

Abstract
The transmembrane aspartate receptor of E. coli and S. typhimurium mediates cellular chemotaxis toward aspartate by regulating the activity of the cytoplasmic histidine kinase, CheA. Ligand binding results in transduction of a conformational signal through the membrane to the cytoplasmic domain where both kinase regulation and adaptation occur. Of particular interest is the linker region, E213 to Q258, which connects and transduces the conformational signal between the cytoplasmic end of the transmembrane signaling helix (α4/TM2) and the major methylation helix of the cytoplasmic domain (α6). This linker is crucial for stable folding and function of the homodimeric receptor. The present study uses cysteine and disulfide scanning mutagenesis to investigate the secondary structure and packing surfaces within the linker region. Chemical reactivity assays reveal that the linker consists of three distinct subdomains: two α-helices termed α4 and α5 and, between them, an ordered region of undetermined secondary structure. When cysteine is scanned through the helices, characteristic repeating patterns of solvent exposure and burial are observed. Activity assays, both in vivo and in vitro, indicate that each helix possesses a buried packing face that is crucial for proper receptor function. The interhelical subdomain is at least partially buried and is also crucial for proper receptor function. Disulfide scanning places helix α4 distal to the central axis of the homodimer, while helix α5 is found to lie at the subunit interface. Finally, sequence alignments suggest that all three linker subdomains are highly conserved among the large subfamily of histidine kinase-coupled sensory receptors that possess methylation sites for use in covalent adaptation.
The mechanism of signal transduction across lipid bilayers is a subject of intense interest since this process initiates many cellular signaling pathways. The chemosensory aspartate receptor of E. coli and S. typhimurium represents a large family of transmembrane receptors that provide extra-cellular regulation of both prokaryotic and eukaryotic histidine kinases (1–7). A large group of these receptors, including the aspartate receptor, serve as chemo-, photo-, osmo-, and thermosensors for cellular taxis and exhibit regions of high conservation in their cytoplasmic domains (1, 8). Family members characterized thus far are stable homodimers and transmit signals via intramolecular conformational changes that propagate through the membrane to the cytoplasm. The fact that chimeric receptors containing the periplasmic ligand binding domain of the aspartate receptor coupled to the cytoplasmic signaling domain of other chemoreceptors are functional suggests that the mechanism of transmembrane signaling is widely conserved within this family (9–12). It has also been demonstrated that a chimeric receptor replacing the cytoplasmic domain of the aspartate receptor with the kinase domain of the human insulin receptor exhibits aspartate regulation of tyrosine kinase activity (13, 14), suggesting that the mechanism of transmembrane signaling might be even more general.
The aspartate receptor is a component of the bacterial chemotaxis pathway, which allows the cell to migrate in gradients of attractant and repellent compounds (15, 16). This receptor, along with the histidine kinase CheA and the coupling protein CheW, forms a ternary complex that serves as the primary source of signal generation during a chemotactic response. The ternary complex is stable on the order of tens of minutes, regardless of the ligand binding state of the receptor (17–19). Attractant binding to the periplasmic domain of the receptor modulates the autophosphorylation activity of the cytoplasmic histidine kinase CheA (20, 21) and thereby controls the subsequent phosphotransfer to the response regulator CheY (7, 22). Phospho-activated CheY dissociates from the ternary complex and diffuses to the motor, where it regulates the direction of flagellar rotation by interaction with the motor switch components (23).
The receptor is a symmetric homodimer of 60 kDa subunits, and the structures of the periplasmic and trans-membrane domains are well characterized (24–29). The crystal structure of the periplasmic domain has revealed that each subunit forms a four-helix bundle exposed to the periplasm, with a pair of symmetric aspartate binding sites at the subunit interface (24–26). These sites show negative cooperativity such that binding of a single aspartate at one site conformationally occludes the other site (30, 31). The N- and C-terminal helices of each subunit (α1/TM1 and α4/TM2)1 extend through the plasma membrane as a four-helix bundle in which α1/TM1 and α1′/TM1′ are directly packed at the subunit interface with α4/TM2 and α4′/TM2′ lying on either side (27–29). Aspartate binding causes a small (~1.6 Å) piston-like displacement of the helix α4/TM2 (the signaling helix) in one subunit, breaking the symmetry of the dimer (32). Studies of the related chemoreceptor for ribose and galactose have demonstrated a similar piston displacement of the signaling helix (33–37), adding to the evidence provided by chimeric receptors that the transmembrane signal has a common mechanism among receptors that regulate histidine kinases.
Structurally, the cytoplasmic domain of the chemotaxis receptors is not as well understood as the periplasmic domain. Circular dichroism and hydrodynamic studies have indicated that the isolated domain is highly α-helical and elongated in shape (38–40). The soluble isolated domain exists in several oligomeric forms as well as being highly dynamic or perhaps molten globule in structure (38, 39, 41). Thus, studies of this domain in the native, full-length receptor are needed to extend and complement work focusing on the isolated domain. Cysteine and disulfide scanning mutagenesis is an ideal approach for such studies since it is independent of protein size, and can be carried out on the intact receptor in isolated bacterial membranes. Cysteine scanning has been used previously to identify secondary structural elements in various proteins (42–44), while disulfide scanning has been used to probe packing arrangements (27, 29, 34, 37, 45). Cysteine and disulfide scanning was recently employed to characterize the major adaptation helix (α6) of the cytoplasmic domain, which possesses regulatory glutamates that are the target of methyl-esterification by the adaptation branch of the chemotaxis pathway (46).
The current study focuses on the linker region of the cytoplasmic domain. This linker connects and transmits the conformational signal between the transmembrane signaling helix (α4/TM2) and the previously characterized major adaptation helix α6 (46) shown in schematic form in Figure 1. The linker is defined to begin at Arg-213, proposed to be the first cytoplasmic residue of the signaling helix, and extend through the major proteolysis site in the cytoplasmic domain at Arg-259. Evidence for the functional importance of the linker has come from work on heterodimers of the aspartate receptor in which the cytoplasmic domain of one subunit is truncated at several different points (47, 48), revealing that some or all of the linker residues 229–259 are necessary for receptor function. It follows that the linker is involved in critical inter- or intrasubunit interactions that maintain receptor activity. Moreover, four signal-locking point mutations have been identified in the linker of the homologous serine receptor (49, 50), providing further evidence for the functional significance of this region. Finally, a total of 22 of the 47 linker positions exhibit sequence identities spanning most of the 7 confirmed E. coli and S. typhimurium chemotaxis receptors, indicating selective pressure to maintain a conserved linker structure (8, 51). The present study is aimed at uncovering the basic secondary structural elements of the linker region as well as the residues that play the most important roles in transmembrane signaling.
Figure 1.

Schematic diagram of the full membrane-bound aspartate receptor. The structures of the periplasmic and transmembrane domains have been characterized by crystallographic and disulfide studies (24, 27–29, 46). The cylinders represent helices that were experimentally defined by those studies. Black circles represent sites of adaptive methylation, three of which lie on the major methylation helix α6 and one closer to the C-terminus (believed to be close in space to the first three). The linker region is defined as a segment that begins at R213 where the transmembrane signaling helix (α4/TM2) emerges into the cytoplasm, and extends to the major proteolytic hotspot R259 [arrow, (38)] just N-terminal to the methylation helix α6. The present cysteine and disulfide scanning study encompasses the boxed region including the entire linker (positions R213 to R259).
To probe the linker, cysteine substitutions are introduced at each consecutive position from I212 to S259. With this library of single cysteine-substituted receptors, the chemical reactivity at each position is measured to determine solvent exposure and identify secondary structural elements. The results reveal two segments that form stable helices termed α4 and α5 in both signaling states of the linker. Activity assays are used to measure the effects of each substitution on signaling of the receptor in vivo and in vitro, indicating that the buried packing surface of each helix plays a functional role in receptor signaling. Disulfide scanning places the more C-terminal helix α5 at the dimer interface where it packs against the symmetric compliment helix α5′ in the other subunit. A third segment of the linker, lying between the α4 and α5 helices, is found to have an ordered structure that is partially buried, although a specific secondary structure has not yet been determined. Models for the packing and roles of the newly defined structures are discussed. Finally, the present results are fully consistent with a prior cysteine and disulfide scanning study of residues 250–309, a range that included the C-terminal end of helix α5 as well as the major adaptation helix α6 (46).
