

Abstract
Fibroblast growth factor 23 null mice (Fgf-23−/−) have a short lifespan and show numerous biochemical and morphological features consistent with premature aging-like phenotypes, including kyphosis, severe muscle wasting, hypogonadism, osteopenia, emphysema, uncoordinated movement, T cell dysregulation, and atrophy of the intestinal villi, skin, thymus, and spleen. Furthermore, increased vitamin D activities in homozygous mutants are associated with severe atherosclerosis and widespread soft tissue calcifications; ablation of vitamin D activity from Fgf-23−/− mice, by genetically deleting the 1α(OH)ase gene, eliminates atherosclerosis and ectopic calcifications and significantly rescues premature aging-like features of Fgf-23−/− mice, resulting in prolonged survival of Fgf-23−/− 1α(OH)ase−/− double mutants. Our results indicate a novel role of Fgf-23 in developing premature aging-like features through regulating vitamin D homeostasis. Finally, our data support a new model of interactions among Fgf-23, vitamin D, and klotho, a gene described as being associated with premature aging process.
Keywords: rescue, klotho, phosphate
Aging is a complex biological process controlled by multiple genetic and environmental factors (1–4). Studies involving molecular mechanisms of human aging and its progression are challenging, as it takes decades to develop some of the age-related features. Since extensive subsets of age-associated phenotypes define human aging, the availability of animal models exhibiting multiple aging features are useful, not only to analyze the molecular mechanisms of age-related changes in various organs, but also for the in vivo screening of molecules that counteract age-associated syndromes including anti-oxidant agents and hormones (5, 6). DNA damage through oxidative stress, among others, is thought to be an important contributing factor in aging, and has been extensively studied in animals (1, 3, 7–11). However, the potential role of humoral factor(s) regulating the aging process has not been studied in similar depth and detail. In this study, we show that genetic ablation of Fgf-23 results in a syndrome that resembles premature aging.
FGF-23, a recently identified member of the FGF family, is involved in regulating phosphate homeostasis (12). FGF-23 is the only member of the FGF family that contains a proconvertase processing site. Cleavage of the intact 30 kDa-secreted protein results in two smaller fragments of ~18 kDa (amino fragment) and 12 kDa (carboxy fragment) of yet unknown function. Although various studies have shown that FGF-23 can exert certain activities through binding to the known receptors for other members of the FGF family (13, 14), the search for a novel receptor that specifically mediates the actions of FGF-23 is ongoing. Genetic modifications of FGF-23 in animals have shown phenotypes similar to those of human diseases. Transgenic mice overexpressing FGF-23 exhibit hypophosphatemia, reduced serum 1,25(OH)2D3 levels, and rickets (15–17), while Fgf-23 null mice exhibit hyperphosphatemia, elevated serum 1,25(OH)2D3 levels with abnormal skeletogenesis (17, 18). FGF-23 transgenic mice mimic autosomal dominant hypophosphatemic rickets (ADHR), X-linked hypophosphatemia (XLH), and oncogenic osteomalacia (OOM) in humans, while the Fgf-23 null mice mimic the features consistent with human familial tumoral calcinosis (FTC) (12, 19, 20).
In this study, using in vivo genetic manipulation approaches, we present a novel role of Fgf-23 in premature aging and show that the premature aging-like phenotype in Fgf-23−/− mice is partly mediated through increased vitamin D activities.
MATERIALS AND METHODS
Experimental mice
We recently generated Fgf-23 null mice (Fgf-23−/−) (17) and 1α hydroxylase gene knockout animals (21). We crossbred heterozygous Fgf-23 and 1α(OH)ase mutants to obtain compound heterozygous animals, which were then interbred to generate desired double homozygous mutants [Fgf-23−/−/1α(OH)ase−/−]. Routine PCR using genomic DNA, extracted from tail clips, was performed for genotyping of the various groups of mice (17, 21). Mice were fed a standard diet ad libidum containing 0.7% calcium, 0.6% phosphorus, and 3.2 IU/g vitamin D3 (PicoLab Mouse Diet 20–5058). Animals were maintained in accordance with the NIH Guide for the Care and Use of Laboratory Animals and were employed using protocols approved by the institution’s subcommittee on animal care (IACUC).
Macroscopic phenotype
The total body weight of all mice was taken every 3–5 days starting at 2.5 wk of age until death. Survival of various groups of animals was recorded until death of control, Fgf-23−/−, and double mutant Fgf-23−/−/1α(OH)ase−/− mice.
Biochemical measurements
Blood was obtained either by retro-orbital or tail bleeding of 3-, 6-, 9- and 11-wk-old wild-type, Fgf-23−/−, and Fgf-23−/−/1α(OH)ase−/− littermates. Serum was isolated by centrifugation at 3000 g for 10 min and stored at −80°C. Serum phosphorus and serum calcium were determined by colorometric measurements using the Stanbio Phosphorus Liqui-UV Test and Calcium (Arsenazo) LiquiColor Test, respectively. Total blood of 4-wk-old mice was used to determine routine hematological parameters such as cell counts.
Skeletal analyses
Skeletal changes in Fgf-23−/− mice and their control littermates were analyzed by X-ray, quantitative CT (pQCT) and PIXImus measurements. Alizarin red S staining of total body skeletons, routine histology, and von Kossa staining were executed as described in our earlier studies (22).
