-bound form of CheY and mechanism of phosphoryl transfer in bacterial chemotaxis. Biochemistry. 1993;32:13375–80. doi: 10.1021/bi00212a001. [DOI] [PubMed] [Google Scholar]
- Stock AM, Mottonen JM, Stock JB, Schutt CE. Three-dimensional structure of CheY, the response regulator of bacterial chemotaxis. Nature. 1989;337:745–49. doi: 10.1038/337745a0. [DOI] [PubMed] [Google Scholar]
- Stock AM, Mowbray SL. Bacterial chemotaxis—a field in motion. Curr Opin Struct Biol. 1995;5:744–51. doi: 10.1016/0959-440x(95)80006-9. [DOI] [PubMed] [Google Scholar]
- Stock AM, Stock JB. Purification and characterization of the CheZ protein of bacterial chemotaxis. J Bacteriol. 1987;169:3301–11. doi: 10.1128/jb.169.7.3301-3311.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stock JB, Surette M. Chemotaxis. In: Neidhardt FC, editor. Escherichia coli and Salmonella typhimurium: Cellular and Molecular Biology. 2 Washington, DC: ASM Press; 1996. pp. 123–45. [Google Scholar]
- Stock AM, Wylie DC, Mottonen JM, Lupas AM, Ninfa EG, et al. Phospho-proteins involved in bacterial signal transduction. Cold Spring Harbor Symp Quant Biol. 1988;53:49–57. doi: 10.1101/sqb.1988.053.01.009. [DOI] [PubMed] [Google Scholar]
- Stock JB, Lukat GS, Stock AM. Bacterial chemotaxis and the molecular logic of intracellular signal transduction networks. Annu Rev Biophys Biophys Chem. 1991;20:109–36. doi: 10.1146/annurev.bb.20.060191.000545. [DOI] [PubMed] [Google Scholar]
- Stoddard BL, Koshland DE. Prediction of the structure of a receptor protein complex using a binary docking method. Nature. 1992;358:774–76. doi: 10.1038/358774a0. [DOI] [PubMed] [Google Scholar]
- Surette M, Levit M, Liu Y, Lukat G, Ninfa EG, et al. Dimerization is required for the activity of the protein histidine kinase CheA that mediates signal-transduction in bacterial chemotaxis. J Biol Chem. 1996;271:939–45. doi: 10.1074/jbc.271.2.939. [DOI] [PubMed] [Google Scholar]
- Surette M, Stock JB. Role of alpha-helical coiled-coil interactions in receptor dimerization, signaling, and adaptation during bacterial chemotaxis. J Biol Chem. 1996;271:17966–73. doi: 10.1074/jbc.271.30.17966. [DOI] [PubMed] [Google Scholar]
- Swanson RV, Alex LA, Simon MI. Histidine and aspartate phosphorylation: two component systems and the limits of homology. Trends Biochem Sci. 1994;19:485–90. doi: 10.1016/0968-0004(94)90135-x. [DOI] [PubMed] [Google Scholar]
- Swanson RV, Bourret RB, Simon MI. Intermolecular complementation of the kinase activity of CheA. Mol Microbiol. 1993a;8:435–41. doi: 10.1111/j.1365-2958.1993.tb01588.x. [DOI] [PubMed] [Google Scholar]
- Swanson RV, Lowry DF, Matsumura P, McEvoy MM, Simon MI, Dahlquist FW. Localized perturbations in CheY structure monitored by NMR identify a CheA binding interface. Nat Struct Biol. 1995;2:906–10. doi: 10.1038/nsb1095-906. [DOI] [PubMed] [Google Scholar]
- Swanson RV, Sanna MG, Simon MI. Thermostable chemotaxis proteins from the hyperthermophilic bacterium Thermotoga maritima. J Bacteriol. 1996;178:484–89. doi: 10.1128/jb.178.2.484-489.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson RV, Schuster SC, Simon MI. Expression of CheA fragments which define domains encoding kinase, phosphotransfer, and CheY binding activities. Biochemistry. 1993b;32:7623–29. doi: 10.1021/bi00081a004. [DOI] [PubMed] [Google Scholar]
