

Abstract
The ability of heparin to block proliferation of vascular smooth muscle cells has been well documented. It is clear that heparin treatment can decrease the level of ERK activity in vascular smooth muscle cells that are sensitive to heparin. In this study, the mechanism by which heparin induces decreases in ERK activity was investigated by evaluating the dual specificity phosphatase, MKP-1, in heparin treated cells. Heparin induced MKP-1 synthesis in a time and concentration dependent manner. The time-course of MKP-1 expression correlated with the decrease in ERK activity. Over the same time frame, heparin treatment did not result in decreases in MEK-1 activity which could have, along with constitutive phosphatase activity, accounted for the decrease in ERK activity. Antibodies against a heparin receptor also induced the synthesis of MKP-1 along with decreasing ERK activity. Blocking either phosphatase activity or synthesis also blocked heparin-induced decreases in ERK activity. Consistent with a role for MKP-1, a nuclear phosphatase, heparin treated cells exhibited decreases in nuclear ERK activity more rapidly than cells not treated with heparin. The data support MKP-1 as a heparin-induced phosphatase that dephosphorylates ERK, decreasing ERK activity, in vascular smooth muscle cells.
Keywords: MKP-1, vascular smooth muscle, heparin, ERK
Vascular smooth muscle cell (VSMC) proliferation induced by growth factors released from injured endothelial cells and/or stimulated platelets is a significant component in the process of atherosclerosis and also plays a role in the restenosis that often occurs after angioplasty [Ross, 1999]. A major pathway leading from growth factor receptors into the cell is the mitogen activated protein kinase (MAPK) pathway. The proliferation specific MAPKs (referred to as ERK1 and ERK2 for extracellular signal-regulated kinase), are proline directed serine/threonine kinases activated by a kinase termed MEK1 (MAPK/ERK kinase) which phosphorylates ERK on both threonine and tyrosine [Garrington and Johnson, 1999]. Active ERK has been shown to accumulate in the nuclei of stimulated and cycling cells where the enzyme plays a key role in G1 progression [Brunet et al., 1999]. Both the strength and duration of the ERK activation are important for cell cycle progression [Stork, 2002]. Among other effects, ERK activity appears to induce several G1 cell cycle phase-related changes including increased cyclin D1 expression [Lavoie et al., 1996], cyclin-dependent kinase 2 (CDK2) accumulation and movement to the nucleus [Lents et al., 2002], and decreases in p27kip1 protein levels [Sakakibara et al., 2005].
Heparin has been widely studied as a potential mechanism to neutralize or reverse many steps in the atherogenic process. Among heparin’s actions in the vasculature is its ability to decrease proliferation of heparin-sensitive VSMCs by blocking progression through G1 [Reilly et al., 1989]. In VSMCs, heparin treatment results in inhibition of a protein kinase C-dependent pathway involved in cell cycle progression, blocks second messenger pathways for FOS expression [Ottlinger et al., 1993] and decreases ERK activation [Ottlinger et al., 1993; Pukac et al., 1997; Savage et al., 2001]. There is also evidence that the G1 phase increase in p27kip1 protein levels resulting from heparin-induced ERK activity decreases is critical for the heparin-induced decreases in vascular smooth muscle cell proliferation because the stable accumulated p27kip1 prevents activation of CDK2 [Fasciano et al., 2005].
Endothelial cells carry heparan sulfate proteoglycans that have anticoagulant properties [Marcum et al., 1986], raising the effective concentration of heparin-like materials adjacent to the VSMCs. These heparan sulfate chains have been shown to act similarly to heparin in blocking smooth muscle cell proliferation [Castellot et al., 1981]. Since wounded endothelial cell layers alter their synthesis of glycosaminoglycan chains in favor of chondroitin sulfate [Kinsella and Wight, 1986], the heparan sulfate in the basal lamina of damaged vessels is lower in concentration.
Despite the documentation of heparin effects in cultured VSMCs, relatively little is known about the mechanism(s) by which heparin alters ERK activity and VSMC proliferation. One possible mechanism by which heparin could function is through heparin’s interactions with growth factors [Chua et al., 2004]. However, the lack of changes in PDGF receptor kinase activity and the lag in MAPK activity decreases [Pukac et al., 1997; Savage et al., 2001] suggest that this is not the only mechanism by which heparin down-regulates MAPK activity. Heparin binding to smooth muscle cells has been known for some time [Castellot et al., 1985] and the identification of a putative heparin receptor provided another possible site for heparin interaction [Patton et al., 1995]. Evidence from our laboratory indicates that antibodies to this heparin receptor can mimic heparin by decreasing ERK activity and DNA synthesis [Savage et al., 2001].
The lag time between ERK activation and inactivation indicates that heparin’s effects on ERK involve down-regulation of activity rather than simply a block in activation [Pukac et al., 1997; Savage et al., 2001]. This suggests that one or more phosphatases are involved in the heparin-induced ERK activity decrease. MKP-1 is the original member of a family of dual-specificity phosphatases that can remove phosphates from tyrosine and threonine in ERK and related kinases [Kondoh and Nishida, 2007]. Three different families of MKP enzymes have been cloned and characterized, for a review see [Kondoh and Nishida, 2007]. Many of these phosphatases are localized in the nuclei of cells, inactivating the MAPK enzymes only after they move to the nuclei. MKP-1 is the most thoroughly studied member of the MKP family. It is an immediate-early gene, being rapidly upregulated in response to ERK pathway activation and resulting in ERK inactivation in VSMCs after its concentration rises [Duff et al., 1993]. Therefore, MKP-1 limits the extent of ERK activity, consistent with a role in the cell cycle. It is also short-lived, allowing ERK activity to increase again later in the cycle [Duff et al., 1995]. In addition to its identification as an immediate-early gene, MKP-1 can be induced by other signals such as insulin [Begum, 1998], atrial natriuretic peptide [Furst et al., 2005], adhesion to fibronectin [Kim and Corson, 2000], glucocorticoid hormones [Wu et al., 2005], arachidonic acid [Metzler et al., 1998], contact inhibition [Wayne et al., 2006] and the endocannabinoid, anandamide [Eljaschewitch et al., 2006]. Evidence from many studies, including the use of knockout mice, has supported a conclusion that MKP-1 is a critical player in inflammatory situations [Li et al., 2009].
Insulin and angiotensin, not normally mitogenic to VSMC, both result in increased VSMC ERK activation and rapid MKP-1 synthesis [Begum, 1998; Duff et al., 1993]. Similar rapid MKP-1 synthesis after oxidative stress limits the extent of Jun N-terminal kinase (JNK) activity [Teng et al., 2007], and both vascular endothelial growth factor (VEGF) and thrombin induce MKP-1 in endothelial cells limiting the extent of ERK and JNK activity [Kinney et al., 2008]. Contact-inhibited fibroblasts express higher levels of MKP-1, MKP-2 and MKP-3 than subconfluent cells, while cells whose proliferation is not contact inhibited show no change of MKP expression upon reaching confluence [Wayne et al., 2006]. In addition, phosphorylation by MAPKs [Brondello et al., 1999] can result in MKP-1 activation and increased half-life, while extensive phosphorylation by ERK leads to ubiquitin-mediated degradation [Lin et al., 2003] as does PKCδ phosphorylation [Choi et al., 2006]. However, HSP70 association can protect MKP-1 from proteasome-dependent degradation [Zheng et al., 2006] as can stabilization by HuR and NF90 binding [Kuwano et al., 2008]. The interactions between ERK and MKP-1 appear to be a finely tuned control system that allows either ERK inactivation or MKP-1 degradation depending on the amount of ERK activation in the system [Lin and Yang, 2006].