Histological analyses
Assorted fixatives as 4% paraformaldehyde, 10% buffered formalin, 95% ethanol, and Carnoy’s solution were used to fix the tissues before embedding in paraffin. Four to six micrometer paraffin sections of diverse tissues were produced on a Microm HM 360 microtome (Microm, Walldorf, Germany) and subsequently mounted on SuperFrost Plus slides. Sections were routinely stained with hematoxylin and eosin, periodic acid-schiff (PAS), periodic acid-Schiff methenamine silver (PAM), Masson’s trichome, von Kossa, and Congo red. Histological examination was carried out by light microscopy.
Immunohistochemical analyses
Immunohistochemical staining was performed as described previously (23). Briefly, tissue sections were deparaffinized with xylene, rinsed thoroughly with 95% ethanol, and then soaked in 0.3% hydrogen peroxide in methanol for 15 min at room temperature to inactivate endogenous peroxidase activity. The tissue sections were incubated with 10% goat serum for 30 min, before the addition of the primary antibody against α-smooth muscle actin (α-SMA) with a dilution of 1:100 (Dako, Glostrup, Denmark). Slides were washed with phosphate buffered saline and processed further using histofine SAB-PO Kit (Nichirei, Tokyo, Japan), according to the instructions provided by the manufacturer, and then reacted with 3,3′-diaminobenzindine and H2O2.
Proliferation and apoptosis
To evaluate the extent of proliferation in the different groups of mice, intraperitoneal injection of BrdU (100 μg/g body wt.) was performed 2 h before death. Tissues were fixed routinely in 10% buffered formalin and processed for paraffin embedding. Sections were stained for BrdU by incubating at 4°C overnight using the mouse anti-BrdU antibody and were visualized with a BrdU-labeling kit (Zymed Laboratories). The extent of apoptosis was determined by TUNEL assays using a commercial kit (Roche Diagnostics), according to the manufacturer’s instructions.
Quantitative real-time PCR
Total RNA isolated from kidneys of various group of mice was used to detect relative expression of 1α(OH)lase and klotho mRNA, as described earlier (24). Real-time PCR was performed in duplicate, and all the reactions were controlled by standards (nontemplate control and standard positive control). The quantity of mRNA was calculated by normalizing the CT (threshold cycle value) of 1α(OH)lase or klotho to the CT of the housekeeping gene GAPDH of the same RNA sample, according to the following formula: the average GAPDH CT value (each multiplex PCR is done in duplicate) was subtracted from the average 1α(OH)lase or klotho CT value, this result represent the ΔCT. This ΔCT is specific and can be compared with the ΔCT of a control calibration sample. The subtraction of control ΔCT from the ΔCT of 1α(OH)lase or klotho samples is referred as ΔΔCT. The relative expression of 1α(OH)lase or klotho (in comparison to control littermates) in renal tissues isolated from Fgf-23−/− mice and Fgf-23/1α(OH)ase was determined by 2−ΔΔCT. The sequences of the primers used were as follows: 1α(OH)lase (forward 5′-ACAAGGACACCTAAACCGAACAC-3′; reverse 5′-AGCTACTGA CTGGTCCTATCACAGAA-3′), klotho (forward 5′-TGGCTTTCCTCCTTTACCTG-3′; reverse 5′-GCCGACACTGGG TTTTGT-3′), and GAPDH (forward 5′-ACTGAGGACCAG GTTGTC-3′; reverse 5′-TGCTGTAGC CGTATTCATTG-3′).
In vitro T cell proliferation assay
T cell proliferation was examined after stimulation of cells with concanavalin A (Con A) or phytohemagglutinin (PHA-P) as a mitogen, using routinely established protocol (25). Briefly, T cells isolated from thymus, lymph nodes, and spleen of control and Fgf-23−/− mice at 6 wk of age were cultured in vitro. The medium used was RPMI-1640 with 10% fetal calf serum, 2.5 mM HEPES buffer, penicillin/streptomycin 100 U/100 μg/ml, and 5 × 10−5 M 2-mercaptoethanol. Cultures were incubated at 37° in an atmosphere of 5% CO2. Cells were cultured in 96-well culture plates, each well containing 2.5 × 10−5 viable cells in 200 μl of medium. Wells were set up in quadruplicate, four wells with no stimulus, four wells with Con A at 5 μg/ml final concentration, and four wells with PHA-P at 10 μg/ml final concentration, for each animal. After 48 h of culture, −0.5 μCi of 3H thymidine in 25 μl of medium was added to each well, and the plates were harvested onto fiber glass filter 24 h later. Radioactive uptake (cpm) was counted by scintillation counter. The results were expressed as Δcpm, which represents the difference of counts of sample with Con A to the corresponding control counts obtained without Con A.
RESULTS
Premature aging-like phenotypes of the Fgf-23−/− mouse
We recently generated two independent Fgf-23−/− mouse lines by replacing the entire coding region of the Fgf-23 gene with lacZ and the neomycin resistance gene (17). In contrast to the normal phenotype of heterozygous mice, homozygous mutants exhibit multiple features resembling premature aging; these mice develop normally until 2 wk of age and are macroscopically indistinguishable from their wild-type littermates. However, visible growth retardation is apparent from 3 wk onwards, associated with uncoordinated sluggish movements, followed by premature lethality between 4 to 13 wk. Fgf-23−/− mice can easily be recognized by their small size and marked kyphosis (Fig. 1A). The genital organs of both sexes of Fgf-23−/− mice are severely atrophic as observed by both macroscopic (Fig. 1D and E) and microscopic (Fig. 2A and B) examinations; the severe hypogonadism in both sexes results in infertility, a major consequence of aging in both humans and experimental animals (8, 26).
