

Abstract
Background
A number of reports have suggested that many of the problems currently associated with the use of microneedle (MN) arrays for transdermal drug delivery could be addressed by using drug-loaded MN arrays prepared by moulding hot melts of carbohydrate materials.
Methods
In this study, we explored the processing, handling, and storage of MN arrays prepared from galactose with a view to clinical application.
Results
Galactose required a high processing temperature (160°C), and molten galactose was difficult to work with. Substantial losses of the model drugs 5-aminolevulinic acid (ALA) and bovine serum albumin were incurred during processing. While relatively small forces caused significant reductions in MN height when applied to an aluminium block, this was not observed during their relatively facile insertion into heat-stripped epidermis. Drug release experiments using ALA-loaded MN arrays revealed that less than 0.05% of the total drug loading was released across a model silicone membrane. Similarly, only low amounts of ALA (approximately 0.13%) and undetectable amounts of bovine serum albumin were delivered when galactose arrays were combined with aqueous vehicles. Microscopic inspection of the membrane following release studies revealed that no holes could be observed in the membrane, indicating that the partially dissolved galactose sealed the MN-induced holes, thus limiting drug delivery. Indeed, depth penetration studies into excised porcine skin revealed that there was no significant increase in ALA delivery using galactose MN arrays, compared to control (P value < 0.05). Galactose MNs were unstable at ambient relative humidities and became adhesive.
Conclusion
The processing difficulties and instability encountered in this study are likely to preclude successful clinical application of carbohydrate MNs. The findings of this study are of particular importance to those in the pharmaceutical industry involved in the design and formulation of transdermal drug delivery systems based on dissolving MN arrays. It is hoped that we have illustrated conclusively the difficulties inherent in the processing and storage of carbohydrate-based dissolving MNs and that those in the industry will now follow alternative approaches.
Keywords: Carbohydrate, galactose, microneedles, processing, stability
Introduction
Microneedle (MN) arrays are minimally invasive devices that can be used to bypass the stratum corneum (SC) barrier and thus achieve enhanced transdermal drug delivery. MNs (50–900 μm in height, up to 100 MNs/cm2) in various geometries have been produced from silicon and metal using recently developed microfabrication techniques1,2. Such MNs are applied to the skin surface and pierce the epidermis (devoid of nociceptors), creating microscopic holes through which drugs diffuse to the dermal microcirculation. MNs are long enough to penetrate to the dermis but are short and narrow enough to avoid stimulation of dermal nerves. Solid MNs puncture skin prior to application of a drug-loaded patch or are precoated with drug prior to insertion. Hollow bore MNs allow diffusion or pressure-driven flow of drugs through a central lumen. The Macroflux® system (Zosano Pharma, Fremont, CA, USA) incorporates a 2-cm2 array of titanium MNs, which can be coated with drug for bolus administration or used in combination with a drug reservoir3. This system does, however, require a specialized device to stretch the skin, allowing drug to diffuse down spaces in the SC created around the MNs.
A particular advantage of MN technology is that drug substances with high molecular weights and/or highly water-soluble drugs can be efficiently delivered transdermally1,2. The rate of drug delivery can now be almost exclusively controlled by the drug delivery system rather than the SC. MNs have been shown to enhance transdermal delivery of a wide range of molecules, including anthrax vaccine4, β-galactosidase5, calcein1,6-9, bovine serum albumin (BSA)6-9, desmospressin3, diclofenac10-12, erythropoietin13,14, methyl nicotinate12, ovalbumin15,16, and plasmid DNA5,17. The most widely studied drug with regards to MN-mediated delivery is insulin, with enhanced skin permeation reported in vitro6,18,19 and in vivo6,10-14,19,20.
MN arrays do not cause pain on application and no reports of development of skin infection exist. However, their use is associated with a number of problems: silicon is not an FDA-approved biomaterial, and broken silicon or metal MNs could cause skin problems2; solid noncoated needles require a two-step application process, which is undesirable2,21; accurately coating MNs is difficult, and these only deliver a very small amount (<1 mg) of drug as a bolus only2; hollow MNs have generally only one outlet and may become blocked by compressed dermal tissue11. In addition, there is a possibility that MN-created holes in the SC may close relatively quickly (within 5–15 minutes), thus limiting the time period for drug delivery.
Recently, a number of reports13,14,22-24 have suggested that many of the above problems could be addressed by using drug-loaded MN arrays prepared by moulding hot melts of carbohydrate materials using silicon or metal MNs as master templates. Such MNs should dissolve upon skin insertion to release their drug payload. In our experience, carbohydrate materials require high processing temperatures to produce hot melts, which are extremely viscous and resistant to flow. In addition, the materials produced upon cooling are extremely hygroscopic, which causes difficulties with storage and handling. In this study, we aimed to explore the processing, handling, and storage of MN arrays prepared from a model carbohydrate material with a view to clinical application.