Many reports of ERK inactivation identify MKP-1 or other MKP family members as involved in decreasing ERK activity during cell cycle progression and after specific signals that do not result in continued ERK activity. Recently, it has even become clear that different dual-specificity phosphatases can cause dephosphorylated ERK to remain in the nucleus [Caunt et al., 2008]. Since cell proliferation involves ERK movement to the nucleus, induction of the nuclear enzyme MKP-1 is a logical mechanism by which heparin could inactivate VSMC ERK and thereby suppress cell proliferation. The focus of the present study was to determine whether heparin induces the synthesis of MKP-1 in VSMCs and whether the increased MKP-1 is important for heparin-induced decreases in ERK activity.
EXPERIMENTAL PROCEDURES
Cell culture
VSMCs from porcine aortas were obtained and cultured as described previously [Savage et al., 2001]. For some experiments, porcine aortic VSMCs were purchased from Cell Applications (San Diego, CA). Briefly, cells were grown in DMEM (Dulbecco’s Modified Eagle’s Medium, Sigma) with 10% heat-inactivated fetal bovine serum (Atlanta Biologicals, BioSource, or Invitrogen) and 100 μg/ml streptomycin/100 U/ml penicillin (Sigma). Cell lines were examined for the presence of smooth muscle actin and myosin to ascertain their smooth muscle phenotype [Savage et al., 2001]. A7r5 (rat vascular cells with smooth muscle cell characteristics, #CRL 1444) were obtained from ATCC and cultured as recommended. This cloned line was employed for reproducibility. Hybridoma cell lines producing antibodies against the putative heparin receptor were grown and antibodies isolated as described previously [Patton et al., 1995; Savage et al., 2001].
PMA activation and heparin treatments
Cells were cultured as above and a single line of cells was used for a given experiment. Cells were plated at about 30% to 50% confluence and allowed to initiate growth. When the cultures reached 60–70% confluence, cells were starved by changing the culture media to 0.4% serum for porcine cells or serum-free for A7r5 cells. Typically, cells were starved for about 48 hrs. and then activated with 50 ng/ml PMA or addition of serum from one to ten percent for various times as noted. Pre-treatment with heparin (at concentrations noted in the text) typically occurred 10 min. prior to activation. In some experiments, anti-heparin receptor antibodies were added in place of heparin as previously described [Savage et al., 2001]. Vanadate treatment, when used, was 100 μM sodium ortho-vanadate added 5 min. prior to addition of PMA. Doxorubicin, previously reported to decrease MKP-1 synthesis [Small et al., 2003], was used at various concentrations as noted in the text. Doxorubicin was added prior to addition of heparin for various times as noted in the text. Activation was consistently carried out with serum in doxorubicin experiments.
SiRNA treatments were carried out as recommended by the supplier. Serum-induced activation of siRNA treated cells occurred in the recommended siRNA procedures (Santa Cruz Biotechnology). Therefore, heparin effects in the siRNA treated cells were consistently evaluated after addition of heparin to cells in serum containing “recovery” media.
Immunoblotting and active enzyme detection
Western blot analysis of smooth muscle cell samples was carried out as previously described [Savage et al., 2001]. Relative protein levels were determined by laser scanning densitometry of the developed blots, and statistical analyses were carried out as described previously [Savage et al., 2001]. Anti-MKP-1 antibodies were obtained from Santa Cruz Biotechnology. ERK activity was determined either by using anti-phospho-ERK antibody (Cell Signaling) as the primary antibody or using pan ERK antibody and the gel-shift of active ERK as described previously [Savage et al., 2001]. A non-specific protein band recognized by extravidin-alkaline phosphatase was used to confirm identical loading of samples and was employed to match density between different experiments (e.g. in Figure 1) [Savage et al., 2001]. Graphs represent averages from three experiments unless otherwise noted.
Figure 1. Heparin effects on MKP-1 levels in VSMCs.

Porcine VSMCs were grown to 70% confluence and starved for 48 hr. After starvation, cells were incubated with 100 μg/ml heparin (panel A) for various times. Cells were harvested as described in Methods and MKP-1 levels determined by Western blot analysis using anti-MKP-1 antibody. Panel A shows a representative blot of data from the five experiments included in the graph plotted below indicating increased MKP-1 levels over time of heparin treatment (panel B). Panel C, illustrates effects of heparin on porcine VSMCs over a heparin concentration range. Panel D illustrates the results when starved A7r5 cells were treated for 10 min with heparin at a wide range of concentrations.
Immunofluorescence
For fluorescence microscopy, cells were seeded onto sterile glass coverslips in individual wells of 6-well plates. Cells were grown, starved, and stimulated as for cells in standard culture dishes. In order to reproducibly stain ERK and MKP-1 in the nucleus, the cells were treated with 30 μg/ml digitonin by adding digitonin to the treatment media 5 min. before harvest. The cells were fixed with ice-cold methanol for 5 min., placed in primary antibody (at the concentrations recommended by the suppliers) at 4°C for 12 to 24 hr, incubated with secondary antibody conjugated to a fluorescent tag (Jackson) as noted in the text, and mounted using Mowoil (Calbiochem, San Diego, CA). The stained cells were examined and photographed under epi-fluorescence with a NIKON Optiphot™ microscope or a Zeiss LSM 500 confocal microscope as described previously [Hamel et al., 2006].
RESULTS
Heparin induces MKP-1 expression
To determine whether the heparin-induced differences in ERK activation [Savage et al., 2001] might be explained by increased levels of MKP-1, VSMCs were treated with heparin for various time points up to 60 min. Figure 1, panel A, illustrates an increase in MKP-1 levels detected within ten min. after heparin treatment and a continuing rise in MKP-1 levels for most of the hour. The graph (panel B) illustrates data compiled from five experiments. Similar increases in MKP-1 were observed in heparin-treated porcine aortic smooth muscle cells (Figure 1C). The heparin effect on MKP-1 levels was concentration dependent in VSMCs (Figure 1C). An expanded heparin range (Figure 1D) in A7r5 cells shows that the concentration dependence is true in both cell types and is maximal at about 300 μg/ml heparin. A ten min. time point was chosen for the concentration dependence because the time frame indicated that a low amount of induction could be observed. At longer times, closer to the maximal time point, it might have been difficult to observe further increased MKP-1 induction at higher concentrations of heparin.