MATERIALS AND METHODS
Materials
Receptor-containing membranes were isolated from E. coli RP3808 (Δ(cheA-cheZ)DE2209 tsr-1 leuB6 his-4 eda-50 rpsL136 [thi-1 Δ(gal-attl)DE99 ara-14 lacY1 mtl-1 xyl-5 tonA31 tsx-78]/mks/) (52) expressing the plasmid pSCF6 (29). Chemotactic swarm assays were carried out using the same plasmid in E. coli RP8611 (Δ(tsr)DE7028 Δ(tar-tap)DE5201 zbd:Tn5 Δ(trg)DE100 leuB6 his-4 rpsL136 [thi-1 ara-14 lacY1 mtl-1 xyl-5 tonA31 tsx-78]/CP362 of G. Hazelbauer via F. Dahlquist, pa/). Both strains above were kindly provided by John S. Parkinson (University of Utah) (52). Strains and plasmids used to express CheA (HB101/pMO4) and CheW (HB101/pME5) were kindly provided by Jeff Stock (Princeton University). The strain and plasmid used to express CheY (RBB455/pRBB40) were provided by Bob Bourret (University of North Carolina). 5-Iodoacetamidofluorescein was purchased from Molecular Probes, Inc. [γ-32P]ATP (6000 Ci/mmol) was obtained from Amersham. Mutagenic oligonucleotides were purchased from Life Technologies, Gibco-BRL.
Protein Engineering
Mutagenesis was performed on the phagemid pSCF6 which contains the gene for the S. typhimurium aspartate receptor under the control of its natural promoter (29). The Kunkel method (53) for site-directed mutagenesis was used with modifications described by the Mutagene mutagenesis kit (BioRad). Mutated plasmids were transformed into E. coli LM1035 for amplification and purified using the QIAprep spin column kit (Qiagen). The mutations were confirmed by PCR DNA sequencing with 32P-labeled primers and Sequitherm DNA polymerase (Epicenter Technologies).
Preparation of Receptor-Containing Cell Membranes
Wild-type and all mutant plasmids were transformed into E. coli RP3808 for receptor expression (mutant plasmids for positions 250–259 were graciously provided by Randal B. Bass) (46). Cultures were first inoculated in 2 mL of Luria Broth, then diluted 1/250 into 500 mL of Vogel Bonner Citrate minimal growth medium (54) containing 0.70% glycerol, 200 μg/mL MgSO4·7H2O, 2000 μg/mL citric acid·H2O, 10 000 μg/mL K2HPO4, 3500 μg/mL NaNH4·HPO4·4H2O, 40 μg/mL D,L-histidine, 20 μg/mL L-methionine, 20 μg/mL L-leucine, 20 μg/mL L-threonine, 1 μg/mL thiamin, and 150 μg/mL ampicillin. Cultures were incubated at 30 °C, shaking at 225 rpm for 17–18 h, and then harvested by centrifugation [Beckman JA-10 rotor at 8000 rpm (11300g) for 10 min].
Receptor-containing bacterial membranes were isolated using a procedure previously described by Chervitz and Falke (28) with several modifications. The cell pellets were resuspended in 5 mL of a low-salt buffer containing 100 mM sodium phosphate, pH 7.0 with NaOH, 10% glycerol, 10 mM EDTA, 50 mM DTT, 0.5 mM PMSF, and 1 mM 1,10-phenanthroline. The cells were then lysed by sonication (Heat Systems W-385 Sonicator) in an ice–NaCl–water bath with a macro tip (Mysonix, Inc.) 3 times for 17 s at ~75% maximum power, with 45 s cooling intervals between repetitions. The resulting solution was centrifuged [Beckman TLA100.3 rotor at 15 000 rpm (12000g) for 20 min] in order to pellet cell debris and any unbroken cells. After decanting, membranes were pelleted by ultracentrifugation [Beckman TLA100.3 rotor at 100 000 rpm (540000g) for 12 min]. The membranes were resuspended in 0.6 mL of a high-salt buffer containing 20 mM sodium phosphate, pH 7.0 with NaOH, 2 M KCl, 10% glycerol, 10 mM EDTA, 5 mM DTT, 0.5 mM PMSF, and 1 mM 1,10-phenanthroline. Resuspension was achieved by sonication with a 1/8 in. microtip 3 times for 17 s, at 25% maximum power, with 45 s cooling intervals. Resuspended membranes were diluted to 2.8 mL with the same buffer before being pelleted as above. The high-salt wash was then repeated as above except that the buffer contained no DTT or 1,10-phenanthroline. Next, the membranes were washed as above in the final buffer, containing 20 mM sodium phosphate, pH 7.0 with NaOH, 10% glycerol, 0.1 mM EDTA, and 0.5 mM PMSF, and pelleted once more. The membrane pellets were finally resuspended in 300 μL of final buffer, aliquoted, frozen in liquid nitrogen, and stored at −70 °C.
The total protein yield was determined by a BCA assay (55). To determine the fraction of total protein consisting of the aspartate receptor, membrane components were resolved on a 10% SDS–PAGE gel with an acrylamide:bis-(acrylamide) ratio of 40:0.2. The relative intensities of the Coomassie-stained protein bands were quantitated by laser densitometry (Ultroscan XL, LKB). The receptor typically comprised 10–20% of the total protein. Depending on the total protein, final aspartate receptor concentrations varied between 12 and 40 μM.
Preparation of Soluble Chemotaxis Components
The soluble chemotaxis components CheA, CheW, and CheY were individually purified using the strains and plasmids indicated above and protocols that have been described in detail previously (56–58).
Chemical Reactivity Assays
Receptor-containing membrane samples were diluted to 5 μM receptor monomer in 10 mM sodium phosphate, pH 6.5 with HCl, 50 mM NaCl, 50 mM KCl, 1 mM EDTA, as described previously (46). The above buffer was prepared with and without 2.5 mM aspartate. Reactions were initiated by adding 5-iodoacetamidofluorescein (IAF) from a 5 mM stock in N,N′-dimethylformamide to a final concentration of 250 μM. Each reaction was incubated at 25 °C for 5 min; then a 20 μL aliquot was removed and quenched by mixing with β-mercaptoethanol to 80 mM in order to destroy the remaining IAF. The remaining 20 μL of reaction was brought to 2% SDS and heated to 95 °C to promote unfolding and complete reaction of all cysteines. After 3 min, this reaction was quenched as above. Neither a longer incubation nor subsequent addition of IAF caused the denatured aliquot to react further, indicating that the labeling reaction had reached completion. Both aliquots were resolved on a 10% SDS–PAGE gel with an acrylamide:bis(acrylamide) ratio of 40: 0.2, after addition of 5 μL of 4× Laemmli nonreducing sample buffer (250 mM Tris, pH 6.8 with HCl, 4% SDS, 40% glycerol). The fluorescence of the aspartate receptor band was quantitated by imaging the gel with a digital camera and UV transilluminator system (Alpha Innotech Corp.) using a 470 nm long-pass filter. Quantitation software was used to integrate the fluorescence intensity volume of each band. The same digital camera system was used to quantitate the protein in each receptor band after Coomassie staining which allowed normalization of fluorescence to protein. The normalized fluorescence of the folded sample from the 5 min, 25 °C incubation was then divided by the normalized fluorescence of the denatured reaction, yielding the relative labeling termed “chemical reactivity”. To confirm the specificity of cysteine labeling, the above reaction was carried out on wild-type protein (lacking cysteine), yielding no significant receptor fluorescence. The fact that cysteine-containing receptors denatured either immediately or after the 5 min incubation yielded the same extent of labeling indicated that no competing reactions occurred during the labeling incubation.