Figure 1.

Premature aging features of Fgf-23−/− mice. A) Mutant mouse (top) shows small body size, reduced muscle mass, and kyphosis when compared with normal control littermate (bottom) at 6 wk of age. B) Number of lymphocytes in peripheral blood of Fgf-23−/− animals (n=9, blue bar) was significantly decreased (P<0.0001), when compared with controls (n=13, red bar). C) Mitogenic response of T cells isolated from thymus, lymph nodes, or spleen to concavanalin A. Shown are mean values (quadruplicates) of controls (n=3, red circles) and Fgf-23−/− animals (n=2, blue squares). Red and blue cross-lines represent mean of 12 measurements in controls (red lines) and 8 measurements in mutants (blue lines). D) Atrophy of mutant uterus and ovary (right) when compared with normal (left) at 6 wk of age leads to hypogonadism and infertility in females. E) Atrophied testes of Fgf-23−/− animals (right) also causes infertility in Fgf-23−/− males. F) Quantitative assessment of the weight of spleen and thymus (n=3, P<0.05). Data are presented as fold over control (=1) after each organ weight was normalized to total body weight of matching animal. Macroscopic picture of an atrophied thymus (G) and spleen (H) of Fgf-23−/− animals (right) when compared with normal control littermates (left).
Figure 2.

Macroscopic and microscopic features of various organs at 6 wk of age (n=8). Hematoxilin- and eosin stained sections of wild-type (A) and Fgf-23−/− (B) testes and of control (C) and Fgf-23−/− (D) lungs (A–D: ×10). Mutant lung exhibits typical emphysematous features as observed during aging. E) Toluidine blue staining of an Fgf-23−/− kidney at 6 wk of age. Arrows depict kidney stones found only in the mutants. F) Congo-red staining indicates amyloid deposition found in small and medium sized arteries of the mutant kidney and heart (H). G) von Kossa staining demonstrates severe calcification of the aortic wall in Fgf-23−/− animals. Macroscopic image of small intestine of wild-type (I) and Fgf-23−/− animal (J) (n=6), showing ballooning of mutant intestine. Hematoxilin/eosin staining of cross sections of wild-type (K) and Fgf-23−/− littermate (L) (K and L: ×5). Reduced height of intestinal villi, and atrophy of intestinal mucosa are shown. Immunohistochemical evaluation by α-smooth muscle actin antibody staining presents dramatic reduction in vascularization and smooth muscle coat in intestine of mutant animals (N), when compared with normal littermates M). BrdU staining demonstrates striking diminution of proliferative cells in mutants (P) vs. wild-type (O) mice (M to P: ×10). R) Increased apoptosis in the mutant skin is visible by positive TUNEL staining mostly located in hair follicles when compared with normal (Q). BrdU labeling of the skin resulted in dramatic decrease in proliferative cells in Fgf-23−/− (T) vs. normal (S) littermate (Q to T: ×10).
Effects of Fgf-23 ablation on lung and lymphoid system
Fgf-23−/− mice show typical features of emphysema in the lungs (Fig. 2D), which appear as early as 3 wk of age and are consistent with similar emphysematous changes documented in aged populations (27). In addition, severe atrophy of thymus, spleen, and lymph nodes can be noted in Fgf-23−/− mice when compared with controls (Fig. 1G and H); total peripheral blood lymphocyte counts are also significantly decreased in homozygous mutant mice (Fig. 1B). To test the functional activity of the mutant T cells, an in vitro proliferation assay was carried out in which cells isolated from lymph nodes, spleen, and thymus were stimulated with mitogens including Con A (Fig. 1C) or PHA-P (data not shown). T cells derived from Fgf-23−/− mice showed markedly reduced ability to proliferate (Fig. 1C), a feature usually characteristic of human aging (28).
Abnormal calcification in Fgf-23 null mice
Widespread thickening of vessel walls with extensive calcifications are observed in Fgf-23−/− mice by 6 wk of age (Fig. 2G); small and medium sized arteries in the kidneys are most extensively affected (Fig. 3). Atherosclerosis is a common age-associated vascular change, and there is a growing body of evidence that increased thickening and stiffness of arteries with endothelial dysfunction in apparently healthy elderly persons poses a higher risk for developing cardiovascular diseases (29). Determining the exact role of FGF-23 in premature age-associated vascular changes may help identify new targets for intervention to prevent or delay the course of the atherosclerotic process. Fgf-23−/− mice also exhibit widespread soft tissue calcifications in lung, kidney, skeletal muscle, skin, urinary bladder, testes, cardiac muscle, and arterial walls, as detected by von-Kossa staining (Fig. 3A, C, and E and data not shown). Soft tissue calcifications appear as early as 6 wk postnatally and progress with age. In general, the distribution of ectopic calcification in Fgf-23−/− mice is compatible with that of premature aging. Furthermore, there are amyloid accumulations in such tissues as kidney and heart (Fig. 2F and H). Detailed skeletal analyses of Fgf-23−/− mice by radiographic analysis show uneven mineral distribution and reduced radio-density in the bones suggesting osteopenia. Autoradiographic studies of both forelimbs and of hindlimbs showed that the bone mineral density (BMD) was strikingly reduced in Fgf-23−/− mice, compared with their control littermates. Dual-energy X-ray absorptiometry (DEXA) analysis showed similar reduction of BMD in 3, 6, and 11 wk of Fgf-23−/− mice; an observation again validated by detection of significant decrease in volumetric BMD of the femoral shaft (control 426.6+21.4 vs. knockout 262.7+25) and the femoral metaphysis (control 350.4+36.2 vs. knockout 236.9+15.1) in Fgf-23−/− mice by pQCT (17). Histomorphometric findings in these mutant mice also bear a resemblance to those found in senile osteoporosis in humans (30).