Heparin-induced decreases in ERK activity are observed in the nucleus
VSMCs treated with heparin expressed MKP-1 primarily in the nucleus as expected. By 10 min. of heparin treatment additional nuclear staining was observed and the maximal increase was at about 30 min (Figure 2A,B). If heparin-induced MKP-1 is responsible for the heparin-induced decrease in ERK activity, a loss of active ERK in the nuclei should occur in heparin treated cells. Therefore, VSMC activated with PMA were examined by immunofluorescence for the location of ERK and active ERK. Figure 2 illustrates the initial accumulation of active ERK in the nuclei of cells treated with PMA (C,D,E) or PMA with heparin (F,G,H). Heparin-treated cells typically exhibit similar patterns of total ERK localization at 5 min. with relatively low levels of active ERK in the cytoplasm and nucleus, although there may be somewhat less active ERK in the nuclei of heparin-treated cells. After 15 min. of PMA treatment, many heparin-treated cells have decreased levels of active ERK in their nuclei compared to active ERK in the nuclei of cells treated with PMA alone. This pattern of decreased nuclear active ERK in heparin-treated cells continues through 30 min (e.g. arrows in 2H with heparin compared to 2E without heparin). During this same period there was no obvious change in overall ERK distribution in PMA-treated cells, and some clearing of nuclear ERK in heparin-treated cells as detected by a PAN ERK antibody (not shown). Little active ERK was detected in cells not stimulated with PMA (C) and staining was not seen in cells treated with secondary antibodies only. The relative levels of active ERK in the nuclei and cytoplasm of activated cells varied between experiments and between different smooth muscle cell cultures. However, the population of heparin-treated cells always exhibited lower levels of active ERK in their nuclei relative to their cytoplasm, while the accumulation of active ERK in the nuclei of cells activated without heparin was always observed.
Figure 2. MKP-1 and active ERK in heparin treated VSMCs.

A7r5 cells were seeded on coverslips, grown to 60% confluence and starved as described in Methods. Cells were treated with heparin at 100 μg/ml for varying times (A - no treatment, B - 30 min.). Other cells were treated with heparin at 200 μg/ml (F–H) or left untreated (C–E) followed by PMA treatment after 10 min. Incubation in PMA was for 15 min (D, G) or 30 min (E, H). All cells were treated with digitonin for the last five min of the treatment to facilitate nuclear staining. Samples A and B were fixed and stained using a primary antibody against MKP-1 and a lissamine rhodamine-labeled secondary antibody. Anti-active MAPK from Cellular Signaling (C–H) antibodies were used as the primary antibody followed by FITC-labeled anti-rabbit secondary antibodies. Stained cells were mounted as described in Methods. Photographs were taken at 40× magnification, converted to grayscale using Fireworks, and reproduced at the same magnifications.
Heparin does not increase MEK1 activity
MEK1 has been identified as the upstream activator of ERK. To determine whether heparin also decreased the level of MEK activity in the same time frame as it altered ERK activity, Western Blots of treated cell samples prepared as above were analyzed for MEK activity. Figure 3A illustrates levels of active MEK1 in heparin-treated and untreated cells activated with PMA. There was no significant difference in the level of phospho-MEK1 in heparin-treated cells over the time period when ERK activity was decreased. Similar results were obtained with porcine cells. A representative experiment is shown in Figure 3B, where the cells were activated with serum instead of PMA. Over the time frame studied, heparin has no effect on MEK1 activity levels in either PMA or serum-treated VSMCs.
Figure 3. Heparin effects on active MEK.
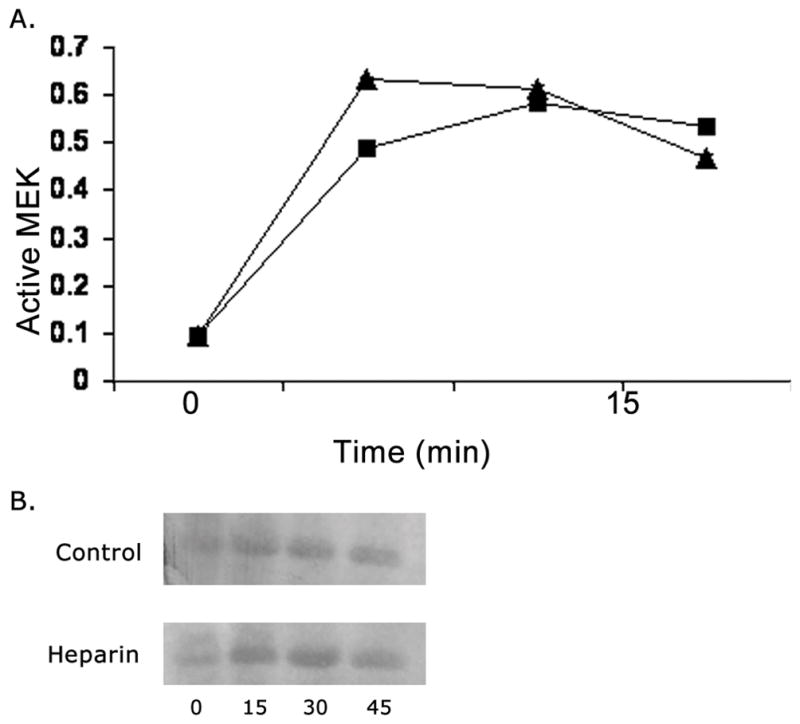
Panel A: A7r5 cells were grown, starved, untreated or treated with 100 μg/ml heparin, activated with PMA and analyzed by Western blotting using an antibody against active MEK. Squares indicate heparin treated cells, while triangles show data from cells not treated with heparin. The time starts with PMA addition. Each point is the average from three experiments, error bars fall within the data points. Panel B: Experiments identical to those in panel A were carried out with porcine VSMCs, but using serum for activation. A representative blot is illustrated.
Anti-heparin receptor antibodies mimic heparin by inducing MKP-1 expression
Previous studies with anti-heparin receptor antibodies indicated that the antibodies caused a decrease in VSMC ERK activity similar to that induced by heparin [Savage et al., 2001]. To determine whether these antibodies also induced MKP-1 increases, cells were treated with heparin or anti-heparin receptor antibodies at concentrations shown to be effective in decreasing ERK activity. In these experiments the antibody-treated cells had higher levels of MKP-1 at 10 min. of treatment than untreated controls (Figure 4). Cells treated with 100 μg/ml heparin for 10 min. exhibited higher levels of MKP-1 than the controls, but lower levels than shown in the antibody-treated cells. However, as shown in Figure 1, 100 μg/ml heparin did not induce maximum levels of MKP-1.
Figure 4. Anti-heparin receptor antibody effects on MKP-1 levels.