Analysis of Signaling Function in Vivo
Chemotactic swarm assays (15) were carried out by first transforming each mutant pSCF6 plasmid into E. coli RP8611 which has been deleted for wild-type aspartate receptor. Starter cultures were grown in Luria Broth at 37 °C with shaking for 5 h; then 5 μL was spotted onto 0.23% agar minimal plates containing Vogel Bonner Citrate medium supplemented with 0.1% glycerol, 20 mM lactate, 40 μg/mL D,L-histidine, 20 μg/mL L-leucine, 1 μg/mL thiamin, and 100 μg/mL ampicillin (59). These plates were prepared with or without 0.1 mM L-aspartate. Plates were incubated at 30 °C, and colony diameters were measured at 3–4 h intervals, starting approximately 18 h after spotting. Swarm rates were determined by least-squares linear best-fit to the slope of diameter as a function of time. To account for pseudotaxis (60) and any other non-aspartate-specific taxis, the rate of swarming in the absence of aspartate was subtracted from the rate of swarming in the presence of aspartate. The resulting aspartate-specific swarm rate for each engineered receptor was normalized to the corresponding rate for the wild-type receptor (typically 0.6 mm/h), which was determined in parallel as a positive control. Cells transformed with vector lacking the receptor gene, as a negative control, exhibited at least 10-fold slower expansion of the colony diameter, and no aspartate-specific swarming.
Analysis of Signaling Function in Vitro
To observe more specifically the ability of each receptor to regulate the activity of CheA, a coupled phosphorylation assay was used to quantitate the transfer of radiolabeled phosphate from the receptor–CheA–CheW ternary complex to CheY (20, 61). The procedure was a modification of that described in detail previously (29). Briefly, isolated E. coli membranes containing 6 μM receptor monomer, after being either oxidized or reduced as described below, were combined with the purified proteins CheW (2 μM), CheA (0.25 μM monomer), and CheY (10 μM) in 50 mM Tris, pH 7.5 with HCl, 50 mM KCl, and 5 mM MgCl2 and incubated at 23 °C for 30 min to equilibrate the receptor–kinase complex. The reaction was initiated by the addition of [γ-32P]ATP (4000–8000 cpm/pmol) to a final concentration of 0.1 mM. Reactions were terminated at 10 s by mixing with phospho-quench (2× Laemmli nonreducing sample buffer plus 25 mM EDTA). The quenched reactions were resolved on a Laemmli gel consisting of a stacking gel of 9.6% acrylamide and 0.048% bis(acrylamide) and a separatory gel of 16% acrylamide and 0.5% bis(acrylamide), as well as 15% urea. The gels were dried immediately after electrophoresis, and the phospho-CheY band was quantitated by phosphorimaging. The resulting extents for engineered receptors were normalized to that for the wild-type receptor, measured in parallel as a positive control, yielding a phosphotransfer rate relative to the wild-type receptor–kinase complex.
Oxidation and Reduction of Receptors
To ensure that the engineered receptors fully disulfide-linked or reduced for phosphorylation assays, membranes containing 12 μM receptor monomer were either (1) oxidized by addition of the redox catalyst Cu(II)(1,10-phenanthroline)3 (200 μM) in the presence of ambient-dissolved oxygen for 20 min at 37 °C (62) or (2) reduced by addition of DTT (50 mM) to ensure elimination of disulfides. Oxidation and reduction reactions were carried out in the final buffer (see above receptor preparation) without the addition of PMSF.
Other experiments measured the efficiency of disulfide formation as an indicator of sulfhydryl proximity. These experiments used receptor-containing membranes in the same oxidation reaction as above, to which 5 mM EDTA was also added to slow the rate of metal-catalyzed disulfide formation (63). The oxidation reaction was initiated by addition of 1 mM Cu(II)(1,10-phenanthroline)3 in the presence of ambient oxygen for 10 s at 25 °C. Aliquots of all oxidation and reduction reactions were analyzed by electrophoresis (described under Chemical Reactivity Assays) in order to observe the extent of dimer formation for each engineered receptor.
Error Determination
The error bars shown in each assay represent the standard deviation of the mean for ≥3 values.
RESULTS
Engineering and Expression of Cysteine-Containing Receptors
Single-cysteine substitutions were engineered at each consecutive position from I212 through R259, which encompasses the entire linker. Since the wild-type receptor contains no intrinsic cysteine residues, each substituted cysteine was unique. The engineered receptors were expressed in the E. coli strain RP3808 which lacks the wild-type aspartate and serine chemoreceptors as well as the soluble components of the chemotaxis pathway (52). Because the adaptation enzymes CheR and CheB were not present, the methylation state of the receptor population was homogeneous. Expression of the engineered receptors in RP3808 was generally between 25 and 100% that of wild type except for four mutant receptors, P219C, I230C, K237C, and S242C, which showed no detectable accumulation of membrane-bound receptor under the conditions described. It follows that these four side chains are located at positions that are critical to receptor assembly or stability.
Use of Chemical Reactivity To Determine Relative Solvent Exposure
The reactivity of an engineered cysteine toward a bulky, aqueous probe yields direct information on the solvent exposure of the corresponding position. In the present study, the water-soluble alkylating reagent 5-iodoacetamidofluorescein (IAF) was used as a cysteine-specific probe. The overall experimental design described by Danielson et al. (46) was followed with modifications to optimize for the higher average reactivity of the linker region. Briefly, 5 μM receptor in isolated E. coli membranes was incubated with 250 μM IAF at 25 °C for 5 min. One aliquot (the folded sample) was removed and quenched with β-mercaptoethanol, while the second aliquot (the unfolded sample) was removed simultaneously, but denatured with SDS and heat for 3 min to allow complete reaction before quenching. The ratio of receptor labeling in the two samples (folded/unfolded), termed the chemical reactivity, was quantitated by integrating the fluorescence intensity of the receptor band after electrophoresis on SDS–polyacrylamide gels.
Following measurement of chemical reactivities for a large set of consecutive cysteines, classic amphiphilic secondary structures were identified by their characteristic repeating patterns of buried or exposed residues: 3.6 residues per repeat for a canonical α-helix; 3.5 residues per repeat for a coiled-coil α-helix, and 2 residues per repeat for a β-strand. As a positive control, the chemical reactivities of positions 95 through 103 in periplasmic helix α2 were measured and compared to the calculated solvent accessibilities revealed by the known crystal structure of this region (64). Figure 2 presents this comparison, which yields excellent agreement between the chemical reactivities measured in the membrane-bound receptor and the solvent accessibility observed in the high-resolution structure of the isolated periplasmic domain. Both methods reveal that ligand binding has little or no effect on the position of helix α2 (Figure 2).
Figure 2.
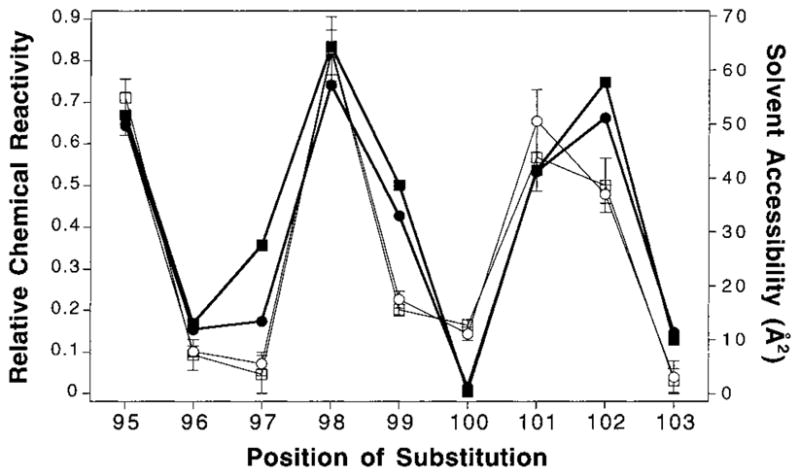
Comparison of chemical reactivity and calculated solvent accessibility in the known helix α2 of the periplasmic domain. Chemical reactivities at 25 °C were measured for engineered cysteines at positions 95 through 103 in the periplasmic domain of the membrane-bound receptor using 5-iodoacetamidofluorescein as a probe, and were normalized to the corresponding reactivities of the unfolded receptor (see text). In addition, the solvent accessibility of each position in the available crystal structure of the periplasmic domain (24) was calculated using the method of Richards (64). The correlation between these data is shown by plotting the relative chemical reactivities measured with and without saturating aspartate (open squares and open circles, respectively, fine line) along with the calculated solvent accessibility of each position in the aspartate-occupied and apostructures of the periplasmic domain (closed squares and closed circles, respectively, bold line).