Figure 3.

Ectopic calcifications. Von Kossa staining of paraffin sections of kidney (A), heart (C; muscle and valves), and lung of Fgf-23−/− (E) animals demonstrating severe calcifications of these organs. Images presented in B, D, and F show complete elimination of abnormal calcification pattern in littermates that are deficient for both Fgf-23 and 1α(OH)ase gene (n=8; A to F: ×10).
Effects of Fgf-23 ablation on skin and intestine
The hair of Fgf-23−/− mice is sparser than that in control littermates. A reduced number of hair follicles with marked decrease in dermal and epidermal thickness and barely detectable subcutaneous fat are consistently observed in mutant mice (Fig. 4H). There is less proliferative activity (as determined by BrDU labeling) (Fig. 2T) and increased rate of apoptosis (as determined by TUNEL staining) (Fig. 2R) in the hair follicles of Fgf-23−/− mice, compared with controls (Fig. 4Q and S), a phenomenon consistent with aging (31, 32). Similarly, reduction in the villus height of the intestinal mucosa, resulting in reduced mucosal surface area is a frequently observed feature during advanced age (33). Fgf-23−/− mice show focal areas of intestinal distention (Fig. 2J), with marked atrophy of intestinal mucosa (Fig. 2L); less vascularization and very thin intestinal muscle layers are also documented in the Fgf-23−/− mice, as determined by immunohistochemical staining of intestinal sections by α-SMA (Fig. 2N). BrdU staining shows decreased proliferation of intestinal cells in Fgf-23−/− mice (Fig. 2P), an additional feature of age-associated intestinal changes. Figure 2I, K, M, and O represents corresponding images of wild-type controls.
Figure 4.
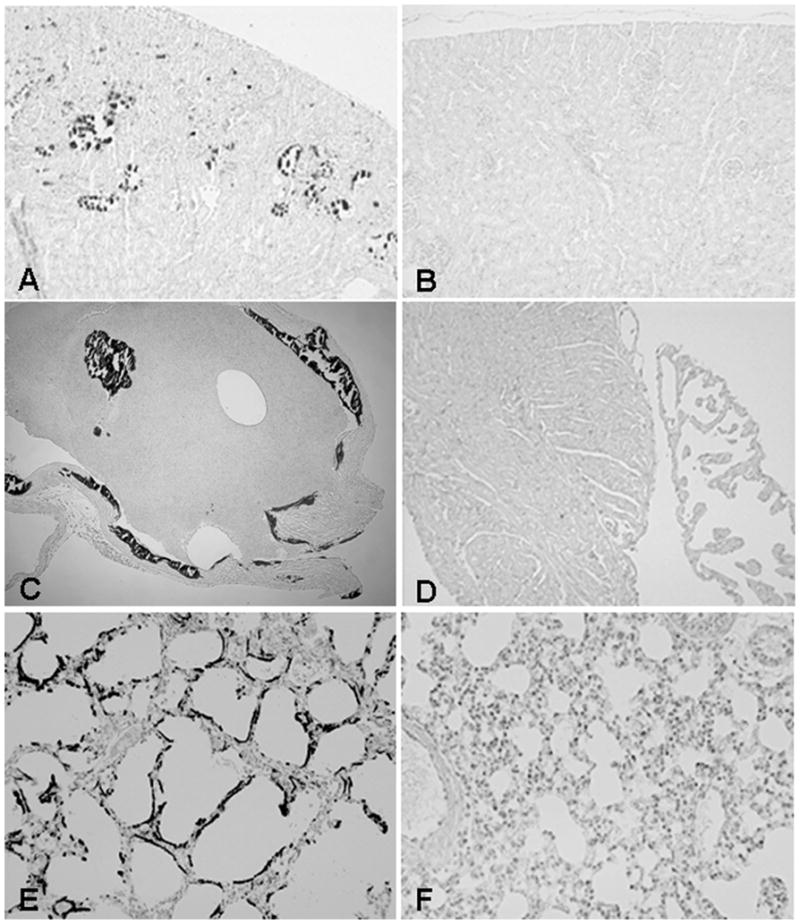
A) Macroscopic image of a wild-type (WT), Fgf-23−/−, and Fgf-23−/−/1α(OH)ase−/− double mutant at ~6 wk of age. B) Body weight curve and survival curve (C) of control, Fgf-23−/− and Fgf-23−/−/1α (OH)ase−/− mice (n>15) showing gain of weight and prolonged lifespan in compound mutants. Hematoxilin/eosin staining of intestinal sections of wild-type (D), Fgf-23−/− (F), and Fgf-23−/−/1α(OH)ase−/− double knockout animals. Single Fgf-23−/− mice exhibit (E) severe atrophy of intestine, which is significantly improved by deletion of the 1α(OH)ase gene. Similar improvement was also noted in the skin section of Fgf-23−/−/1α(OH)ase−/− double knockout animals, compared with Fgf-23−/− mice; wild-type (G), Fgf-23−/− (H), and Fgf-23−/−/1α(OH)ase−/− (I) double knockout animals (D to I: ×10).