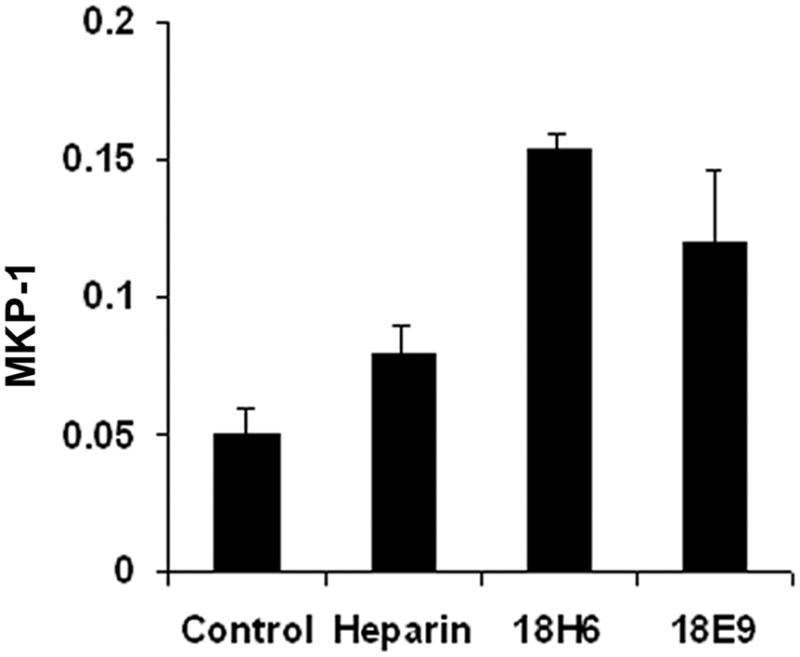
A7r5 cells were grown to 60% confluence, starved, and treated for 10 min. with 100 μg/ml heparin, 0.8 μg/ml antibody 18H6, 0.9 μg/ml antibody 18E9, or left untreated. MKP-1 levels were determined by Western blotting and represent the averages of three trials.
Blocking phosphatase activity protects ERK activity from heparin-induced decreases
To confirm that phosphatase activity is required for heparin-induced ERK activity decreases, we blocked dual specificity and tyrosine phosphatases with vanadate. Cells were treated with vanadate prior to heparin and PMA treatments. The porcine cells rapidly rounded up in the presence of vanadate and were not evaluated. The A7r5 cells also rounded up in the presence of vanadate. However, that process did not begin until almost 30 min. after vanadate addition. Therefore, A7r5 cells were tested at a 15 min. time point. At 15 min. after PMA stimulation, heparin-treated cells typically have significantly less active ERK than those activated without heparin treatment [Savage et al., 2001]. Vanadate had no significant effect on the ability of PMA to activate ERK, although total levels of phospho-ERK were often slightly higher in vanadate-treated cells than cells not treated with vanadate. PMA-activated cells treated with both heparin and vanadate had levels of phospho-ERK similar to the levels in activated cells without heparin (Figure 5). The cells treated with heparin and PMA had significantly less phospho-ERK than PMA alone, PMA with vanadate and PMA with heparin and vanadate (p < 0.05 in each case). Sanguinarine was reported in the literature to specifically decrease MKP-1 activity [Vogt et al., 2005], but also resulted in cell loss beyond about 15 min of incubation (data not shown).
Figure 5. Vanadate intereferes with heparin effects on MAPK activity.
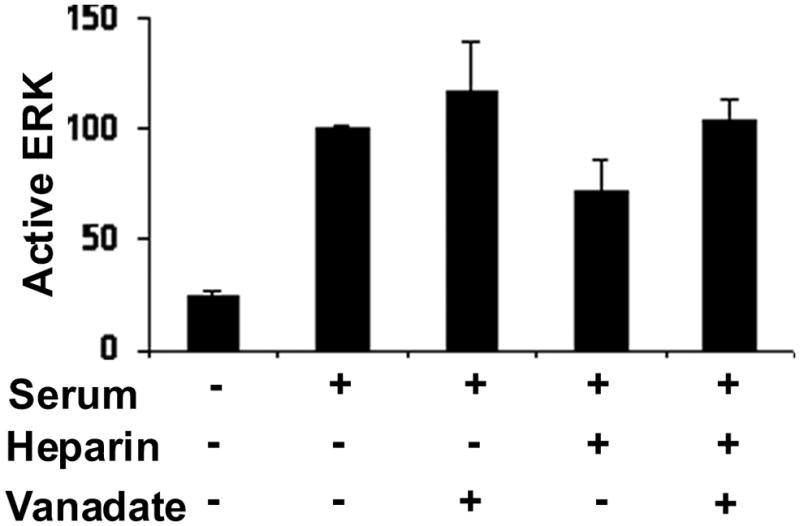
A7r5 cells were grown to approximately 60% confluence, starved and treated with 100 μg/ml heparin, 100 μM sodium vanadate, and activated with PMA for 15 min or left untreated as described in Methods. Active ERK was determined by Western blotting as described in the Methods. The absorbance determined for the PMA samples in each experiment was set to 100% and the other values were calculated as a percent of the PMA only samples to facilitate compiling the data from four experiments. The differences between PMA and PMA with vanadate and PMA with heparin and vanadate are not significant. All three are significantly different from PMA and heparin (p<0.05 for each case).
MKP-1 synthesis is required for heparin-induced decreases in ERK activation
Blocking phosphatase activity was accompanied by increased ERK activation in heparin-treated cells. However, the effects of blocking phosphatase activity were more dramatic than simple increases in ERK phosphorylation. Therefore, we evaluated the effects of blocking MKP-1 synthesis on heparin-induced decreases in ERK activity. For these experiments, the cells were more confluent than in the Figure 1 studies, so the basal MKP-1 levels are higher than in Figure 1. Doxorubicin has been shown to block MKP-1 synthesis [Small et al., 2003] and was used in the present study for the same purpose. Treatment of A7r5 cells with doxorubicin decreased MKP-1 synthesis induced by heparin in a concentration-dependent manner (Figure 6, panel A). Further, in the absence of doxorubicin, heparin decreased the amount of active ERK as seen previously, even with the higher basal MKP-1 levels. However, in the presence of doxorubicin, heparin-treated cells exhibited almost the same levels of active ERK as did the cells activated without heparin (Figure 6, panel B). Anti-heparin receptor antibody-treated cells responded to doxorubicin identically to heparin-treated cells (Figure 6, panels C and D) showing decreased ability of the antibodies to induce MKP-1 synthesis and decreased ability to decrease ERK activation.
Figure 6. Doxorubicin blocks MKP-1 synthesis and protects ERK activity.