Overall, these control experiments confirm that solvent exposure is the main determinant of chemical reactivity under the assay conditions used. By contrast, the local electrostatic and steric environments of each cysteine substitution have only minor effects on chemical reactivities. It follows that chemical reactivity measurements can accurately identify buried and exposed positions in the aspartate receptor.
Relative Solvent Exposure of Linker Positions in Both Signaling States
The chemical reactivities of engineered cysteines at positions 212 through 259 were measured using the same assay and are displayed in Figure 3. Breaks in the plot indicate positions where cysteine substitution blocked expression or accumulation of membrane-bound receptor, thereby preventing analysis. Interestingly, the average chemical reactivity is seen to gradually decrease as the cysteine is scanned from the N-terminal to C-terminal end of the linker. For this reason, it is necessary to divide the linker into three segments (Figure 3) so that local criteria of solvent exposure and burial can be defined as follows. For each segment, the range of chemical reactivity is defined by the highest and lowest measured reactivities in that segment. This range is then used to classify the chemical reactivities of individual residues as buried (lower 30% of the range), intermediate (mid 30% of the range), and exposed (upper 40% of the range). An important advantage of such local classification is that it maximizes the probability of identifying local periodic fluctuations of exposure and burial arising from amphiphilic secondary structure.
Figure 3.

Chemical reactivity of cysteine scanning positions in the linker region of the cytoplasmic domain. Chemical reactivities at 25 °C were measured for engineered cysteines in the membrane-bound receptor using 5-iodoacetamidofluorescein as a probe, and were normalized to the corresponding reactivities of the unfolded receptor (see text). Shown are the chemical reactivities obtained in the presence (solid circles, connected by line) or absence (open circles, no line) of saturating aspartate. The gaps at positions 219, 230, 237, and 242 represent positions where cysteine substitution blocked expression or accumulation of the receptor. The three major segments (I–III) defined by vertical dashed lines represent the three distinct structural subdomains of the linker: (i) residues 213 through 228; (ii) residues 229 through 242; and (iii) residues 243 through 258. Criteria for exposure were defined within each segment as buried (lower 30%), intermediate (mid 30%), and exposed (upper 40%) where the highest and lowest reactivities in each segment define the upper and lower limits of the range, respectively (see Results).
The first subdomain begins approximately where the transmembrane signaling helix α4/TM2 reaches the cytoplasm, R213, and extends through R228. Chemical reactivities in this region exhibit an oscillating pattern with a periodicity characteristic of an α-helix possessing an exposed face and a buried face (see Figure 6 under Discussion). No evidence of β-strand periodicity was detected in this segment. The simplest interpretation is that this region represents the cytoplasmic extension of the signaling helix α4/TM2.
Figure 6.

Helical wheel projections of the two proposed helices displaying the chemical reactivity of each cysteine substitution. Filled boxes indicate highly buried positions, and open boxes indicate highly exposed positions (defined in Figure 3), revealing the strong amphiphilic nature of both modeled helices. (A) Helix α4 displays strong segregation of buried and exposed residues to opposite faces of the canonical 3.6 residue per turn helix. It should be noted that R213 is located at the membrane interface, and that the current data do not distinguish whether protection of this residue from solvent results from the defined packing interaction or from partial burial in the membrane. (B) Helix α5 also displays clear buried and exposed faces, and is modeled as a 3.5 residue per turn helix (see Discussion). The a and d positions of this helix are proposed to form a coiled-coil interaction with the symmetric helix α5′ at the dimer interface. Parentheses indicate substitutions that block receptor expression or accumulation in the membrane.
In the middle segment corresponding to residues 229 through 242, the range of measured chemical reactivities is over 2-fold smaller than in the other segments, yielding little oscillation and no information regarding secondary structure. The lack of highly exposed positions, however, suggests that this segment has an ordered structure and is at least partially shielded from solvent. Moreover, this segment is unusually important for receptor assembly or stability since cysteine substitution at 3 of its 13 positions prevents detectable accumulation of membrane-bound receptor (see above).
The final segment, positions 243 through 258, displays a clear oscillation between buried and exposed residues, again with periodicity indicating α-helical structure (see Figure 6 under Discussion). This segment, termed helix α5, can be modeled as either a 3.6 residue per turn canonical helix or a 3.5 residue per turn helix involved in an unidentified coiled-coil interaction. The proposed C-terminal end of helix α5 falls at the major proteolytic hotspot in the C-terminal domain, R259, that has been previously described (38).
Chemical reactivities were measured throughout the linker in both the absence and presence of 1 mM aspartate in order to probe for a ligand-induced conformational change. Previous studies have shown that 1 mM aspartate is sufficient to saturate the receptor with ligand, even in mutant receptors possessing engineered cytoplasmic cysteines (46). Although a number of small changes greater than error were observed at positions N218, R222, T240, E249, G252, and S260, most linker positions show no detectable change in solvent exposure upon ligand binding. The small magnitude (≤20%) and limited range of the observed exposure changes indicate that the ligand-induced conformational rearrangement of the linker is quite subtle. Such a picture is consistent with previous findings for the periplasmic and transmembrane regions of the receptor, which also revealed subtle rearrangements of less than 2 Å (32, 35).
Identification of Residues Crucial to Receptor Function in Vivo
The effect of each cysteine substitution on chemotactic ability was assessed in order to determine which residues play a critical role in receptor function in the complete chemotaxis signaling pathway. Each mutant receptor was first transformed into E. coli RP8611, a strain that has been deleted for the wild-type aspartate receptor (65) such that chemotaxis toward aspartate can be restored only by the presence of a functional receptor. Starter cultures were spotted onto minimal medium soft agar plates containing or lacking 0.1 mM aspartate (29). Outward expansion of the colonies depended largely on the ability of each culture to chemotax up the self-created radial aspartate gradient. The rate of expansion of each colony was measured over a period of 14–16 h, and the difference in chemotaxis rates observed on plates with and without aspartate was reported as the aspartate-specific swarm rate normalized to that of a wild-type control. It should be noted that this assay is designed to detect the receptor mutations that cause the most serious functional defects, since receptor overexpression and the adaptation pathway will compensate for small perturbations.
Figure 4A summarizes the normalized aspartate-specific swarm rates for all of the cysteine-substituted receptors tested in the scanned region 212–259. Swarm rates of the cysteine-containing receptors varied from 0 to nearly 2-fold that of wild type, reflecting a wide array of effects of individual substitutions although the majority of positions were non-perturbing. Highly inhibited receptors were defined as those exhibiting a swarm rate less than 30% that of wild-type receptor. Of the 48 cysteine substitutions, 11 yielded highly inhibited receptors. Of these 11, 4 (P219C, I230C, K237C, S242C) did not accumulate in E. coli membranes, as described above, while the remaining 7 yielded expression levels similar to wild type. Focusing on the five inhibitory substitutions within the two newly defined cytoplasmic helices reveals that all of them fall on the buried packing faces (L217C, I227C, G246C, L250C, Q258C), indicating that these interfaces are critical to receptor function in vivo (see Figure 7 in Discussion).
Figure 4.