Generation of Fgf-23−/−/1α(OH)ase−/− compound mutants
The expression of the 1α(OH)ase gene was significantly elevated, as determined by quantitative real-time PCR in Fgf-23−/− mice (data not shown); such an increased renal expression of 1α(OH)ase in Fgf-23−/− mice is also reflected by increased serum levels of 1,25 dihydroxyvitamin D3 (368.1±226.3 pg/ml in knockout mice; 56.4±13.8 pg/ml in wild-type mice) (17). To determine whether increased levels of vitamin D in Fgf-23−/− mice could mediate some of the premature aging features documented in Fgf-23−/− mice, we generated Fgf-23−/−/1α(OH)ase−/− double knockout mice, by cross breeding heterozygous Fgf-23 mutants with 1α(OH)ase mutants. In contrast to Fgf-23−/− animals, Fgf-23−/−/1α(OH)ase−/− double knockout mice were indistinguishable from wild-type mice, in terms of their appearance, body weight, and physical activities (Fig. 4); serum calcium and phosphate levels were reversed when compared with Fgf-23−/− littermates, resulting in the absence of calcifications in all examined tissues, including in the kidney (Fig. 3B), heart (Fig. 3D), and lung (Fig. 3F). The serum calcium level in control mice was 8.5 + 0.42mg/dl, was increased in Fgf-23−/− mice (12.18 mg/dl), but decreased in Fgf-23−/−/1α(OH)ase−/− double knockout mice (6.3+0.34mg/dl). Similarly, serum phosphate levels in control mice was 8.93+0.0 5mg/dl and was significantly higher in Fgf-23−/− mice (19.63+2.72mg/dl), while markedly reduced in Fgf-23−/−/1α(OH)ase−/− double knockout mice (6.36+0.17mg/dl). The size and shape of thymus, intestine, and genital organs in double knockout mice were similar to those of wild-type littermates (data not shown). The pathological changes in skin (Fig. 4H) and intestine (Fig. 4E) of Fgf-23−/− mice were dramatically ameliorated and rescued in Fgf-23−/−/1α(OH)ase−/− double mutants (Fig. 4F and I). The fact that Fgf-23−/−/1α(OH)ase−/− double mutants ameliorate systemic premature aging-like features, dramatically improve lifespan (Fig. 4C), and regain fertility, suggests that the induction of “premature aging-like features” in Fgf-23−/− mice is partly mediated through abnormal metabolism of vitamin D.
Expression of klotho in Fgf-23 null mice
Mice homozygous for hypomorphic alleles of the klotho gene exhibit a number of premature aging features, similar to above-described features of Fgf-23−/− mice; these include kyphosis, ectopic calcifications, atherosclerosis, hypogonadism, emphysema, osteopenia, generalized organ atrophy, and short lifespan (34). We, therefore, studied the level of klotho expression in Fgf-23−/− mice. By quantitative real-time PCR, we found less expression of klotho in the kidney of both Fgf-23−/− mice and Fgf-23−/−/1α(OH)ase−/− compound mutants (Fig. 5A).
Figure 5.
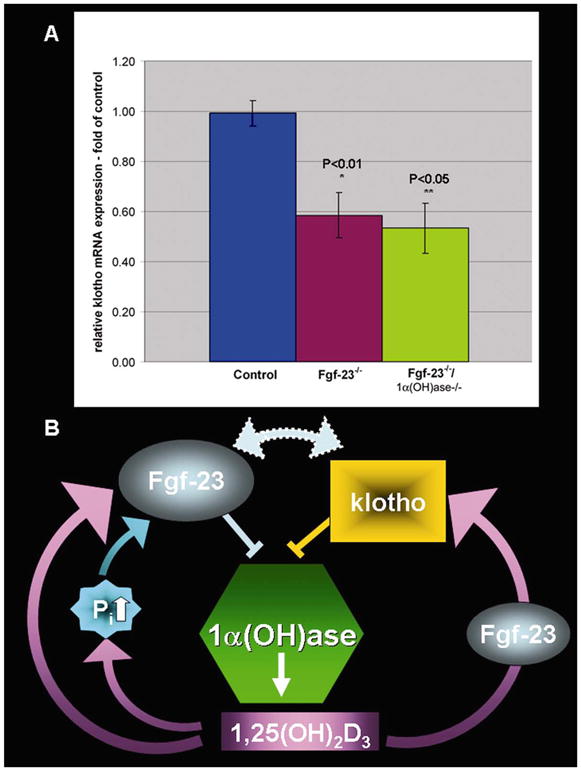
A) Quantitative real-time PCR was performed for klotho expression in kidney (n>3). Relative expression of klotho mRNA in kidneys of Fgf-23−/− animals (P<0.01) and in Fgf-23−/−/1α(OH)ase−/− double knockout animals (P<0.05) is significantly decreased when compared with controls. Data shown is the klotho mRNA expression as fold over control (=1). B) Schematic representation of a model showing the possible interactions among Fgf-23, vitamin D, and klotho. Fgf-23 activity is required for vitamin-D mediated expression of klotho.
DISCUSSION
We present here a novel role of Fgf-23 in aging and longevity. Our results showed that genetically ablated Fgf-23 mice develop extensive premature aging-like features, which include, but are not limited to reduced lifespan, infertility, osteopenia, atherosclerosis, and atrophy of the skin and other internal organs, bearing the resemblance to that observed during human aging. All these extensive age-associated phenotypes were caused by the disruption of a single gene, Fgf-23, a secreted protein that appears to exert its effects through binding with cell surface receptors; Fgf-23−/− mice, along with klotho mutant mice, thus are very different from described previously premature-aging models that are mostly due to dysregulation of nuclear proteins (1, 3, 7, 8). Despite the fact that Fgf-23−/− mice show systemic premature aging-like phenotypes, the expression of the FGF-23 gene is very restricted and mainly documented in bone (osteoblasts and osteocytes). Generalized involvement of tissues and organs outside the skeletal system in Fgf-23−/− mice suggests that these premature aging-like features are likely to be noncell-autonomous phenomena, exerted through the pleiotropic hormonal function of Fgf-23.