Materials and methods
Chemicals
5-Aminolevulinic acid (ALA), hydrochloride salt, was obtained from Crawford Pharmaceuticals (Milton Keynes, UK). Acetyl acetone and formaldehyde 37% (w/w) solution in water, galactose, and BSA were obtained from Sigma Aldrich (Dorset, UK). Silastic® 9280/60E silicone elastomer was obtained from Dow Corning (Wiesbaden, Germany). Radiolabeled 5-ALA solution (3.7 MBq/mL) was supplied by PerkinElmer Life Sciences (Beaconsfield, Bucks, UK). All other chemicals were of analytical reagent grade.
Silicon microneedle arrays
Silicon MN arrays were prepared using a previously reported approach25. Briefly, a silicon wafer was coated with a nitride and oxide layer using a low-pressure chemical vapor deposition. This layer was then lithographically patterned using plasma etching. The patterned wafer was subsequently etched using 29% (w/v) potassium hydroxide solution at a temperature of 79°C in a water bath with constant agitation. The aspect ratio of the resulting MNs was 3:2 (height: base diameter). In a final step, the MNs were surface coated with titanium, as an adhesive layer, and then platinum by evaporation.
Clearly, individual MNs could not be seen with the naked eye. Scanning electron microscope (SEM) imaging (JEOL JSM840 scanning electron microscope; Jeol, Tokyo, Japan) revealed that the MNs were arranged on the silicon chips as 6 × 7 arrays, meaning there were 42 MNs in each array (Figure 1a and c). Each MN was approximately 270 μm in height, with a diameter of 240 μm at the base and an interspacing of 750 μm between rows of MNs (Figure 1b and d). Figure 1e shows a digital photograph of a typical silicon MN array. Using a digital micrometer, the dimensions of each array were determined to be approximately 5.6 mm in length by 4.8 mm in breadth.
Figure 1.

SEM images taken of a typical silicon MN array from directly above (a), of a single MN from above (b), of an array from the side (c), and of an individual MN from the side (d). Digital photograph of a typical silicon MN array (e). For SEM, individual MN arrays were mounted onto aluminium stubs using double-sided adhesive tape and coated in gold (Polaron® E5150 sputter coater; Quorum Technologies, Ringmer, UK). Specimens were then visualized using a JEOL JSM840 scanning electron microscope (Jeol) and images captured on Ilford FP4 black and white roll film (Jessops, Leicester, UK), which was then developed and digitally scanned.
Moulding of galactose microneedle arrays
Silicone elastomer micromoulds were prepared using a silicon MN array as a master template. The silicon array was mounted on a Teflon® slab using cyanoacrylate adhesive. The slab was then fixed to the center of an aluminium stub (Figure 2a). Custom-made aluminium containers were fashioned so that that the aluminium stubs fitted snugly into them. The silicone elastomer was carefully poured into the aluminium container so that the level of the silicone was approximately 5 mm above the surface of the MN tips. An aluminium lid was used to secure the device. To eliminate any entrapped air bubbles from the silicone, the containers were centrifuged for 15 minutes at 3000 × g. The silicone was then cured by placing the container in an oven overnight at 40°C, following which it was allowed to cool to room temperature. The silicone elastomer mould was then removed from the aluminium container by gently pressing a metal rod against an opening situated at the base of the device. The silicone mould was then carefully peeled away from the silicon array, leaving the master silicon template intact for further use.
Figure 2.

Diagrammatic representation of the steps involved in the preparation of galactose microneedles, showing first production of silicone master moulds from silicon microneedle arrays (a) and second preparation of galactose microneedle arrays from silicone master moulds (b).
Approximately 10 g of powdered galactose was heated at 160°C (Gallenkamp hotbox oven with fan; Sanyo-Gallenkamp PLC, Leicester, UK) until it completely melted. Aluminium containers containing the silicone moulds were placed into centrifuge tubes and also heated to approximately the same temperature. The molten galactose was then carefully poured into a silicone mould and the aluminium lid screwed on (Figure 2b). To ensure that that the galactose reached the tips of the MN mould, the containers were centrifuged for 15 minutes at 3000 × g (Jouan C312 laboratory centrifuge; DJB Labcare, Bukinghamshire, UK). Upon cooling, the silicone mould containing the galactose MN array was removed from the aluminium holder as described above.
The baseplate of the galactose arrays produced by the micromoulding technique described above was found to be approximately 5–8 mm thick (Figure 3d). To reduce the thickness of the baseplate, arrays were placed in a custom-made aluminium block that contained hollow channels of varying depth (Figure 3a–b). The galactose baseplate was carefully ground down to a thickness of approximately 0.75 mm using a high-speed abrasive sanding disk fitted to an electric rotary tool (Dremel® Lithium-Ion 10.8 V; Dremel UK, Middlesex, UK) (Figure 3c). Reducing the baseplate thickness below 0.75 mm caused it to shatter.
Figure 3.