Effects of cysteine substitutions on activity in vivo and in vitro. (A) Rates of aspartate-specific chemotactic swarming relative to wild-type receptor (29). Plotted are the chemotactic swarm rates of an E. coli strain overexpressing each of the cysteine-substituted receptors, measured at 30 °C on minimal medium soft agar plates. The open circles indicate positions where cysteine substitution is known to prevent detectable accumulation of membrane-bound receptor. Receptors that yielded swarm rates below 0.3 (dashed line) are designated as inhibitory. For each mutant receptor, the plotted aspartate-specific swarm rate was determined as the difference in swarm rates measured in the presence and absence of aspartate, normalized to the corresponding rate difference measured for wild-type receptor (typically 0.6 mm/h) (B) Rates of CheY phosphorylation by the reduced receptor–CheA–CheW ternary complex (29). The core of the chemotaxis pathway was reconstituted using wild-type receptor and each of the substituted receptors with separately purified ternary complex components CheA and CheW, and the response regulator CheY. The ability of each receptor to regulate the histidine kinase activity of CheA was measured by monitoring the rate of formation of [32P]-phospho-CheY at 25 °C, normalized to the wild-type rate in the absence of aspartate. The wild-type receptor yields a normalized rate of 1.0 in the absence of aspartate (closed circles, fine line) and is down-regulated effectively to 0.0 by the presence of saturating aspartate (open circles, bold line). Mutations that yield over 150% (upper line) or less than 10% (lower line) of the wild-type rate in the absence of aspartate are designated super-activating or inhibitory, respectively.
Figure 7.

Helical wheel projections of (A) helix α4 and (B) helix α5 displaying effects of cysteine substitutions on receptor activity. Positions where substitution inhibits chemotactic swarming in vivo are shown in black boxes. The open box at P219 indicates a cysteine substitution that prevents expression or accumulation of the receptor. Black squares indicate substitutions that inhibit kinase activation, and open squares indicate those that result in kinase super-activation (defined in Figure 4). The star at position 227 indicates constitutive activation of the kinase upon substitution.
Identification of Residues Crucial to Receptor Function in Vitro
A more sensitive functional assay was carried out in order to test the effects of cysteine substitution specifically on the ability to regulate CheA kinase activity (20, 29, 61). Isolated membranes containing the reduced receptor were mixed with purified proteins to reconstitute the ternary complex (receptor, CheA, and CheW) as well as the response regulator CheY. Kinase activity was assessed by the rate of phosphotransfer from the ternary complex to CheY, observing [32P]phospho-CheY formation by SDS–PAGE. CheY was added in molar excess to ensure that receptor-regulated autophosphorylation of CheA in the ternary complex was the rate-limiting step in this reaction. The assay was carried out in the presence and absence of saturating aspartate to observe the ability of each receptor to switch between signaling states.
Figure 4B is a summary of the effects of cysteine substitution on the kinase activity of the ternary complex in both the apo- and aspartate-bound states, normalized to wild-type activity. The wild-type receptor activates CheA in the absence of aspartate and down-regulates the kinase over 100-fold upon binding aspartate. Inhibitory substitutions were defined as those that retained less than 10% of the wild-type kinase activation in the absence of aspartate. Of the 10 cysteine substitutions that blocked kinase activation, 5 fall in helical regions, 4 fall in the segment between the helices, and 1 is at the membrane interface. Four of the five inhibitory helical positions (L217, L220, G246, I247) are located on the hydrophobic, buried faces of the two helical regions defined by chemical reactivity patterns, further indicating that the packing interfaces of both helices play an important role in the proper signaling function of the aspartate receptor. Notably, the highest density of inhibitory cysteines is found within the region between the two helices where four consecutive substitutions (G233C, D234C, L235C, T236C) block kinase activation, indicating the functional importance of this region. Finally, the I227C mutation on the buried surface of helix α4 has a unique effect on kinase regulation: it causes constitutive receptor activation such that aspartate binding reduces kinase activity by less than 28%. This finding indicates that the I227 residue is critical for normal kinase down-regulation, and explains the observation that the I227C substitution inhibits chemotactic swarming in vivo, despite retaining the ability to stimulate kinase activity.
The analyses of receptor activity described above have each involved the reduced state of the cysteine-containing receptor, maintained either by (i) the reducing environment of the cytoplasm in the chemotactic swarm assay or (ii) chemical reduction of the receptor in the receptor-coupled kinase assay. To probe the effects of intersubunit disulfide bonds on receptor activity, isolated membranes containing the engineered receptors were oxidized prior to reconstitution of the complex, yielding 50–80% covalent dimer formation. After this treatment, the wild-type receptor remained active, but nearly all of the disulfide-containing receptors were unable to activate the ternary complex, indicating that intersubunit disulfide formation in the linker region is generally highly perturbing to receptor function. The only two exceptions were T253C and H256C which retain ~45% of wild-type activity. Both positions are in α5, placing this helix near the subunit interface.
Efficiency of Disulfide Formation as a Measure of Sulfhydryl Proximity
The rate of disulfide formation is sensitive to the frequency of sulfhydryl collisions, which generally increases with proximity in the average structure (66–69). As previously noted, the cytoplasmic domain is quite dynamic, which leads to rapid intersubunit disulfide formation for most engineered cysteine positions (41, 46). Recently, however, a mild oxidation system capable of identifying the most rapidly formed disulfides in the cytoplasmic domain has been described (63). Mild oxidation of the cysteines scanned in the present study reveals four positions that form intersubunit disulfides significantly faster than all others: S247, S250, S254, and S257 shown in Figure 5. All four of the noted cysteines lie on the buried face of helix α5, providing additional evidence that this helix packs against its symmetric counterpart at the dimer interface.
Figure 5.

Efficiency of disulfide formation in the linker region. Formation of each disulfide bond between a symmetric pair of engineered cysteines in the two subunits of the homodimer was catalyzed by the addition of Cu(II)(1,10-phenanthroline)3 to a mild oxidation system (see text). Reactions at 25 °C were quenched after 10 s, and intersubunit disulfide formation was detected by a gel shift in SDS–PAGE. Plotted is the fraction of disulfide formed (dimer/total) for each cysteine substitution in the linker region. The notations a and d above the data refer to positions in the conserved heptad repeating coiled-coil motif seen in sequence alignments (8, 51). The dashed lines indicate the boundaries of the three major subdomains discussed, while breaks in the plot indicate cysteine substitutions that blocked receptor expression or accumulation.
DISCUSSION
The present study further demonstrates the utility of cysteine and disulfide scanning mutagenesis in probing basic secondary structural elements and functionally important regions of an unknown structure. This study reveals that the linker region of the aspartate receptor is comprised of three structurally distinct segments. Two of these segments (213–228, 243–258) have been identified as surface-exposed α-helices by distinct repeating patterns of exposed and buried residues in chemical reactivity assays. Both of these α-helices (α4 and α5) are strongly amphiphilic, which is clearly illustrated by mapping the substitutions onto helical wheel projections as in Figure 6. The helical projections for both α4 and α5 display the strong segregation of nonpolar residues to the buried faces of the helices and charged residues to the exposed faces. Furthermore, Figure 7 shows that the cysteine substitutions that perturb receptor function are highly segregated to the buried helix faces, indicating that the packed face of each α-helix is crucial for normal receptor activity.
Between the α4 and α5 helices, a stretch of 14 residues (229–242) is proposed to form a well-structured, partially buried subdomain based on its relatively low average solvent accessibility of the engineered cysteines (~40% of the denatured control) and its lack of highly exposed residues. The secondary structure of this region is not defined by the present results. The fact that three substitutions in this region prevent detectable accumulation of membrane-bound receptor in vivo, while four others yield nonfunctional receptors in vitro, suggests that this subdomain is critical to the proper folding, stability, and function of the receptor. Figure 8 summarizes the three segments of the linker and proposes a structural model that is consistent with current evidence. In this structure, helices α5 and α6 lie at the dimer interface while helix α4 and the interhelical subdomain are more distal from the central axis.
Figure 8.

Model of the secondary structural features of the cytoplasmic linker region, as suggested by the currently available data (see Discussion). Helix α4 continues from the transmembrane signaling helix α4/TM2 into the cytoplasm, where it is distal to the central axis of the dimer. This helix is followed by an ordered region of undefined secondary structure (oval) that is also distal to the central axis and becomes helix α5 at its C-terminus. Finally, the two symmetric α5 and α5′ helices of the dimer pack against each other to form a coiled-coil at the subunit interface.