A7r5 cells were grown to 75% confluence and starved. Cells were pre-treated with doxorubicin (for one hour) at varying concentrations and then treated with heparin, at 200 μg/ml for 30 min., to induce MKP-1 synthesis (panel A). The level of MKP-1 present with no doxorubicin (but with heparin) was set as 1.0. Other cells were treated with or without doxorubicin (one hr at 10 μM) followed by addition of 200 μg/ml heparin for 10 min. prior to activation with serum for varying times, and ERK activity was determined by western blotting with antibodies recognizing phosphorylated ERK (panel B). Samples without heparin that were activated for 20 min. were set equal to 1.0. Similar cell samples to those in Panel A were treated with or without doxorubicin and 18E9 anti-heparin receptor antibodies (0.8 μg/ml) and MKP-1 levels were determined (panel C). Doxorubicin and anti-heparin receptor antibodies were used to treat cells analogous to experiments in panel B followed by serum treatment (panel D) for evaluation of active ERK levels. In panels B and D, black bars represent cells treated with doxorubicin, but without heparin (B) or antibodies (D). Dark grey bars represent no doxorubicin and no heparin (B) or antibodies (D). Light grey bars represent cells treated with doxorubicin and heparin (B) or antibodies (D). White bars represent cells treated without doxorubicin, but with heparin (B) or antibodies (D). In each panel, averages from four separate experiments are plotted. The results were analyzed by ANOVA. In panels A and C, doxorubicin treatment results in significant decreases in MKP-1 levels (p<0.05). In panels B and D, doxorubicin treated samples showed no significant difference in activation with heparin or antibodies compared to serum activation, while without doxorubicin, both heparin (panel B) and anti-heparin receptor antibody (panel D) treatment resulted in significant decreases in ERK activity (p<0.05).
To ensure that the effects of doxorubicin were due to inhibition of MKP-1 synthesis rather than some other doxorubicin response, cells were treated with siRNA to specifically eliminate MKP-1 expression. First, we confirmed the MKP-1 siRNA ability to block heparin-induced MKP-1 synthesis (Figure 7A, light bars vs. control siRNA, dark bars). The recommended protocol involves a recovery period in serum-containing media after the siRNA treatment in serum-free media. Therefore, we altered the protocol for heparin treatment so that it occurred in the serum-containing media rather than prior to serum treatment of starved cells. Heparin treatment of the cells in serum-containing media also resulted in decreased ERK activation. A7r5 cells treated with control siRNA had reduced ERK activity after heparin treatment (Figure 7B, dark bars) while identical cells with MKP-1 siRNA had ERK activity levels that remained high despite treatment with heparin (Figure 7B, light bars). Porcine VSMC treated with siRNA to MKP-1 also failed to accumulate MKP-1 protein, while those treated with control siRNA did accumulate MKP-1 in response to heparin (Figure 7C). When MKP-1 siRNA treated porcine cells were treated with 200 μg/ml heparin followed by a fresh addition of 1% serum five minutes later, ERK activity was much higher in the MKP-1 siRNA treated cells than in the control siRNA treated cells (Figure 7D).
Figure 7. Inhibition of MKP-1 induction using siRNA protects ERK activity.

A7r5 cells were grown to 75% confluence and placed into transfection media for siRNA treatment as recommended. After transfection with either MKP-1 siRNA (light grey bars) or control siRNA (black bars), cells were placed back into regular growth media for recovery as recommended. After 24 hours recovery, cells were treated with 200 μg/ml heparin for varying times, harvested, and Western blots were developed for MKP-1 (panel A) and active ERK (panel B). Panel A represents a single experiment where the 30 min. MKP-1 level in cells with control siRNA was set as 1.0. The experiment in panel B was repeated four times and averages from these experiments are shown along with standard deviations. The active ERK levels in cells with MKP-1 siRNA were set at 1.0 for purposes of comparison and data analysis for the four experiments. The control siRNA samples contain significantly less active ERK than those samples treated with MKP-1 siRNA (p<0.05). Porcine VSMC treated with MKP-1 siRNA or control siRNA were treated with 200 μg/ml heparin for times noted and Western blots were developed with antibodies to MKP-1 (panel C). Panel D illustrates blotting for active ERK. Times shown represent time after heparin addition. To increase ERK activity, fresh serum was added (1% of total volume) 5 min after heparin. Antibodies against phospho-ERK were used for development.
DISCUSSION
Research on inactivation of MAPK enzymes indicates that specific phosphatases are synthesized as immediate early genes [Sun et al., 1993]. These phosphatases can selectively inactivate kinases in the MAPK cascades returning the cell to an inactivated state. Much interest has been focused on the dual-specificity family of phosphatases whose members can dephosphorylate both threonine and tyrosine residues in members of the MAPK family [Dickinson and Keyse, 2006].
Published data indicate that heparin can negatively influence the ability of VSMCs to divide after specific growth factor stimulation. Heparin-induced decreases in VSMC proliferation have been determined to be mediated by stabilization and increased levels of p27kip1 protein [Fasciano et al., 2005]. ERK activity had previously been known to decrease p27kip1 levels [Sakakibara et al., 2005], but the identification of p27kip1 as an ERK target that was important for heparin effects on proliferation had not been clear. Understanding how heparin could dampen ERK activity has been complicated by the ability of heparin to interact with growth factors in stimulatory (and possibly inhibitory) manners. In addition, evidence suggests that heparin can block the ERK activity after direct stimulation of protein kinase C, bypassing the growth factors entirely [Ottlinger et al., 1993]. Heparin treatment also results in delayed inactivation of ERK [Pukac et al., 1997; Savage et al., 2001]. Together, these data indicate that heparin may influence cell growth through interactions with proteins other than growth factors. The identification of a putative heparin receptor [Patton et al., 1995] and evidence that antibodies against that receptor mimic heparin effects [Savage et al., 2001] support the hypothesis that the ERK pathway is intersected within the cell by a heparin-induced signal.
Therefore, the possibility that heparin could induce MKP-1 increases was examined. Heparin treatment of vascular smooth muscle cells results in the time and concentration dependent increase of MKP-1 protein, a dual specificity phosphatase that normally is expressed as an immediate early gene (Figure 1). Under growth factor stimulation conditions, the ERK activity typically continues long enough to result in synthesis of immediate early gene products including MKP-1. ERK activity is then decreased and the cells either return to a resting state or continue through the cell cycle depending upon whether the cell was stimulated sufficiently to pass the G1 restriction point. In heparin-treated cells, initial growth factor activation of ERK in the cytoplasm may result in ERK phosphorylation of cytoplasmic proteins and transport of the ERK to the nuclei as usual. However, once in the nucleus, the ERK would be inactivated by the heparin-induced MKP-1 before it could phosphorylate sufficient transcription factors and other targets resulting in decreased p27kip1 levels. Immunofluorescent data indicate that levels of active ERK decrease in heparin-treated cells (Figure 2). In cells that are activated by serum or PMA and treated with heparin it was possible to observe cells with very low levels of active ERK in the nucleus consistent with ERK inactivation occurring in the nucleus (Figure 2).