Fgf-23−/− mice show increased vitamin D activities and abnormal mineral ion homeostasis leading to widespread soft tissue calcifications (17, 35). Interestingly, abolition of vitamin D activities from Fgf-23−/− mice, by genetical ablation of the 1α(OH)lase gene, rescued a number of systemic aging phenotypes, suggesting that “premature aging-like phenotypes” of Fgf-23−/− mice are partly exerted through abnormal regulation of vitamin D.
Fgf-23−/− mice show extremely high biochemical and morphological similarities with mice homozygous for hypomorphic alleles of the klotho gene (34). We found a decreased expression of klotho in kidney of Fgf-23−/− mice. Based on the strikingly similar biochemical and morphological phenotypes of Fgf-23−/− and klotho mutant mice (34) and reduced expression of klotho in Fgf-23−/− mice, we presume that part of the premature aging process may be regulated through a common humoral signaling pathway in both these strains of mice. Similar to Fgf-23−/− mice, klotho mutant mice display abnormal mineral ion homeostasis and increased serum levels of 1,25 dihydroxyvitamin D3; lowering serum levels of vitamin D by dietary restriction results in alleviation of some of the aging phenotypes in klotho mutants, suggesting that they are downstream events resulting from elevated 1,25 dihydroxyvitamin D3 (36); these observations are very similar to the rescue of premature aging-like phenotypes of Fgf-23−/− mice by genetic ablation of the 1α(OH)ase gene. From current results, and taking into account earlier studies, we propose a new model of interactions among Fgf-23, vitamin D, and klotho (Fig. 5B). Both Fgf-23 and klotho are negative regulators of vitamin D metabolism by suppressing the expression of the 1α(OH)ase gene. It has been shown that 1,25 dihydroxyvitamin D3 can induce klotho, as demonstrated by up-regulation of renal expression of klotho following 1,25 dihydroxyvitamin D3 injection into wild-type mice (36). However, despite significantly increased levels of 1,25 dihydroxyvitamin D3, we found decreased renal expression of klotho in Fgf-23−/− mice, suggesting that interactions of vitamin D and klotho are mediated through Fgf-23. More importantly, in Fgf-23−/−/1α(OH)ase−/− compound mutants, where synthesis of the active form of vitamin D was completely abolished, expression level of klotho was similar to those found in Fgf-23−/− mice, again emphasizing that vitamin D-mediated expression of klotho is regulated through Fgf-23. Furthermore, when we injected control mice with recombinant FGF-23, we did not find any significant changes in the renal expression of klotho compared with the vehicle treated mice, although these FGF-23-treated mice did show low level of serum phosphate, compared with vehicle treated control mice. However, when Fgf-23−/− mice were treated with FGF-23, we found induction of renal expression of klotho; despite high levels of active vitamin D, renal expression of klotho is low in Fgf-23−/− mice; however, as soon as these mice were exposed to exogenous FGF-23, we could observe induction of klotho expression.
Another aspect that needs further study is to determine the role of serum phosphate on the expression of klotho. In our study, the expression of klotho in both Fgf-23−/− mice and Fgf-23−/−/1α(OH)ase−/− compound mutants was very similar, despite the fact that Fgf-23−/− mice are hyperphosphatemic and Fgf-23−/−/1α(OH)ase−/− compound mutants are being hypophosphatemic; it is therefore unlikely that less expression of klotho is influenced by serum phosphate, at least in our two studied models. The influence of serum calcium, phosphate, and PTH on the expression of klotho in normal and abnormal mineral ion homeostasis needs further investigation. However, the premature aging features observed in Fgf-23−/− mice are unlikely to be due to reduced expression of klotho; therefore, to clarify this issue, we are planning further studies to determine whether pharmacological or genetic restoration of klotho has any effect on the aging phenotypes in Fgf-23−/−. Our proposed model (Fig. 5B) should provide the basis for further molecular studies to establish the exact mechanisms by which Fgf-23 interacts with klotho to regulate mineral ion homeostasis and to produce premature aging-like phenotypes.
In conclusion, our results provide novel genetic links among Fgf-23, vitamin D, and multiple premature aging-like phenotypes; the result that genetic ablation of Fgf-23 leads to premature aging with reduced lifespan supports the novel concept that a humoral factor(s) can regulate the aging process. Finally, Fgf-23−/− mice may provide an important tool to study the effects of genetic or pharmacological interventions to delay premature aging process. A better understanding of intrinsic factors influencing aging processes will lead to better insights into the mechanisms by which extrinsic factors can have differential effects on aging individuals.
Acknowledgments
We would like to thank Drs. Henry Kronenberg and Bjorn Olsen for critically reading the manuscript and Dr. Reinhold Erben for helpful suggestions, Dr. Jean Eastcott for technical assistance, and Dr. Roderick Bronson for pathological consultations. All studies were supported by funding provided to B. Lanske by the Harvard School of Dental Medicine.