The transmembrane signal causes a quite subtle rearrangement of the proposed structure, in the membrane-bound receptor, since only minor aspartate-induced changes in solvent exposure are detected by the chemical reactivity assay. At present, however, we cannot rule out a more dramatic signal-induced conformational change in the full receptor–kinase complex, since, for technical reasons, the chemical reactivity assays are carried out in the absence of CheA and CheW. Yet previous results for the periplasmic and transmembrane domains indicate that these regions undergo a quite subtle ligand-induced conformational change even in the full receptor–kinase ternary complex (1, 24, 32), so the proposed small magnitude of conformational rearrangement in the linker seems plausible. Following is a detailed discussion of the structural information available for each of the linker subdomains.
The Cytoplasmic Region of Helix α4
The first helix of the cytoplasmic domain, α4, is proposed to be an extension of the continuous transmembrane signaling helix α4/TM2, from the membrane interface, R213, into the cytoplasm to approximately R228. The periodicity of buried and exposed residues identifies this as a canonical 3.6 residue per turn α-helix (Figure 6A). The only discrepancy in the canonical pattern is position L220, which is identified as an exposed position by the chemical reactivity assay but falls on the buried face of the helix (Figure 6A). The simplest explanation for this apparent discrepancy is that the native leucine side chain is essential for stabilizing the helix packing interaction, such that the L220C substitution disrupts the packing surface and exposes the mutant side chain to solvent. This picture predicts that the L220C mutation will perturb receptor function, and indeed the mutant receptor is unable to generate kinase activation in vitro (Figures 4B and 7B).
In short, all the available evidence is consistent with the identification of residues R213 through R228 as the cytoplasmic extension helix α4/TM2. This cytoplasmic extension is a well-conserved structural element since four of its six packing face positions (L216, L217, L220, I227) exhibit a hydrophobic side chain in all seven of the E. coli and S. typhimurium chemotaxis receptors, while a fifth buried position (I224) is hydrophobic in six of the seven chemoreceptors. The precise range of the buried packing face on the cytoplasmic extension is not yet clear: in the aspartate receptor, it may begin as early as position R213 or as late as L216 (Figure 6A). The low chemical reactivity of R213C (Figure 3) could stem either from burial within the membrane or from burial within the packing region.
While considering possible molecular models for α4, attention should be given to the fully conserved proline at position 219, where cysteine substitution prevented detectable expression of the membrane-bound receptor in vivo, suggesting that P219 is critical for proper folding or stability of the cytoplasmic domain. Prolines observed in α-helices of known protein crystal structures introduce a kink into the helix axis ranging from 9° to 40° (70, 71). The kink in α4 due to P219 does not disrupt the observed packing face of the helix since modeling of the α4 residues onto proline-containing helices in the Brookhaven databank revealed that the experimentally determined buried residues remain on the same face of the bent helix throughout the range of possible kink angles (data not shown). Another noteworthy position on helix α4 is the buried residue I227, at which a cysteine substitution causes constitutive activation of the kinase CheA in the reconstituted ternary complex. The native isoleucine residue is proposed to be involved in a key steric or hydrophobic interaction that is required to generate the kinase down-regulating state of the receptor. Further support of the importance of this residue is that it is conserved within the eight bacterial chemoreceptors with only two exceptions, both of which utilize another large hydrophobic residue, phenylalanine.
A final intriguing feature of the cytoplasmic extension of α4 is that the buried packing face is oriented away from the central axis of the receptor dimer, which was previously defined by disulfide mapping studies of α4/TM2 in the transmembrane region (27, 29). Further evidence that the packing face does not lie at the dimer interface is provided by the low efficiencies of intersubunit disulfide formation observed for cysteines in this region (Figure 5). This poses the question of what is packing against the outside face of helix α4 in the cytoplasm. Figure 9 illustrates three possible arrangements that could provide the observed protection against chemical labeling whereby: (1) another element of the linker or cytoplasmic domain packs against the outer face of α4; or (2) the buried face of α4 is protected by intermolecular interactions between receptor dimers, which are known to cluster under certain conditions; or (3) a break in helix α4 enables its protected face to pack against the cytoplasmic face of the membrane. Currently, the first possibility seems most plausible since receptor clustering does not occur in the absence of CheA and CheW (72) (corresponding to the labeling assay conditions), and there are no obvious helix-breaking residues near the cytoplasmic end of the transmembrane helix α4/TM2.
Figure 9.
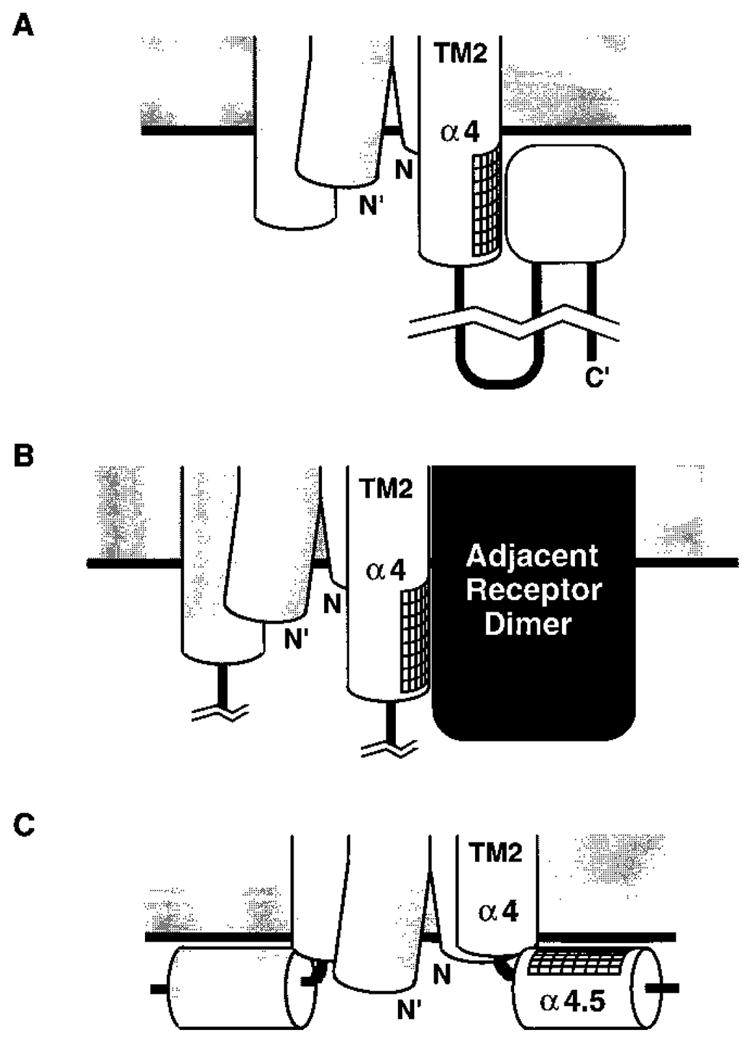
Possible packing arrangements that are consistent with the experimentally determined buried face of helix α4. The fact that helix α4 displays a packing face oriented away from the dimer interface could be explained by: (A) the packing of an undetermined element within the cytoplasmic domain against α4; (B) the packing of another receptor dimer against α4; or (C) the packing of α4 against the surface of the membrane, which would require a break in the helix. Currently, model (A) seems most plausible (see Discussion).
The Interhelical Region
The segment lying C-terminal to helix α4, spanning residues E229–S242, did not yield a periodic solvent exposure pattern in the chemical reactivity assays. As a result, no specific secondary structure could be proposed or ruled out for this region of the linker. The lack of highly reactive positions in the chemical labeling assays suggests that this region is at least partially buried and thus has a well ordered structure, although it does not appear to be located at the dimer interface since the efficiency of intersubunit disulfide formation is low in this region (Figure 5). This structure is unlikely to be a simple extension of helix α4 since, in all seven chemoreceptors, there are one or two glycine residues that would likely break the helix near the proposed C-terminus of helix α4, corresponding to S232 and G233 in the S. typhimurium aspartate receptor or G232 and G233 in the E. coli aspartate receptor.