Further confirmation that heparin induction of MKP-1 is involved in heparin responses includes the fact that the MEK1 (the dual function kinase responsible for activating ERK) activation is not decreased by heparin treatment. Levels of activated MEK1 rise upon PMA or growth factor treatment of the starved cells, but over the time course examined in our studies are not any lower in cells treated with heparin than in activated cells without heparin treatment. Data in Figure 3 indicate no significant difference in the levels of active MEK between heparin-treated, activated, and control activated cells over 45 min. To confirm that tyrosine or dual-specificity phosphatase activity was important in the lower levels of activated ERK induced by heparin, cells were treated with vanadate (a general tyrosine phosphatase inhibitor). Under these conditions cells activated with PMA and treated with heparin had levels of phospho-ERK similar to PMA treated cells without heparin (Figure 5). Because vanadate inhibits many phosphatases, these data indicate phosphatase activity is important for heparin effects, but don’t specifically indicate MKP-1 involvement. Blocking MKP-1 synthesis by doxorubicin or, specifically, using MKP-1 siRNA resulted in a loss of heparin effects on ERK activation (Figures 6 and 7). This information supports the idea that inactivation of ERK is an important aspect of the mechanism by which ERK activity is altered.
Some other signals that result in the decrease of MAPK activity (or induction of ERK activity followed by rapid decrease) also increase MKP-1 levels. These include insulin [Begum, 1998], angiotensin [Duff et al., 1995], glucocorticoid hormones [Wu et al., 2005] and ANP [Furst et al., 2005]. In the heparin situation evaluated here, the vanadate, doxorubicin, and MKP-1 siRNA sensitivity of the heparin response, the specific nuclear effect of heparin on ERK activity and the MKP-1 induction by heparin and the anti-heparin receptor antibodies indicate that MKP-1 is an important player in the heparin-induced decrease in ERK activation. Recent reports indicate that doxorubicin treatment, which blocks MKP-1 induction, results in increased MAPK activity and accompanying changes in physiology indicating that MKP-1 induction can be important in modulating physiologically relevant MAPK signaling [Poizat et al., 2005]. While significant attention has been paid to evidence that MKP-1 knockout tissues have little decrease in active ERK levels, much of this work has been focused on tissues and cells involved in inflammatory responses as reviewed [Li et al., 2009], other evidence indicates that insulin-sensitive tissues and tissues where MKP-2 increases can compensate for the MKP-1 loss may show no decrease in active ERK. One example is evidence for a role of MKP-1 in diet-induced obesity [Wu et al., 2006].
Our data support the conclusion that heparin affects ERK activity levels in heparin-sensitive vascular smooth muscle cells, at least in part, by inducing the synthesis of the dual-specificity phosphatase MKP-1. This explanation fits well with evidence that rapid ERK-dependent phosphorylation is not altered by treatment of cells with heparin as there is a lag between ERK activation and heparin-induced inactivation of the ERK as has also been shown previously [Pukac et al., 1997; Savage et al., 2001]. It is possible that additional steps in the signal transduction pathway between growth factors and the nucleus are also turned off by heparin treatment. As an example, Raf inactivation in heparin-treated VSMCs was noted previously [Pukac et al., 1997] and non-steroidal anti-inflammatory drugs were shown to decrease Raf activation and increase MKP-1 production in lung cancer cells [Pan et al., 2008]. The mechanism by which Raf is inactivated in heparin treated cells has yet to be reported. In the present study, the inactivation of MEK that would need to rapidly follow Raf inactivation, if Raf inactivation in response to heparin was totally responsible for the heparin effects on ERK activity, was not observed. However, there appeared to be a slight decrease in the active MEK1 at the 45 min. time point. Our data support an alternative possibility to Raf activity being the primary target for heparin effects in VSMCs. This alternate hypothesis is that heparin treatment causes inactivation of several enzymes in the ERK pathway. Further studies will be required to determine whether MEK1 activity does decrease more rapidly in heparin-treated cells and the mechanism(s) by which Raf or other pathway components are also targets for heparin effects.
Our data indicate that there is a mechanism(s) to induce the synthesis of MKP-1 without the need for activation of transcription factors normally responsible for immediate early gene induction. Insulin [Begum, 1998], arachidonic acid [Metzler et al., 1998], angiotensin II [Duff et al., 1993], oxidative stress [Teng et al., 2007], glucocorticoid hormones [Wu et al., 2005] and atrial natriuretic peptide [Furst et al., 2005] all resulted in the synthesis of MKP-1 in various cell types. The mechanisms by which these other molecules induce MKP-1 synthesis include heat shock elements, changes in stability, cAMP and cGMP sensitive activation, glucocorticoid receptors, p53-induced transcription and Jak2 kinase-induced responses [Li et al., 2003; Wong et al., 2005; Wu et al., 2005]. The proximal promoter for MKP-1 includes multiple cis-acting elements indicating a complex array of possibilities by which heparin might function [Ryser et al., 2004]. Additional studies must be carried out to determine which of these mechanisms is involved in increasing MKP-1 levels in response to heparin. Such studies should also explore the possibility that a coordinately regulated set of inactivation steps for the ERK pathway exists.
Acknowledgments
The early MAPK localization studies of Joseph Enama and John P. Grason and technical support of Felix Molina are gratefully acknowledged. We also gratefully acknowledge the help of Maria Brace with figure preparation.
The abbreviations used are
- CDK2
cyclin-dependent kinase 2
- DMEM
Dulbecco’s Modified Eagle’s Medium
- ERK
extracellular signal-regulated kinase
- FITC
fluorescein isothio-cyanate
- JNK
Jun N-terminal kinase
- MAPK
mitogen activated protein kinase
- MEK1
MAPK/ERK kinase
- PMA
phorbol-12-myristate-13-acetate
- VEGF
vascular endothelial growth factor
- VSMC
vascular smooth muscle cell
Footnotes
Contract Grant Sponsor: NIH; Contract grant number: HL54269 to LJL-K.