- Tang H, Blair DF. Regulated underexpression of the FliM protein of Escherichia coli and evidence for a location in the flagellar motor distinct from the MotA/MotB torque generators. J Bacteriol. 1995;177:3485–95. doi: 10.1128/jb.177.12.3485-3495.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatsuno I, Homma M, Oosawa K, Kawagishi I. Signaling by the Escherichia coli aspartate chemoreceptor Tar with a single cytoplasmic domain per dimer. Science. 1996;274:423–25. doi: 10.1126/science.274.5286.423. [DOI] [PubMed] [Google Scholar]
- Tawa P, Stewart RC. Kinetics of CheA autophosphorylation and dephosphorylation reactions. Biochemistry. 1994;33:7917–24. doi: 10.1021/bi00191a019. [DOI] [PubMed] [Google Scholar]
- Terwilliger TC, Koshland DE. Sites of methyl esterification and deamidation on the aspartate receptor involved in chemotaxis. J Biol Chem. 1984;259:7719–25. [PubMed] [Google Scholar]
- Terwilliger TC, Wang JY, Koshland DE. Kinetics of receptor modification. The multiply methylated aspartate receptors involved in bacterial chemotaxis. J Biol Chem. 1986;261:10814–20. [PubMed] [Google Scholar]
- Tisa LS, Adler J. Calcium ions sre involved in Escherichia coli chemotaxis. Proc Natl Acad Sci USA. 1992;89:11804–8. doi: 10.1073/pnas.89.24.11804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toker AS, Kihara M, Macnab RM. Deletion analysis of the FliM flagellar switch protein of Salmonella typhimurium. J Bacteriol. 1996;178:7069–79. doi: 10.1128/jb.178.24.7069-7079.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volz K, Matsumura P. Crystal structure of Escherichia coli CheY refined at 1.7 Å resolution. J Biol Chem. 1991;266:15511–19. doi: 10.2210/pdb3chy/pdb. [DOI] [PubMed] [Google Scholar]
- Vyas NK, Vyas MN, Quiocho FA. Sugar and signal-transducer binding sites of the Escherichia coli galactose chemoreceptor protein. Science. 1988;242:1290–95. doi: 10.1126/science.3057628. [DOI] [PubMed] [Google Scholar]
- Vyas NK, Vyas MN, Quiocho FA. Comparison of the periplasmic receptors for L-arabinose, D-glucose, D-galactose, and D-ribose- structural and functional similarity. J Biol Chem. 1991;266:5226–37. [PubMed] [Google Scholar]
- Wang H, Matsumura P. Characterization of the CheAs/CheZ complex. A specific interaction resulting in enhanced dephosphorylating activity on CheY-phosphate. Mol Microbiol. 1996;19:695–703. doi: 10.1046/j.1365-2958.1996.393934.x. [DOI] [PubMed] [Google Scholar]
- Wang H, Matsumura P. Phosphorylating and dephosphorylating protein complexes in bacterial chemotaxis. J Bacteriol. 1997;179:287–89. doi: 10.1128/jb.179.1.287-289.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JX, Balazs YS, Thompson LK. Solid-state REDOR NMR distance measurements at the ligand site of a bacterial chemotaxis membrane receptor. Biochemistry. 1997;36:1699–703. doi: 10.1021/bi962578k. [DOI] [PubMed] [Google Scholar]
- Wang N, Shaulsky G, Escalante R, Loomis WF. A 2-component histidine kinase gene that functions in Dictyostelium development. EMBO J. 1996;15:3890–98. [PMC free article] [PubMed] [Google Scholar]
- Welch M, Oosawa K, Aizawa SI, Eisenbach M. Phosphorylation-dependent binding of a signal molecule to the flagellar switch of bacteria. Proc Natl Acad Sci USA. 1993;90:8787–91. doi: 10.1073/pnas.90.19.8787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch M, Oosawa K, Aizawa SI, Eisenbach M. Effects of phosphorylation, Mg2+, and conformation of the chemotaxis protein CheY on its binding to the flagellar switch protein FliM. Biochemistry. 1994;33:10470–76. doi: 10.1021/bi00200a031. [DOI] [PubMed] [Google Scholar]
- Wells JA. Structural and functional basis for hormone-binding and receptor oligomerization. Curr Opin Cell Biol. 1994;6:163–73. doi: 10.1016/0955-0674(94)90132-5. [DOI] [PubMed] [Google Scholar]