Diagrammatic representation of the process of thinning the galactose baseplate. View of the aluminium block from above (a) and from the side (b). Galactose arrays are placed into the aluminium block and ground down using a high-speed abrasive sanding disk fitted to an electric rotary tool (c). Formed galactose microneedle array immediately after completion of micromoulding and prior to baseplate thickness reduction (d).
Measurement of mechanical strength of galactose arrays
To determine the force necessary for mechanical fracture of galactose MNs, arrays were fixed to the tip of a moveable cuboidal probe (length 5 cm, cross-sectional area 1.5 cm2) of a TA-XT2 Texture Analyser (Stable Micro Systems, Surrey, UK) using double-sided adhesive tape. An axial compression load was then applied. The test station pressed the arrays against a flat block of aluminium at a rate 0.5 mm/s with defined forces for 30 seconds. Pretest and posttest speed was 1 mm/s, and the trigger force was set at 0.049 N. All MNs of each array were visually examined using a digital microscope (GXMGE-5 USB Digital Microscope; Laboratory Analysis Ltd., Devon, UK) before and after fracture testing and changes in height recorded.
Measurement of skin insertion force for galactose arrays
Heat-stripped piglet epidermis was employed to determine the force required for insertion of galactose MNs into skin. Stillborn piglets were obtained from a local farmer and used within 12 hours of birth. First, all subcutaneous fat was removed. Second, skin samples were incubated at 60°C in water for 2 minutes, and subsequently, the epidermis was gently removed from the dermal layer and shaved using a disposable razor26. Arrays of galactose MNs were again fixed to the tip of the moveable cuboidal probe of the Texture Analyser. The test station, in compression mode, then pressed arrays against piglet epidermis at a speed of 0.5 mm/s for 30 seconds with known forces ranging from 0.05 to 0.267 N/MN. Following MN removal, methylene blue solution (500 μg/mL) was applied to the epidermis for 15 minutes and the solution then gently wiped off, first with dry tissue paper and then with saline and alcohol swabs. The surface of the stained skin was inspected using the digital microscope. To provide additional visual evidence of MN insertion, photographs of representative patterns of dye staining on epidermal surface were also taken (Nikon Coolpix 900 digital camera; Nikon, Tokyo, Japan).
Stability studies
Galactose arrays were stored at 20°C under three different conditions of relative humidity (RH), namely, 0%, 43%, and 85%. These conditions were obtained by storing the arrays in evacuated heat-sealed foils containing silica gel, storing them in desiccation chambers containing saturated solutions of potassium carbonate or potassium chloride, respectively. Arrays were removed at regular intervals and visualized using the digital microscopy. No array was returned to the container after visualization.
Drug incorporation and release
Two model drugs representative of classes that may benefit from transdermal administration using MN arrays were used. These were the small hydrophilic amino acid 5-ALA, a precursor of the potent photosensitizer protoporphyrin IX used in photodynamic therapy, and the large hydrophilic protein BSA. Importantly, the physicochemical properties of both substances militate against their passive transdermal transport.
ALA or BSA powder was added directly to molten galactose, which was then moulded into MNs as described above. The amounts of each drug added were such that, upon thinning of the baseplate of the formed arrays, the theoretical BSA loading was 5 mg/42-MN array and the theoretical ALA loadings were 5 and 19 mg/42-MN array. Drug content of galactose MN arrays was determined by high-performance liquid chromatography (HPLC) as described below following dissolution of formed arrays in deionized water.
Release of ALA and BSA from galactose MN arrays across Silescol® membranes was investigated in vitro using modified Franz cells (modified Franz diffusion cells, FDC-400 flat flange, 15 mm orifice diameter, mounted in triplicate on an FDCD diffusion drive console providing synchronous stirring at 600 rpm; Crown Glass Co. Inc., Sommerville, NJ, USA). Using gentle finger pressure, MN arrays were pressed into the center of the membrane. With the array in place, the punctured membranes were carefully mounted onto the Franz cells, ensuring that the punctured area was directly over the orifice of the receptor compartment. Using a long needle, samples (0.10 mL) were removed from the receptor compartment at defined time intervals, and ALA and BSA analysis was performed by HPLC as described below. The receptor phase was phosphate-buffered saline (pH 7.4) containing 1% (w/v) sodium dodecyl sulfate for BSA and 0.1 M borate buffer (pH 5) (Pharmacopoeia Helvetica) for ALA.
Analysis of ALA and BSA in solution
ALA analysis was performed by a validated fluorimetric HPLC method following derivatization with acetyl acetone and formaldehyde, as described previously27. BSA was analyzed using HPLC (Agilent 1200® Binary Pump, Agilent 1200®, Standard Autosampler, Agilent 1200® Variable Wavelength Detector; Agilent Technologies UK Ltd., Stockport, UK), this time with UV detection at 280 nm. The column used was a Biosep®-SEC-S 3000 (300 × 7.80 mm2 with 5-μm packing; Phenomenex, Torrance, CA, USA). The injection volume was 100 μL, and the mobile phase was 0.05 M ammonium acetate. The flow rate was 1 mL/min.