The importance of this subdomain for both receptor folding and function is revealed by point mutations. In the related serine receptor, three of the four signal-locking point mutations of the linker are found in the interhelical region (50). In the present study, three substitutions (I230C, K237C, and S242C) yield receptors that fail to accumulate in the membrane during expression in vivo, indicating these residues play a critical role in folding or stability (Figure 4A). Moreover, a stretch of four consecutive cysteine substitutions (G233C, D234C, L235C, T236C) diminishes the ability of receptor to activate the histidine kinase CheA (Figure 4B). Finally, 5 of the 13 residues within the region are 60–100% conserved among the 7 known E. coli and S. typhimurium chemoreceptor sequences (I230, A231, G233, D234, L235), and 3 of these side chains (G233, D234, L235) are conserved in at least 80% of the 56 known sequences of the chemo-, photo-, osmo-, and thermoreceptor superfamily (8, 70). It follows that the interhelical region is crucial for regulation and is a signature element of the conserved cytoplasmic domain motif.
Cytoplasmic Helix α5
The C-terminal helix of the linker, α5, is proposed to begin at G243 and continue through Q258. The pattern of solvent exposure and burial in this segment is clearly helical, although the periodicity cannot be distinguished between 3.5 and 3.6 residues per turn. The helix displays a heptad repeating pattern of hydrophobic residues (a b c d e f g, where residues a and d are hydrophobic) which is highly conserved throughout the 56 member superfamily (8, 51, 73, 74). Thus, the segment was modeled as a 3.5 residue per turn helix that participates in a coiled-coil structure (Figure 6B). The simplest packing model is one in which the symmetric α5 and α5′ helices of the dimer form a coiled-coil by packing against each other at the subunit interface. This helix–helix interaction explains the four positions in α5 (247, 250, 254, and 257) that yield the highest extents of disulfide formation in the linker, since all four of these positions coincide with the a and d contact positions of the coiled-coil motif (Figure 5). The observed conservation of the heptad repeat further indicates that the α5-α5′ coiled-coil is likely to be a characteristic structural element of the cytoplasmic domain motif in the full receptor superfamily.
In conclusion, the current study provides an experimentally based picture of the secondary structure of the conserved linker region which is critical to receptor stability and function. The present results define two helices separated by an ordered interhelical segment as well as specific positions that are crucial to the characteristic fold and function of the linker motif. These findings provide the foundation for future studies designed to further elucidate the structure of the interhelical segment, the detailed packing arrangement of the helices and interhelical region, and the nature of the signal-induced conformational change within the linker.
Acknowledgments
We thank Randal B. Bass and Mark A. Danielson for helpful discussions on experimentation and comments on the manuscript.
Footnotes
Support provided by NIH Grant GM40731.
Abbreviations: TM, transmembrane; BCA, bicinchoninic acid; DTT, dithiothreitol; PMSF, phenylmethanesulfonyl fluoride; IAF, 5-iodoacetamidofluorescein; PAGE, polyacrylamide gel electrophoresis.
References
- 1.Falke JJ, Bass RB, Butler SL, Chervitz SA, Danielson MA. Annu Rev Cell Dev Biol. 1997;13:457. doi: 10.1146/annurev.cellbio.13.1.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wurgler-Murphy SM, Saito H. Trends Biochem Sci. 1997;22:172. doi: 10.1016/s0968-0004(97)01036-0. [DOI] [PubMed] [Google Scholar]
- 3.Stock AM, Mowbray SL. Curr Opin Struct Biol. 1995;5:744. doi: 10.1016/0959-440x(95)80006-9. [DOI] [PubMed] [Google Scholar]
- 4.Stock JB, Surette MG. In: Escherichia coli and Salmonella Cellular and Molecular Biology. Neidhardt FC, editor. ASM Press; Washington, DC: 1996. pp. 1103–1129. [Google Scholar]
- 5.Blair DF. Annu Rev Microbiol. 1995;49:489. doi: 10.1146/annurev.mi.49.100195.002421. [DOI] [PubMed] [Google Scholar]
- 6.Parkinson JS. Cell. 1993;73:857. doi: 10.1016/0092-8674(93)90267-t. [DOI] [PubMed] [Google Scholar]
- 7.Bourret RB, Borkovich KA, Simon MI. Annu Rev Biochem. 1991;60:401. doi: 10.1146/annurev.bi.60.070191.002153. [DOI] [PubMed] [Google Scholar]
- 8.Le Moual H, Koshland DE., Jr J Mol Biol. 1996;261:568. doi: 10.1006/jmbi.1996.0483. [DOI] [PubMed] [Google Scholar]
- 9.Krikos A, Conley MP, Boyd A, Berg HC, Simon MI. Proc Natl Acad Sci USA. 1985;82:1326. doi: 10.1073/pnas.82.5.1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Utsumi R, Brissette RE, Rampersaud A, Forst SA, Oosawa K, Inouye M. Science. 1989;245:1246. doi: 10.1126/science.2476847. [DOI] [PubMed] [Google Scholar]
- 11.Baumgartner JW, Kim C, Brissette RE, Inouye M, Park C, Hazelbauer GL. J Bacteriol. 1994;176:1157. doi: 10.1128/jb.176.4.1157-1163.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weerasuriya S, Schneider BM, Manson MD. J Bacteriol. 1998;180:914. doi: 10.1128/jb.180.4.914-920.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moe GR, Bollag E, Koshland DE., Jr Proc Natl Acad Sci USA. 1989;86:5683. doi: 10.1073/pnas.86.15.5683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Biemann HP, Harmer SL, Koshland DE., Jr J Biol Chem. 1996;271:27927. doi: 10.1074/jbc.271.44.27927. [DOI] [PubMed] [Google Scholar]
- 15.Adler J. Science. 1966;153:708. doi: 10.1126/science.153.3737.708. [DOI] [PubMed] [Google Scholar]
- 16.Macnab RM, Koshland DEJ. Proc Natl Acad Sci USA. 1972;69:2509. doi: 10.1073/pnas.69.9.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gegner JA, Graham DR, Roth AF, Dahlquist FW. Cell. 1992;18:975. doi: 10.1016/0092-8674(92)90247-a. [DOI] [PubMed] [Google Scholar]
- 18.Schuster SC, Swanson RV, Alex LA, Bourret RB, Simon MI. Nature. 1993;365:343. doi: 10.1038/365343a0. [DOI] [PubMed] [Google Scholar]
- 19.Wang H, Matsumura P. J Bacteriol. 1997;179:287. doi: 10.1128/jb.179.1.287-289.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Borkovich KA, Kaplan N, Hess JF, Simon MI. Proc Natl Acad Sci USA. 1989;86:1208. doi: 10.1073/pnas.86.4.1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Surette MG, Levit M, Liu Y, Lukat G, Ninfa EG, Ninfa A, Stock JB. J Biol Chem. 1996;271:939. doi: 10.1074/jbc.271.2.939. [DOI] [PubMed] [Google Scholar]
- 22.Bourret BB, Hess JF, Simon MI. Proc Natl Acad Sci USA. 1990;87:41. doi: 10.1073/pnas.87.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Welch M, Oosawa K, Aizawa S, Eisenbach M. Proc Natl Acad Sci USA. 1993;90:8787. doi: 10.1073/pnas.90.19.8787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Milburn MV, Prive GG, Milligan DL, Scott WG, Yeh J, Jancarik J, Koshland DE, Jr, Kim SH. Science. 1991;254:1342. doi: 10.1126/science.1660187. [DOI] [PubMed] [Google Scholar]