- West AH, Martinez-Hackert E, Stock AM. Crystal structure of the catalytic domain of the chemotaxis receptor methylesterase, CheB. J Mol Biol. 1995;250:276–90. doi: 10.1006/jmbi.1995.0376. [DOI] [PubMed] [Google Scholar]
- Woodward DE, Tyson R, Myerscough MR, Murray JD, Budrene EO, Berg HC. Spatiotemporal patterns generated by Salmonella typhimurium. Biophys J. 1995;68:2181–89. doi: 10.1016/S0006-3495(95)80400-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JR, Li JY, Li GY, Long DG, Weis RM. The receptor binding site for the methyltransferase of bacterial chemotaxis is distinct from the sites of methylation. Biochemistry. 1996;35:4984–93. doi: 10.1021/bi9530189. [DOI] [PubMed] [Google Scholar]
- Wu JR, Long DG, Weis RM. Reversible dissociation and unfolding of the Escherichia coli aspartate receptor cytoplasmic fragment. Biochemistry. 1995;34:3056–65. doi: 10.1021/bi00009a037. [DOI] [PubMed] [Google Scholar]
- Wylie D, Stock AM, Wong C-Y, Stock J. Sensory transduction in bacterial chemotaxis involves phosphotransfer between Che proteins. Biochem Biophys Res Comm. 1988;151:891–96. doi: 10.1016/s0006-291x(88)80365-6. [DOI] [PubMed] [Google Scholar]
- Yamaguchi S, Aizawa S-I, Kihara M, Isomura M, Jones CJ, Macnab RM. Genetic evidence for a switching and energy-transducing complex in the flagellar motor of Salmonella typhimurium. J Bacteriol. 1986;168:1172–79. doi: 10.1128/jb.168.3.1172-1179.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K, Imae Y. Cloning and characterization of the Salmonella typhimurium specific chemoreceptor Tcp for taxis to citrate and from phenol. Proc Natl Acad Sci USA. 1993;90:217–21. doi: 10.1073/pnas.90.1.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh JI, Biemann HP, Pandit J, Koshland DE, Kim S-H. The three-dimensional structure of the ligand-binding domain of a wild-type bacterial chemotaxis receptor. J Biol Chem. 1993;268:9787–92. [PubMed] [Google Scholar]
- Yeh JI, Biemann HP, Prive GG, Pandit J, Koshland DE, Kim SH. High resolution structures of the ligand binding domain of the wild type bacterial aspartate receptor. J Mol Biol. 1996;262:186–201. doi: 10.1006/jmbi.1996.0507. [DOI] [PubMed] [Google Scholar]
- Zhang YH, Conway C, Rosato M, Suh Y, Manson MD. Maltose chemotaxis involves residues in the N-terminal and C-terminal domains on the same face of maltose-binding protein. J Biol Chem. 1992;267:22813–20. [PubMed] [Google Scholar]
- Zhang YH, Mannering DE, Davidson AL, Yao NH, Manson MD. Maltose-binding protein containing an interdomain disulfide bridge confers a dominant-negative phenotype for transport and chemotaxis. J Biol Chem. 1996;271:17881–89. doi: 10.1074/jbc.271.30.17881. [DOI] [PubMed] [Google Scholar]
- Zhao RB, Amsler CD, Matsumura P, Khan S. FliG and FliM distribution in the Salmonella typhimurium cell and flagellar basal bodies. J Bacteriol. 1996;178:258–65. doi: 10.1128/jb.178.1.258-265.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou HJ, Dahlquist FW. Phosphotransfer site of the chemotaxis-specific protein kinase CheA as revealed by NMR. Biochemistry. 1997;36:699–710. doi: 10.1021/bi961663p. [DOI] [PubMed] [Google Scholar]
- Zhou HJ, Lowry DF, Swanson RV, Simon MI, Dahlquist FW. NMR studies of the phosphotransfer domain of the histidine kinase CheA from Escherichia coli. Assignments, secondary structure, general fold, and backbone dynamics. Biochemistry. 1995;34:13858–70. doi: 10.1021/bi00042a018. [DOI] [PubMed] [Google Scholar]