Calibration plots were performed on three separate days by making up fresh BSA calibration standards on each day. Three standard solutions were prepared separately for each BSA concentration on each day of the analyses. These calibration plots were used to produce a representative calibration curve. Least squares linear regression analysis and correlation analysis were performed on the curve produced, enabling determination of the equation of the line, its coefficient of determination, and the residual sum of squares, as recommended by the Journal of Chromatography A28. This representative calibration curve was used to determine the limits of detection (LoD) (Equation 1) and quantification (LoQ) (Equation 1) of the method as described in the International Conference on Harmonisation’s Topic Q2B (1996).
| (1) |
where σ is the standard deviation of the response (peak area) of the data used to construct the regression line and S is the slope of that line. Similarly, the LoQ may be determined using Equation (2)
| (2) |
Skin penetration studies
The distribution of ALA in neonate porcine skin after a 5-hour application period was determined using a modified jacketed Franz-type diffusion cell. The cell was positioned upright and the receptor compartment filled with degassed 0.1 M borate buffer, pH 5 (Pharmacopoeia Helvetica). Full-thickness skin was excised and the subcutaneous fat and connective tissue carefully removed. For MN-treated skin, galactose arrays were pressed into the upper surface of the skin for 30 seconds using gentle finger pressure. With the array in place, the porcine skin was then carefully mounted onto the modified Franz cell with the dermal side facing downwards into the receptor compartment. Using a micropipette, 1 mL of 0.1 M borate buffer (pH 5) containing 19 mg/mL ALA and spiked with 30 μL of 14C-radiolabeled ALA was added to the donor compartment of the Franz cell. For control experiments, the solution was added directly to the surface of the skin, with no MN array in place. Cylindrical biopsies, 5 mm in diameter, were then taken from the skin using a proprietary dermatological biopsy punch (Stiefel Laboratories Ltd., High Wycombe, UK). For MN-treated skin, care was taken to ensure that biopsies were removed from skin where the array had previously been applied. Biopsies were immediately frozen in liquid nitrogen vapor.
ALA was quantified using liquid scintillation spectroscopy, as described previously27. Briefly, the cylinders of frozen tissue were mounted on the stage of a cryostatic microtome (Leica CM1900-1-1; Leica Microsystems, Nussloch, Germany) using tissue embedding fluid (Tissue-TEK®; Sakura Finetech Europe B.V., Zoeterwade, the Netherlands). Frozen tissue cylinders were positioned so that their upper surfaces, to which the solution had been applied, were parallel to the slicing motion of the blade. Slice thickness was set at 50 μm, and five consecutive slices were taken and placed into the same scintillation vial. This procedure was repeated until the entire biopsy was sliced. The total depth of slicing into the tissue was approximately 2.5 mm. Tissue slices containing radiolabeled ALA were dissolved in 1 mL NCS®-II Tissue Solubilizer overnight at 37°C. Scintillation cocktail (5 mL) was added to each vial and mixed. Vials were stored in darkness for 2 hours prior to analysis to reduce chemiluminescence. Samples were counted using a scintillation counter (Tri-Carb® 2300 Liquid Scintillation Analyser; Packard Instrument Company, Meriden, CT, USA), and the counts per minute in each vial were determined over a 20-minute period. The counts per minute was then converted to disintegrations per minute using an internally stored quench correction curve for 14C. Mean disintegrations per minute values from three replicate experiments were converted into mean ALA concentrations. These ALA tissue concentrations were plotted against the mean tissue depth of each five slice replicate. Mean penetration depths were the average depths for a given set of five consecutive slices, such that if 5 × 50 μm2 slices were taken from the top of a tissue sample, then the mean depth plotted against the mean ALA concentration would be 0.125 mm.
Statistical analysis
Where appropriate, data were analyzed using a one-way analysis of variance. Mathematical characterization of the relationships between the x and y variables in the representative calibration plots was performed using least squares linear regression, following analysis of residuals. Comparison between formulations, in terms of skin penetration, was made using the Mann–Whitney U test. In all cases P < 0.05 denoted significance.
Results
Because of the highly viscous nature of molten galactose and its tendency to solidify, preparation of micromoulded MN arrays was not without difficulties. Only two arrays could be prepared at any one time, and micromoulds and centrifuge tubes had to be preheated to prevent solidification during the centrifugation step. As a result of the mould design employed, the galactose MN arrays formed on very thick baseplates. These were trimmed appropriately so as to yield a device that could be more readily incorporated into a transdermal patchtype system. This thinning process required a great deal of care and precision to avoid inadvertent damage to the MN tips. Not only was this process time-consuming, but each individual MN had to be inspected for damage following the procedure.