References
- 1.Johnson FB, Sinclair DA, Guarente L. Molecular biology of aging. Cell. 1999;96:291–302. doi: 10.1016/s0092-8674(00)80567-x. [DOI] [PubMed] [Google Scholar]
- 2.Dolle ME, Giese H, Hopkins CL, Martus HJ, Hausdorff JM, Vijg J. Rapid accumulation of genome rearrangements in liver but not in brain of old mice. Nat Genet. 1997;17:431–434. doi: 10.1038/ng1297-431. [DOI] [PubMed] [Google Scholar]
- 3.Kirkwood TB, Austad SN. Why do we age? Nature. 2000;408:233–238. doi: 10.1038/35041682. [DOI] [PubMed] [Google Scholar]
- 4.de Boer J, Andressoo JO, de Wit J, Huijmans J, Beems RB, van Steeg H, Weeda G, van der Horst GT, van Leeuwen W, Themmen AP, et al. Premature aging in mice deficient in DNA repair and transcription. Science. 2002;296:1276–1279. doi: 10.1126/science.1070174. [DOI] [PubMed] [Google Scholar]
- 5.Delmas D, Jannin B, Latruffe N. Resveratrol: preventing properties against vascular alterations and aging. Mol Nutr Food Res. 2005;49:377–395. doi: 10.1002/mnfr.200400098. [DOI] [PubMed] [Google Scholar]
- 6.Parkes TL, Elia AJ, Dickinson D, Hilliker AJ, Phillips JP, Boulianne GL. Extension of Drosophila lifespan by overexpression of human SOD1 in motorneurons. Nat Genet. 1998;19:171–174. doi: 10.1038/534. [DOI] [PubMed] [Google Scholar]
- 7.Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, Hofer T, Seo AY, Sullivan R, Jobling WA, et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005;309:481–484. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- 8.Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, Bohlooly YM, Gidlof S, Oldfors A, Wibom R, et al. Premature aging in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429:417–423. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- 9.Edwards MG, Sarkar D, Klopp R, Morrow JD, Weindruch R, Prolla TA. Age-related impairment of the transcriptional responses to oxidative stress in the mouse heart. Physiol Genomics. 2003;13:119–127. doi: 10.1152/physiolgenomics.00172.2002. [DOI] [PubMed] [Google Scholar]
- 10.Radak Z, Chung HY, Naito H, Takahashi R, Jung KJ, Kim HJ, Goto S. Age-associated increase in oxidative stress and nuclear factor kappaB activation are attenuated in rat liver by regular exercise. FASEB J. 2004;18:749–750. doi: 10.1096/fj.03-0509fje. [DOI] [PubMed] [Google Scholar]
- 11.Judge S, Jang YM, Smith A, Hagen T, Leeuwenburgh C. Age-associated increases in oxidative stress and antioxidant enzyme activities in cardiac interfibrillar mitochondria: implications for the mitochondrial theory of aging. FASEB J. 2005;19:419–421. doi: 10.1096/fj.04-2622fje. [DOI] [PubMed] [Google Scholar]
- 12.Quarles LD. FGF23, PHEX, and MEPE regulation of phosphate homeostasis and skeletal mineralization. Am J Physiol Endocrinol Metab. 2003;285:E1–E9. doi: 10.1152/ajpendo.00016.2003. [DOI] [PubMed] [Google Scholar]
- 13.Yamashita T, Yoshioka M, Itoh N. Identification of a novel fibroblast growth factor, FGF-23, preferentially expressed in the ventrolateral thalamic nucleus of the brain. Biochem Biophys Res Commun. 2000;277:494–498. doi: 10.1006/bbrc.2000.3696. [DOI] [PubMed] [Google Scholar]
- 14.Yamashita T, Konishi M, Miyake A, Inui K, Itoh N. Fibroblast growth factor (FGF)-23 inhibits renal phosphate reabsorption by activation of the mitogen-activated protein kinase pathway. J Biol Chem. 2002;277:28265–28270. doi: 10.1074/jbc.M202527200. [DOI] [PubMed] [Google Scholar]
- 15.Larsson T, Yu X, Davis SI, Draman MS, Mooney SD, Cullen MJ, White KE. A novel recessive mutation in fibroblast growth factor-23 causes familial tumoral calcinosis. J Clin Endocrinol Metab. 2005;90:2424–2427. doi: 10.1210/jc.2004-2238. [DOI] [PubMed] [Google Scholar]
- 16.Bai X, Miao D, Li J, Goltzman D, Karaplis AC. Transgenic mice overexpressing human fibroblast growth factor 23(R176Q) delineate a putative role for parathyroid hormone in renal phosphate wasting disorders. Endocrinology. 2004;145:5269–5279. doi: 10.1210/en.2004-0233. [DOI] [PubMed] [Google Scholar]
- 17.Sitara D, Razzaque MS, Hesse M, Yoganathan S, Taguchi T, Erben RG, Juppner H, Lanske B. Homozygous ablation of fibroblast growth factor-23 results in hyperphosphatemia and impaired skeletogenesis, and reverses hypophosphatemia in Phex-deficient mice. Matrix Biol. 2004;23:421–432. doi: 10.1016/j.matbio.2004.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shimada T, Kakitani M, Yamazaki Y, Hasegawa H, Takeuchi Y, Fujita T, Fukumoto S, Tomizuka K, Yamashita T. Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J Clin Invest. 2004;113:561–568. doi: 10.1172/JCI19081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Benet-Pages A, Orlik P, Strom TM, Lorenz-Depiereux B. An FGF23 missense mutation causes familial tumoral calcinosis with hyperphosphatemia. Hum Mol Genet. 2005;14:385–390. doi: 10.1093/hmg/ddi034. [DOI] [PubMed] [Google Scholar]