References
- Begum N, Ragolia L, Rienzie J, McCarthy M, Duddy N. Regulation of mitogen-activated protein kinase phosphatase-1 induction by insulin in vascular smooth muscle cells. J Biol Chem. 1998;273:25164–25170. doi: 10.1074/jbc.273.39.25164. [DOI] [PubMed] [Google Scholar]
- Brondello J, Pouyssegur J, McKenzie FR. Reduced MAPK phosphatase-1 degradation after p42/p44MAPK-dependent phosphorylation. Science. 1999;286:2514–2517. doi: 10.1126/science.286.5449.2514. [DOI] [PubMed] [Google Scholar]
- Brunet S, Roux D, Lenomand P, Dowd S, Keyse S, Pouysségur J. Nuclear translocation of p42/p44 mitogen-activated protein kinase is required for growth factor-induced gene expression and cell cycle entry. EMBO J. 1999;18:664–674. doi: 10.1093/emboj/18.3.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellot JJJ, Addonizio ML, Rosenberg R, Karnovsky MJ. Cultured endothelial cells produce a heparinlike inhibitor of smooth muscle cell growth. J Cell Biol. 1981;90:372–379. doi: 10.1083/jcb.90.2.372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellot JJJ, Wong K, Herman B, Hoover RL, Albertini DF, Wright TC, Caleb BL, Karnovsky MJ. Binding and internalization of heparin by vascular smooth muscle cells. J Cell Physiol. 1985;124:13–20. doi: 10.1002/jcp.1041240104. [DOI] [PubMed] [Google Scholar]
- Caunt CJ, Rivers CA, Conway-Campbell BL, Norman MR, McArdle CA. Epidermal growth factor receptor and protein kinase C signaling to ERK2. Spatiotemporal regulation of ERK2 by dual specificity phosphatases. J Biol Chem. 2008;283:6241–6252. doi: 10.1074/jbc.M706624200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi B-H, Hur E-M, Lee J-H, Jun D-J, Kim K-T. Protein kinase Cδ-mediated proteasomal degradation of MAP kinase phosphatase-1 contributes to glutamate-induced neuronal cell death. J Cell Sci. 2006;119:1329–1340. doi: 10.1242/jcs.02837. [DOI] [PubMed] [Google Scholar]
- Chua C, Rahimi N, Forsten-Williams K, Nugent MA. Heparan sulfate proteoglycans function as receptors for fibroblast growth factor-2 activation of extracellular signal-regulated kinases 1 and 2. Circ Res. 2004;94:316–323. doi: 10.1161/01.RES.0000112965.70691.AC. [DOI] [PubMed] [Google Scholar]
- Dickinson R, Keyse S. Diverse physiological functions for dual-specificity MAP kinase phosphatases. J Cell Sci. 2006;119:4607–4615. doi: 10.1242/jcs.03266. [DOI] [PubMed] [Google Scholar]
- Duff JL, Marrero MB, Paxton WG, Charles CH, Lau LF, Bernstein KE, Berk BC. Angiotensin II induces 3CH134, a protein tyrosine phosphatase, in vascular smooth muscle cells. J Biol Chem. 1993;268:26037–26040. [PubMed] [Google Scholar]
- Duff JL, Monia BP, Berk BC. Mitogen-activated protein (MAP) kinase is regulated by the MAP kinase phosphatase (MKP-1) in vascular smooth muscle cells. J Biol Chem. 1995;270:7161–7166. doi: 10.1074/jbc.270.13.7161. [DOI] [PubMed] [Google Scholar]
- Eljaschewitch E, Witting A, Mawrin C, Lee T, Schmidt P, Wolf S, Hoertnagl H, Raine C, Schneider-Stock R, Nitsch R, Ullrich O. The endocannabinoid anandamide protects neurons during CNS inflammation by induction of MKP-1 in microglial cells. Neuron. 2006;49:67–79. doi: 10.1016/j.neuron.2005.11.027. [DOI] [PubMed] [Google Scholar]
- Fasciano S, Patel R, Handy I, Patel C. Regulation of vascular smooth muscle proliferation by heparin. Inhibition of cyclin-dependent kinase 2 activity by p27kip1. J Biol Chem. 2005;280:15682–15689. doi: 10.1074/jbc.M411458200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furst R, Brueckl C, Kuebler W, Zahler S, Krotz F, Gorlach A, Vollmar A, Kiemer A. Atrial natriuretic peptide induces mitogen-activated protein kinase phosphatase-1 in human endothelial cells via Rac1 and NAD(P)H oxidase/Nox2-activation. Circ Res. 2005;96:43–53. doi: 10.1161/01.RES.0000151983.01148.06. [DOI] [PubMed] [Google Scholar]
- Garrington TP, Johnson GL. Organization and regulation of mitogen-activated protein kinase signaling pathways. Curr Opin Cell Biol. 1999;11:211–218. doi: 10.1016/s0955-0674(99)80028-3. [DOI] [PubMed] [Google Scholar]
- Hamel M, Kanyi D, Cipolle MD, Lowe-Krentz LJ. Active stress kinases in proliferating endothelial cells associate with cytoskeletal structures. Endothelium. 2006;13:157–170. doi: 10.1080/10623320600760191. [DOI] [PubMed] [Google Scholar]
- Kim F, Corson MA. Adhesion to fibronectin enhances MKP-1 activation in human endothelial cells. Biochem Biophys Res Commun. 2000;273:539–545. doi: 10.1006/bbrc.2000.2951. [DOI] [PubMed] [Google Scholar]
- Kinney CM, Chandrasekharan U, Mavrakis L, DiCorleto PE. VEGF and thrombin induce MKP-1 through distinct signaling pathways: role of MKP-1 in endothelial cell migration. Am J Physiol Cell Physiol. 2008;294:241–250. doi: 10.1152/ajpcell.00187.2007. [DOI] [PubMed] [Google Scholar]
- Kinsella M, Wight T. Modulation of sulfated proteoglycan synthesis by bovine aortic endothelial cells during migration. J Cell Biol. 1986;102:679–687. doi: 10.1083/jcb.102.3.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondoh K, Nishida E. Regulation of MAP kinases by MAP kinase phosphatases. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 2007;1773:1227–1237. doi: 10.1016/j.bbamcr.2006.12.002. [DOI] [PubMed] [Google Scholar]
- Kuwano Y, Kim H, Abdelmohsen K, Pullmann R, Martindale J, Yang X, Gorospe M. MKP-1 mRNA stabilization and translational control by RNA-binding proteins HuR and NF90. Molec Cell Biol. 2008;28:4562–4575. doi: 10.1128/MCB.00165-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavoie J, L’Allemain G, Brunet A, Muller R, Pouyssegur J. Cyclin D1 expression is regulated positively by the p42/p44 MAPK and negatively by the p38/HOG MAPK pathway. J Biol Chem. 1996;271:20608–20616. doi: 10.1074/jbc.271.34.20608. [DOI] [PubMed] [Google Scholar]
- Lents N, Keenan S, Bellone C, Baldassare J. Stimulation of the Raf/MEK/ERK cascade is necessary and sufficient for activation and Thr-160 phosphorylation of a nuclear-targeted CDK2. J Biol Chem. 2002;277:47489–47475. doi: 10.1074/jbc.M207425200. [DOI] [PubMed] [Google Scholar]
- Li L, Chen S-F, Liu Y. MAP kinase phosphatase-1, a critical negative regulator of the innate immune response. International Journal of Clinical and Experimental Medicine. 2009;2:4867. [PMC free article] [PubMed] [Google Scholar]