Micromoulded galactose MNs initially fully replicated the physical structure of the silicon MNs used to prepare micromoulds. This could, however, only be shown by light microscopy (Figure 4a), with the processing of samples for SEM having a somewhat deleterious effect on the MNs, turning them from pyramidal projections to protrusions more conical in appearance (Figure 4b). Storage of galactose arrays at a RH approximating ambient conditions (43%) led to rapid deformation of the MN structure after only 1 hour (Figure 4c), with complete disappearance of MNs after 6 hours (Figure 4d). This effect was even more pronounced at an elevated RH of 75%, where no MN remained after 1 hour (Figure 4e). In contrast, when the RH was reduced to 0% by using heat-sealed packaging including silica gel as desiccant, no change in MN morphology was observed over a 3-week storage period (Figure 4f). Galactose arrays were observed to develop an increasingly adhesive nature during storage at relative humidities of 43% and 75%. This made their handling, even with forceps, quite problematic. In fact, when an array stored for 1 hour at a RH of 43% was applied to heat-stripped epidermis with an application force of 5 N for 30 seconds using the probe of the Texture Analyser, the force required to remove the array was as high as 1.195 N/cm2. This is comparable to the forces of removal typically observed when using bioadhesive drug delivery systems that are specifically designed to adhere to tissue29. Despite the obvious effects of moisture on galactose MNs, it was not possible to detect a mass increase during storage, even at a RH of 75%.
Figure 4.

Light micrograph of galactose microneedles upon preparation (a). Scanning electron micrographs of the same array (b). Light micrograph of galactose microneedles after storage at a relative humidity of 43% for 1 hour (c) and 6 hours (d). Light micrograph of galactose microneedles after storage at a relative humidity of 75% for 1 hour. Light micrograph of galactose microneedles after storage at a relative humidity of 0% for 3 weeks.
Immersion of galactose arrays in phosphate-buffered saline (pH 7.4) at 32°C (skin temperature) led to progressive dissolution of the MNs and the baseplate, with dissolution complete in only 25 minutes (Figure 5a). Figure 5b shows that as application force is increased, a progressive reduction in height of galactose MNs pressed against an aluminium plate is observed. When the insertion force was greater than 0.2 N/MN, over 90% of the MNs in the array were shown to puncture piglet epidermis. Below this force, insertion was less efficient and appreciably less reproducible. Interestingly, no reduction in MN height was observed following insertion into heat-stripped epidermis, even when the insertion force was as high as 0.267 N/MN (data not shown).
Figure 5.

The dissolution profile of galactose microneedle arrays in PBS, pH 7.4 (a). Means ± SD, n = 6. Influence of axial loads on height reduction of galactose microneedles (b). Means ± SD, n = 6. Influence of applied forces on the percentage of microneedles penetrating heat-stripped epidermis in vitro (c). Means ± SD, n = 6.
The LoD and LoQ determined for the developed HPLC method for BSA were 20.08 and 62.36 μg/mL, respectively. The LoD and LoQ previously determined for the HPLC method for ALA were 0.05 and 0.14 μg/mL, respectively27. Galactose did not interfere with either analytical methods, as no galactose peaks were observed in either set of chromatograms. When ALA or BSA was added to molten galactose, a black material immediately formed. This material was, in both cases, water-insoluble and could not be removed from the glass container. Addition of BSA solutions of appropriate concentrations also yielded this black substance. This approach was more successful with ALA. When an appropriate volume of 19 mg/mL ALA solution in 0.1 M borate buffer (pH 5) was added to molten galactose, no black color was observed, and MNs were formed by micromoulding. Following reduction of baseplate thickness and dissolution in 10 mL of 0.1 M borate buffer (pH 5), the mean percentage recovery of ALA was 67.13% of the theoretical drug loading (19 mg ALA per array + baseplate).
When an intact Silescol® membrane was employed, no BSA was detectable in the receiver compartment over a 24-hour period when the donor compartment contained 1 mL of a solution containing 19 mg BSA. In a similar experiment, approximately 3 ± 0.401 μg of ALA penetrated the unpunctured Silescol® membrane over a 6-hour period. This amounted to approximately 0.016% of the ALA contained in 1 mL of the 19 mg/mL solution. The low amounts penetrating the SC-mimicking membrane were to be expected, given the large size of BSA and the hydrophilicity of ALA.
Freshly prepared galactose MNs were capable of penetrating the Silescol® membrane using only gentle finger pressure (Figure 6a). ALA-loaded MN arrays delivered approximately 5.15 ± 1.45 μg across the Silescol® membrane over 6 hours. This was approximately only 0.04% of the total loading in the MN device.
Figure 6.

Image showing galactose MN puncturing the Silescol® membrane (a). Holes created in Silescol® membrane upon immediate removal of galactose MNs (b). Silescol® membrane following the 6-hour application of galactose MNs (c). Silescol® membrane 6 hours after the removal of galactose MNs. Image a was taken using a JEOL JSM840 scanning electron microscope (Jeol) and images b, c, and d were obtained using an ECLIPSE TE300 inverted microscope with a DXM1200 digital still camera (Nikon, UK Ltd., Kingston upon Thames Surrey, UK).