- 20.Ichikawa S, Lyles KW, Econs MJ. A novel GALNT3 mutation in a pseudoautosomal dominant form of tumoral calcinosis: evidence that the disorder is autosomal recessive. J Clin Endocrinol Metab. 2005;90:2420–2423. doi: 10.1210/jc.2004-2302. [DOI] [PubMed] [Google Scholar]
- 21.Dardenne O, Prud’homme J, Arabian A, Glorieux FH, St-Arnaud R. Targeted inactivation of the 25-hydroxyvitamin D(3)-1(alpha)-hydroxylase gene (CYP27B1) creates an animal model of pseudovitamin D-deficiency rickets. Endocrinology. 2001;142:3135–3141. doi: 10.1210/endo.142.7.8281. [DOI] [PubMed] [Google Scholar]
- 22.McLeod MJ. Differential staining of cartilage and bone in whole mouse fetuses by alcian blue and alizarin red S. Teratology. 1980;22:299–301. doi: 10.1002/tera.1420220306. [DOI] [PubMed] [Google Scholar]
- 23.Razzaque MS, Taguchi T. Collagen-binding heat shock protein (HSP) 47 expression in anti-thymocyte serum (ATS)-induced glomerulonephritis. J Pathol. 1997;183:24–29. doi: 10.1002/(SICI)1096-9896(199709)183:1<24::AID-PATH1106>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 24.Razzaque MS, Foster CS, Ahmed AR. Role of collagen-binding heat shock protein 47 and transforming growth factor-beta1 in conjunctival scarring in ocular cicatricial pemphigoid. Invest Ophthalmol Vis Sci. 2003;44:1616–1621. doi: 10.1167/iovs.02-0644. [DOI] [PubMed] [Google Scholar]
- 25.Kawai T, Eisen-Lev R, Seki M, Eastcott JW, Wilson ME, Taubman MA. Requirement of B7 costimulation for Th1-mediated inflammatory bone resorption in experimental periodontal disease. J Immunol. 2000;164:2102–2109. doi: 10.4049/jimmunol.164.4.2102. [DOI] [PubMed] [Google Scholar]
- 26.Rauser CL, Mueller LD, Rose MR. Aging, fertility, and immortality. Exp Gerontol. 2003;38:27–33. doi: 10.1016/s0531-5565(02)00148-1. [DOI] [PubMed] [Google Scholar]
- 27.Martin CJ, Chihara S, Chang DB. A comparative study of the mechanical properties in aging alveolar wall. Am Rev Respir Dis. 1977;115:981–988. doi: 10.1164/arrd.1977.115.6.981. [DOI] [PubMed] [Google Scholar]
- 28.Taub DD, Longo DL. Insights into thymic aging and regeneration. Immunol Rev. 2005;205:72–93. doi: 10.1111/j.0105-2896.2005.00275.x. [DOI] [PubMed] [Google Scholar]
- 29.Lakatta EG, Levy D. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: Part I: aging arteries: a “set up” for vascular disease. Circulation. 2003;107:139–146. doi: 10.1161/01.cir.0000048892.83521.58. [DOI] [PubMed] [Google Scholar]
- 30.Riggs BL, Wahner HW, Dunn WL, Mazess RB, Offord KP, Melton LJ., III Differential changes in bone mineral density of the appendicular and axial skeleton with aging: relationship to spinal osteoporosis. J Clin Invest. 1981;67:328–335. doi: 10.1172/JCI110039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gilhar A, Ullmann Y, Karry R, Shalaginov R, Assy B, Serafimovich S, Kalish RS. Aging of human epidermis: reversal of aging changes correlates with reversal of keratinocyte fas expression and apoptosis. J Gerontol A Biol Sci Med Sci. 2004;59:411–415. doi: 10.1093/gerona/59.5.b411. [DOI] [PubMed] [Google Scholar]
- 32.Gilhar A, Ullmann Y, Karry R, Shalaginov R, Assy B, Serafimovich S, Kalish RS. Aging of human epidermis: the role of apoptosis, Fas and telomerase. Br J Dermatol. 2004;150:56–63. doi: 10.1111/j.1365-2133.2004.05715.x. [DOI] [PubMed] [Google Scholar]
- 33.Evers BM, Townsend CM, Jr, Thompson JC. Organ physiology of aging. Surg Clin North Am. 1994;74:23–39. doi: 10.1016/s0039-6109(16)46226-2. [DOI] [PubMed] [Google Scholar]
- 34.Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, Ohyama Y, Kurabayashi M, Kaname T, Kume E, et al. Mutation of the mouse klotho gene leads to a syndrome resembling aging. Nature. 1997;390:45–51. doi: 10.1038/36285. [DOI] [PubMed] [Google Scholar]
- 35.Razzaque MS, St-Arnaud R, Taguchi T, Lanske B. FGF-23, vitamin D and calcification: the unholy triad. Nephrol Dial Transplant. 2005;20:2032–2035. doi: 10.1093/ndt/gfh991. [DOI] [PubMed] [Google Scholar]
- 36.Tsujikawa H, Kurotaki Y, Fujimori T, Fukuda K, Nabeshima Y. Klotho, a gene related to a syndrome resembling human premature aging, functions in a negative regulatory circuit of vitamin D endocrine system. Mol Endocrinol. 2003;17:2393–2403. doi: 10.1210/me.2003-0048. [DOI] [PubMed] [Google Scholar]