- Li M, Zhou J-Y, Ge Y, Matherly L, Wu G. The phosphatase MKP1 is a transcriptional target of p53 involved in cell cycle regulation. J Biol Chem. 2003;278:41059–41068. doi: 10.1074/jbc.M307149200. [DOI] [PubMed] [Google Scholar]
- Lin Y-W, Yang J-L. Cooperation of ERK and SCFSkp2 for MKP-1 destruction provides a positive feedback regulation of proliferation signaling. J Biol Chem. 2006;281:915–926. doi: 10.1074/jbc.M508720200. [DOI] [PubMed] [Google Scholar]
- Lin Y, Chuang S, Yang J. ERK1/2 achieves sustained activation by stimulating MAPK phosphatase-1 degradation via the ubiquitin-proteasome pathway. J Biol Chem. 2003;278:21534–21541. doi: 10.1074/jbc.M301854200. [DOI] [PubMed] [Google Scholar]
- Marcum J, Atha D, Fritz L, Nawroth PP, Stern DM, Rosenberg R. Cloned bovine aortic endothelial cells synthesize anticoagulantly active heparan sulfate proteoglycans. J Biol Chem. 1986;261:7507–7517. [PubMed] [Google Scholar]
- Metzler B, Hu Y, Strum G, Wick G, Xu Q. Induction of mitogen-activated protein kinase phosphatase-1 by arachidonic acid in vascular smooth muscle cells. J Biol Chem. 1998;273:33320–33326. doi: 10.1074/jbc.273.50.33320. [DOI] [PubMed] [Google Scholar]
- Ottlinger ME, Pukac LA, Karnovsky MJ. Heparin inhibits mitogen-activated protein kinase activation in intact rat vascular smooth muscle cells. J Biol Chem. 1993;268:19173–19176. [PubMed] [Google Scholar]
- Pan M-R, Chang H-C, Hung W-C. Non-steroidal anti-inflammatory drugs suppress the ERK signaling pathway via block of Ras/c-Raf interaction and activation of MAP kinase phosphatases. Cellular Signalling. 2008;20:1134–1141. doi: 10.1016/j.cellsig.2008.02.004. [DOI] [PubMed] [Google Scholar]
- Patton WAI, Granzow CA, Getts LA, Thomas SC, Zotter LM, Gunzel KA, Lowe-Krentz LJ. Identification of a heparin-binding protein using monoclonal antibodies that block heparin binding to porcine aortic endothelial cells. Biochem J. 1995;311:461–469. doi: 10.1042/bj3110461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poizat C, Puri P, Bai Y, Kedes L. Phosphorylation-dependent degradation of p300 by doxorubicin-activated p38 mitogen-activated protein kinase in cardiac cells. Mol Cell Biol. 2005;25:2673–2678. doi: 10.1128/MCB.25.7.2673-2687.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pukac LA, Carter JE, Ottlinger ME, Karnovsky MJ. Mechanisms of inhibition by heparin of PDGF stimulated MAP kinase activation in vascular smooth muscle cells. J Cell Physiol. 1997;172:69–78. doi: 10.1002/(SICI)1097-4652(199707)172:1<69::AID-JCP8>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Reilly C, Kindy M, Brown K, Rosenberg R, Sonenshein G. Heparin prevents vascular smooth muscle cell progression through the G1 phase of the cell cycle. J Biol Chem. 1989;264:6990–6995. [PubMed] [Google Scholar]
- Ross R. Atherosclerosis. An inflammatory disease. N Engl J Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- Ryser S, Massiha A, Piuz I, Schlegel W. Stimulated initiation of mitogen-activated protein kinase phosphatase-1 (MKP-1) gene transcription involves the synergistic action of multiple cis-acting elements in the proximal promoter. Biochem J. 2004;378:473–484. doi: 10.1042/BJ20031022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakakibara K, Kubota K, Worku B, Ryer E, Miller J, Koff A, Kent C, Liu B. PDGF-BB regulates p27 expression through ERK-dependent RNA turn-over in vascular smooth muscle cells. J Biol Chem. 2005;280:25470–25477. doi: 10.1074/jbc.M502320200. [DOI] [PubMed] [Google Scholar]
- Savage JM, Gilotti AC, Granzow CA, Molina F, Lowe-Krentz LJ. Antibodies against a heparin receptor slow cell proliferation and decrease MAPK activation in vascular smooth muscle cells. J Cell Physiol. 2001;187:283–293. doi: 10.1002/jcp.1076. [DOI] [PubMed] [Google Scholar]
- Small G, Somasundaram S, Moore D, Shi Y, Orlowski R. Repression of mitogen-activated protein kinase (MAPK) phosphatase-1 by anthracyclines contributes to their antiapoptotic activation of p44/42-MAPK. J Pharmacology and experimental therapeutics. 2003;307:861–869. doi: 10.1124/jpet.103.055806. [DOI] [PubMed] [Google Scholar]
- Stork PJ. ERK signaling. Duration, duration, duration. Cell Cycle. 2002:1. doi: 10.4161/cc.1.5.145. [DOI] [PubMed] [Google Scholar]
- Sun H, Charles CH, Lau LF, Tonks NK. MKP-1 (3CH134), an immediate early gene product, is a dual specificity phosphatase that dephosphorylates MAP kinase in vivo. Cell. 1993;75:487–493. doi: 10.1016/0092-8674(93)90383-2. [DOI] [PubMed] [Google Scholar]
- Teng C-H, Huang W-N, Meng T-C. Several dual specificity phosphatases coordinate to control the magnitude and duration of JNK activation in signaling response to oxidative stress. J Biol Chem. 2007;282:28395–28407. doi: 10.1074/jbc.M705142200. [DOI] [PubMed] [Google Scholar]
- Vogt A, Tamewitz A, Skoko J, Sikorski R, Giuliano K, Lazo J. The benzo[c]phenanthridine alkaloid, sanguinarine, is a selective, cell-active inhibitor of mitogen-activated protein kinase phosphatase-1. J Biol Chem. 2005;280:19078–19086. doi: 10.1074/jbc.M501467200. [DOI] [PubMed] [Google Scholar]
- Wayne J, Sielski J, Rizvi A, Georges K, Hutter D. ERK regulation upon contact inhibition in fibroblasts. Molec Cell Biochem. 2006;286:181–189. doi: 10.1007/s11010-005-9089-z. [DOI] [PubMed] [Google Scholar]
- Wong H, Dunsmore K, Page K, Shanley T. Heat shock-mediated regulation of MKP-1. Am J Physiol Cell Physiol. 2005;289:C1152–C1158. doi: 10.1152/ajpcell.00138.2005. [DOI] [PubMed] [Google Scholar]
- Wu JJ, Roth RJ, Anderson EJ, Hong E-G, Lee M-K, Choi CS, Neufer PD, Shulman GI, Kim JK, Bennett AM. Mice lacking MAP kinase phosphatase-1 have enhanced MAP kinase activity and resistance to diet-induced obesity. Cell Metabolism. 2006;4:61–73. doi: 10.1016/j.cmet.2006.05.010. [DOI] [PubMed] [Google Scholar]
- Wu W, Pew T, Zou M, Pang D, Conzen SD. Glucocorticoid Receptor-induced MAPK Phosphatase-1 (MPK-1) Expression Inhibits Paclitaxel-associated MAPK Activation and Contributes to Breast Cancer Cell Survival. J Biol Chem. 2005;280:4117–4124. doi: 10.1074/jbc.M411200200. [DOI] [PubMed] [Google Scholar]
- Zheng Y, Im C-N, Seo J-S. Inhibitory effect of Hsp70 on angiotensin II-induced vascular smooth muscle cell hypertrophy. Exp Mol MEd. 2006;38:509–518. doi: 10.1038/emm.2006.60. [DOI] [PubMed] [Google Scholar]