Galactose MNs were used to puncture Silescol® membranes, left in place, and 1 mL of a 19 mg/mL solution of either BSA or ALA was then added to the donor compartment. This approach did not allow BSA delivery, even over a 24-hour period. However, a marked increase in ALA delivery was achieved, with approximately 24.79 μg (0.13% of the total drug in the donor compartment) being delivered across the membrane over a 5-hour period (Figure 7). The relatively small amount of ALA delivered and the absence of BSA delivery across the membrane may be to do with partially dissolved galactose causing sealing of the MN-induced holes. Figure 6b shows an image of a Silescol® membrane immediately after puncture with galactose MNs. The MN-induced holes are clearly visible. Figure 6c shows a similar membrane that had galactose MNs left in place for 6 hours. No holes are now visible. Figure 6d shows a similar membrane that was punctured with galactose MNs, which were then removed, and the membrane was allowed to stand for 6 hours before viewing. The holes are still evident.
Figure 7.
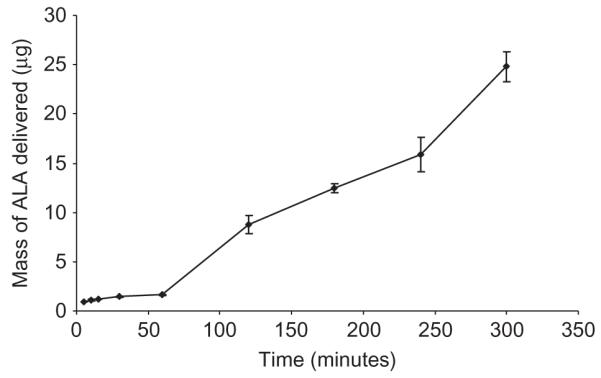
Galactose MNs were used to puncture Silescol® membranes, left in place, and 1 mL of a 19 mg/mL solution of ALA was then added to the donor compartment.
Figure 8 illustrates that the concentration of ALA within excised porcine skin shows depth dependence, with the concentration decreasing as progressive slices are taken down through the tissue biopsy. The highest concentrations are found in tissue slices closest to the site of application. Generally, ALA concentrations throughout the biopsy were reduced when the galactose MN array was left in place, compared to control. Indeed, only 24% of the applied drug was delivered when the array was employed, compared to 28.3% for control skin. However, statistical analysis revealed that this reduction in delivery was not significant (P > 0.2752).
Figure 8.
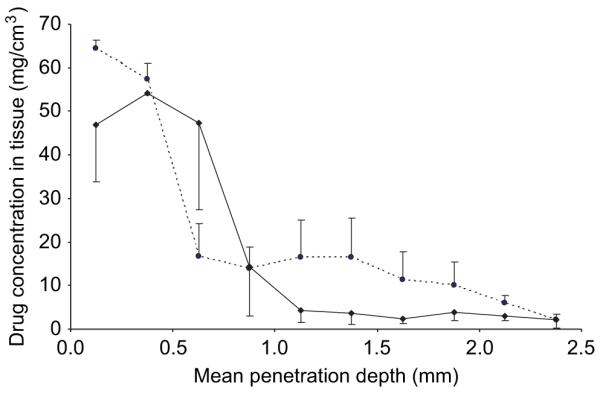
ALA penetration into intact (--●--) and MN-punctured —◆—) porcine skin from a 1 mL solution. The formulation was tailored to deliver 19 mg ALA/mL. Galactose MNs were applied for 30 seconds using finger pressure and left in place for 5 hours. Means ± SD, n = 3.
Discussion
It is without doubt that MN arrays allow greatly enhanced transdermal delivery of drug molecules not normally amenable to administration via this route. That they do so without the cumbersome and often complicated equipment associated with iontophoresis, electroporation and sonophoresis, is a particular advantage of technology, which is likely to really come to the fore with the need to deliver the protein and peptide products of the biotech boom outside the hospital setting. The disadvantages of metal and, particularly, silicon MNs are now well known and may partially explain why no MN-based transdermal drug delivery system is currently marketed. It has been suggested that the use of biodegradable materials for MN production may offer a useful and low-cost alternative to conventional materials. A number of groups have investigated carbohydrates as potential MN materials. When carbohydrates are heated at temperatures around their melting points, they change to yellow-brown-colored substances called caramels. This process, known as caramelization, involves the removal of water from a carbohydrate followed by complex isomerizaton and polymerization steps30. This leads to a hard brittle material sometimes referred to as a ‘candy’.