- 25.Bowie JU, Pakula AA, Simon MI. Acta Crystallogr. 1995;51:145. doi: 10.1107/S0907444994010498. [DOI] [PubMed] [Google Scholar]
- 26.Yeh JI, Biemann HP, Prive GG, Pandit J, Koshland DE, Jr, Kim SH. J Mol Biol. 1996;262:186. doi: 10.1006/jmbi.1996.0507. [DOI] [PubMed] [Google Scholar]
- 27.Pakula AA, Simon MI. Proc Natl Acad Sci USA. 1992;89:4144. doi: 10.1073/pnas.89.9.4144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chervitz SA, Falke JJ. J Biol Chem. 1995;270:24043. doi: 10.1074/jbc.270.41.24043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chervitz SA, Lin CM, Falke JJ. Biochemistry. 1995;34:9722. doi: 10.1021/bi00030a010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Biemann HP, Koshland DE., Jr Biochemistry. 1994;33:629. doi: 10.1021/bi00169a002. [DOI] [PubMed] [Google Scholar]
- 31.Kolodziej AF, Tan T, Koshland DE., Jr Biochemistry. 1996;35:14782. doi: 10.1021/bi961481v. [DOI] [PubMed] [Google Scholar]
- 32.Chervitz SA, Falke JJ. Proc Natl Acad Sci USA. 1996;93:2545. doi: 10.1073/pnas.93.6.2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee GF, Lebert MR, Lilly AA, Hazelbauer GL. Proc Natl Acad Sci USA. 1995;92:3391. doi: 10.1073/pnas.92.8.3391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee GF, Dutton DP, Hazelbauer GL. Proc Natl Acad Sci USA. 1995;92:5416. doi: 10.1073/pnas.92.12.5416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hughson AG, Hazelbauer GL. Proc Natl Acad Sci USA. 1996;93:11546. doi: 10.1073/pnas.93.21.11546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Baumgartner JW, Hazelbauer GL. J Bacteriol. 1996;178:4651. doi: 10.1128/jb.178.15.4651-4660.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hughson AG, Lee GF, Hazelbauer GL. Protein Sci. 1997;6:315. doi: 10.1002/pro.5560060206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mowbray SL, Foster DL, Koshland DE., Jr J Biol Chem. 1985;260:11711. [PubMed] [Google Scholar]
- 39.Long DG, Weis RM. Biochemistry. 1992;31:9904. doi: 10.1021/bi00156a007. [DOI] [PubMed] [Google Scholar]
- 40.Wu JR, Long DG, Weis RM. Biochemistry. 1995;34:3056. doi: 10.1021/bi00009a037. [DOI] [PubMed] [Google Scholar]
- 41.Seeley SK, Weis RM, Thompson LK. Biochemistry. 1996;35:5199. doi: 10.1021/bi9524979. [DOI] [PubMed] [Google Scholar]
- 42.Frillingos S, Sun J, Gonzalez A, Kaback HR. Biochemistry. 1997;36:269. doi: 10.1021/bi9618629. [DOI] [PubMed] [Google Scholar]
- 43.Akabas MH, Stauffer DA, Xu M, Karlin A. Science. 1992;258:307. doi: 10.1126/science.1384130. [DOI] [PubMed] [Google Scholar]
- 44.Vaniwaarden PR, Driessen AJM, Menick DR, Kaback HR, Konings WN. J Biol Chem. 1991;266:15688. [PubMed] [Google Scholar]
- 45.Ganguli S, Wang H, Matsumura P, Volz K. J Biol Chem. 1995;270:17386. [PubMed] [Google Scholar]
- 46.Danielson MA, Bass RB, Falke JJ. J Biol Chem. 1997;272:32878. doi: 10.1074/jbc.272.52.32878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gardina PJ, Manson MD. Science. 1996;274:425. doi: 10.1126/science.274.5286.425. [DOI] [PubMed] [Google Scholar]
- 48.Tatsuno I, Homma M, Oosawa K, Kawagishi I. Science. 1996;274:423. doi: 10.1126/science.274.5286.423. [DOI] [PubMed] [Google Scholar]
- 49.Ames P, Parkinson JS. Cell. 1988;55:817. doi: 10.1016/0092-8674(88)90137-7. [DOI] [PubMed] [Google Scholar]
- 50.Ames P, Chen J, Wolff A, Parkinson JS. Cold Spring Harbor Symp Quantum Biol. 1988;53:59. doi: 10.1101/sqb.1988.053.01.010. [DOI] [PubMed] [Google Scholar]
- 51.Danielson MA. PhD Thesis. Department of Chemistry and Biochemistry, University of Colorado; Boulder, CO: 1997. The molecular mechanism of transmembrane signaling and kinase regulation by the aspartate receptor of bacterial chemotaxis. [Google Scholar]
- 52.Liu JD, Parkinson JS. Proc Natl Acad Sci USA. 1989;86:8703. doi: 10.1073/pnas.86.22.8703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kunkel TA, Bebenek K, McClary J. Methods Enzymol. 1991;204:125. doi: 10.1016/0076-6879(91)04008-c. [DOI] [PubMed] [Google Scholar]
- 54.Vogel HJ, Bonner DM. J Biol Chem. 1956;218:97. [PubMed] [Google Scholar]
- 55.Stoscheck CM. Methods Enzymol. 1990;182:50. doi: 10.1016/0076-6879(90)82008-p. [DOI] [PubMed] [Google Scholar]
- 56.Bourret RB, Drake SK, Chervitz SA, Simon MI, Falke JJ. J Biol Chem. 1993;268:13089. [PMC free article] [PubMed] [Google Scholar]
- 57.Stock A, Chen T, Welsh D, Stock J. Proc Natl Acad Sci USA. 1988;85:1403. doi: 10.1073/pnas.85.5.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stock A, Mottonen J, Chen T, Stock J. J Biol Chem. 1987;262:535. [PubMed] [Google Scholar]
- 59.Weis RM, Koshland DE., Jr Proc Natl Acad Sci USA. 1988;85:83. doi: 10.1073/pnas.85.1.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ames P, Yu YA, Parkinson JS. Mol Microbiol. 1996;19:737. doi: 10.1046/j.1365-2958.1996.408930.x. [DOI] [PubMed] [Google Scholar]
- 61.Ninfa EG, Stock AM, Mowbray SL, Stock JB. J Biol Chem. 1991;266:9764. [PubMed] [Google Scholar]
- 62.Falke JJ, Koshland DE., Jr Science. 1987;237:1596. doi: 10.1126/science.2820061. [DOI] [PubMed] [Google Scholar]
- 63.Chen X, Koshland DE., Jr Biochemistry. 1997;36:11858. doi: 10.1021/bi970911u. [DOI] [PubMed] [Google Scholar]
- 64.Richards FM. Annu Rev Biophys Bioeng. 1977;6:151. doi: 10.1146/annurev.bb.06.060177.001055. [DOI] [PubMed] [Google Scholar]
- 65.Chothia C, Lesk AM. Trends Biochem Sci. 1985;10:116. [Google Scholar]
- 66.Butler SL, Falke JJ. Biochemistry. 1996;35:10595. doi: 10.1021/bi961107v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Careaga CL, Sutherland J, Sabeti J, Falke JJ. Biochemistry. 1995;34:3048. doi: 10.1021/bi00009a036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Careaga CL, Falke JJ. J Mol Biol. 1992;226:1219. doi: 10.1016/0022-2836(92)91063-u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Careaga CL, Falke JJ. Biophys J. 1992;62:209. doi: 10.1016/S0006-3495(92)81806-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sankararamakrishnan S, Vishveshwara S. Int J Pept Protein Res. 1992;39:356. doi: 10.1111/j.1399-3011.1992.tb01595.x. [DOI] [PubMed] [Google Scholar]
- 71.Polinsky A, Goodman M, Williams KA, Deber CM. Biopolymers. 1992;32:399. doi: 10.1002/bip.360320416. [DOI] [PubMed] [Google Scholar]
- 72.Maddock JR, Shapiro L. Science. 1993;259:1717. doi: 10.1126/science.8456299. [DOI] [PubMed] [Google Scholar]
- 73.Lupas A. Trends Biochem Sci. 1996;21:375. [PubMed] [Google Scholar]
- 74.Stock JB, Lukat GS, Stock AM. Annu Rev Biophys Biophys Chem. 1991;20:109. doi: 10.1146/annurev.bb.20.060191.000545. [DOI] [PubMed] [Google Scholar]