Miyano et al.22 described micromoulding of MNs from maltose heated to 140°C. The authors reported that maltose needles dissolved within a few hours in environments where the RH exceeded 50% and that the viscosity of the molten maltose reduced the throughput of MNs during manufacture23. Such MNs were used clinically to deliver ascorbate-2-glucoside into the skin of volunteers. However, the amount of drug incorporated into MNs during manufacture, or drug stability upon storage, was not reported. Micromoulded maltose MNs were also used by Kolli and Banga24, who reported that such MNs enhanced the flux of nicardipine hydrochloride across full-thickness rat skin fourfold in vitro. However, the drug was not incorporated into the MNs but was contained in a liquid reservoir patch that was applied to the skin on top of inserted MNs.
To overcome some of the disadvantages associated with the preparation of carbohydrate MNs by heating, Ito et al.13 reported a novel thread-forming technique for the preparation of needle-like projections from a ‘dextrin glue’ (a mixture of 2 g dextrin and 1 mL water) containing dissolved drug. Needles prepared in this way, and loaded with insulin, were shown to reduce blood glucose levels in vivo13,31. In addition, they were also shown to deliver erythropoietin to mice in vivo14. Clearly, this fabrication method has the advantage of not requiring high temperatures. However, at an average length of 3 mm and base diameter of 0.55 mm, these devices are considerably larger than most true MN systems studied elsewhere. In addition, each needle is a separate entity with no baseplate. Therefore, each needle has to be inserted individually, which is not only cumbersome but presumably requires great expertise or a custom-made applicator.
Until now, there have been no published reports with regards to the mechanical strength of carbohydrate MN arrays, to the forces required to insert them into the skin, or the effects of storage on their morphology. In this study, we used galactose as a model material to explore the processing, handling, and storage of carbohydrate MN arrays. While relatively small forces caused significant reductions in MN height when applied to an aluminium block, this was not observed during their relatively facile insertion into heat-stripped epidermis. This suggests that galactose MNs will not break during insertion into skin but could become damaged by improper handling by untrained personnel, such as a patient in their home. In addition, as shown from storage studies, leaving a galactose MN-based drug delivery system unpackaged under ambient conditions for relatively short time periods could lead to a system that is adhesive but lacks the MNs required for its proper use. Incorporation of drug substances into MNs prepared from hot melts of carbohydrate materials is likely to be problematic. The substantial losses of ALA and BSA seen during processing in this study is likely to be due to reaction between the drugs’ amino groups and the R-hydroxy carbonyl group of galactose, as well as direct drug degradation at the elevated temperature32. A similar drug loss problem has been reported by those producing MNs from hot polymer melts8,9. As well as influencing drug stability, the high processing temperatures employed were also found to have a detrimental affect on the silicone moulds. Consequently, the moulds could only be reused a limited number of times.
Drug release experiments using ALA-loaded MN arrays revealed that less than 0.05% of the total drug loading was released across the model silicone membrane. Similarly, only low amounts of ALA (~0.13%) and undetectable amounts of BSA were delivered when galactose arrays were combined with aqueous vehicles. Microscopic inspection of the membrane following release studies revealed that no holes could be observed in the membrane (Figure 6c), indicating that the partially dissolved galactose sealed the MN-induced holes, thus limiting drug delivery.
Because of the difficulties associated in obtaining excised human skin, neonatal porcine skin was used as an in vitro model for drug penetration studies. Porcine skin has comparable morphological and functional skin characteristics to human skin33 and is generally considered to provide the best in vitro model of all domestic animals34. As described above, MN puncture was not shown to enhance BSA delivery across Silescol®. Consequently, skin studies were only performed with ALA. In addition, it was shown above that almost 35% of ALA was degraded during the manufacture of galactose MN arrays. For skin penetration studies, radiolabeled ALA was employed to facilitate accurate and sensitive drug quantification in tissue. However, if radiolabeled ALA was broken down during the MN manufacturing process, any analysis would be determining significant amounts of the degradation products as well as the actual drug. Hence, rather than incorporating ALA into MN arrays, penetration studies were carried out by using blank galactose arrays to puncture the skin, followed by the application of an ALA solution.
In the field of transdermal drug delivery, skin permeation studies typically determine the amount and rate of drug diffusing across the membrane. However, with regards to the topical delivery of ALA, it is the drug concentration within the skin layers that is of interest. Consequently, in this study, skin was sectioned into 50-μm slices, and the amount of radiolabeled ALA was determined through the skin biopsy. Skin penetration studies revealed that ALA tissue concentrations were generally lower when galactose MN arrays were left in place, compared to control. It was noted that when skin biopsies from MN-treated skin were flash frozen in liquid nitrogen vapor, a brittle crust was evident on the skin surface. This may indicate that the galactose baseplate may not have completely dissolved over the course of the experiment but rather formed an additional layer through which the drug must partition into and diffuse through. These results indicate that carbohydrate MN arrays should not be left in situ but rather should be removed prior to the application of the formulation.
In this study, we have, for the first time, demonstrated the problems inherent in the processing, storage, and use of MN arrays prepared from carbohydrate materials. Such difficulties are likely to preclude their successful clinical application. In particular, it is unlikely that carbohydrate MNs could ever be used appropriately by a patient in their own home. Even if used by trained personnel, these MNs would need to be stored in a sealed container at controlled RH prior to use. Their use would also have to be restricted to piercing the skin to create holes and then being removed, so as not to restrict drug delivery from an applied formulation. However, this leads to an undesirable two-step administration process. Carbohydrate MNs are clearly, therefore, not the solution to the problems posed by use of silicon and metal MNs. This is especially true as MNs produced from hot polymer melts also suffer from substantial drug loss during processing.
The findings of this study are of particular importance to those in the pharmaceutical industry involved in the design and formulation of transdermal drug delivery systems based on dissolving MN arrays. It is hoped that we have illustrated conclusively the difficulties inherent in the processing and storage of carbohydrate-based dissolving MNs and that those in the industry will now follow alternative approaches.
Acknowledgments
This work was supported by BBSRC grant number BB/E020534/1 and the Science Foundation Ireland Tyndall National Access Program project number NAP 156.
Footnotes
Declaration of interest: The authors report no conflicts of interest.
References
- 1.Henry S, McAllister DV, Allen MG, Prausnitz MR. Microfabricated microneedles: A novel approach to transdermal drug delivery. J Pharm Sci. 1998;87:922–5. doi: 10.1021/js980042+. [DOI] [PubMed] [Google Scholar]
- 2.Prausnitz MR. Microneedles for transdermal drug delivery. Adv Drug Deliv Rev. 2004;56:581–87. doi: 10.1016/j.addr.2003.10.023. [DOI] [PubMed] [Google Scholar]
- 3.Cormier M, Johnson B, Ameri M, Nyam K, Libiran L, Zhang DD. Transdermal delivery of desmopressin using a coated microneedle array patch system. J Control Release. 2004;97:503–11. doi: 10.1016/j.jconrel.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 4.Mikszta JA, Dekker JP, Harvey NG, Dean CH, Brittingham JM, Huang J. Microneedle-based intradermal delivery of the anthrax recombinant protective antigen vaccine. Infect Immun. 2006;74:6806–10. doi: 10.1128/IAI.01210-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Coulman SA, Barrow D, Anstey A, Gateley C, Morrissey A, Wilke N. Minimally invasive cutaneous delivery of macromolecules and plasmid DNA via microneedles. Curr Drug Deliv. 2006;3:65–75. doi: 10.2174/156720106775197510. [DOI] [PubMed] [Google Scholar]
- 6.McAllister DV, Wang PM, Davis SP, Park JH, Canatella PJ, Allen MG. Microfabricated needles for transdermal delivery of macromolecules and nanoparticles: Fabrication methods and transport studies. Proc Natl Acad Sci USA. 2003;100:13755–60. doi: 10.1073/pnas.2331316100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xie Y, Xu B, Gao Y. Controlled transdermal delivery of model drug compounds by MEMS microneedle array. Nanomedicine. 2005;1:184–90. doi: 10.1016/j.nano.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 8.Park JH, Allen MG, Prausnitz MR. Biodegradable polymer microneedles: Fabrication, mechanics and transdermal drug delivery. J Control Release. 2005;104:51–66. doi: 10.1016/j.jconrel.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 9.Park JH, Allen MG, Prausnitz MR. Polymer microneedles for controlled-release drug delivery. Pharm Res. 2006;23:1008–19. doi: 10.1007/s11095-006-0028-9. [DOI] [PubMed] [Google Scholar]
- 10.Gardeniers JGE, Luttge R, Berenschot EJW. Silicon micromachined hollow microneedles for transdermal liquid transport. J Microelectromech Syst. 2003;12:855–62. [Google Scholar]
- 11.Martanto W, Davis SP, Holiday NR, Wang J, Gill HS, Prausnitz MR. Transdermal delivery of insulin using microneedles in vivo. Pharm Res. 2004;21:947–52. doi: 10.1023/b:pham.0000029282.44140.2e. [DOI] [PubMed] [Google Scholar]
- 12.Nordquist L, Roxhed N, Griss P, Stemme G. Novel microneedle patches for active insulin delivery are efficient in maintaining glycaemic control: An initial comparison with subcutaneous administration. Pharm Res. 2007;24:1381–8. doi: 10.1007/s11095-007-9256-x. [DOI] [PubMed] [Google Scholar]
- 13.Ito Y, Hagiwara E, Saeki A, Sugioka N, Takada K. Feasibility of microneedles for percutaneous absorption of insulin. Eur J Pharm Sci. 2006;29:82–8. doi: 10.1016/j.ejps.2006.05.011. [DOI] [PubMed] [Google Scholar]
- 14.Ito Y, Yoshimitsu J, Shiroyama K, Sugioka N, Takada K. Self-dissolving microneedles for the percutaneous absorption of EPO in mice. J Drug Target. 2006;14:255–61. doi: 10.1080/10611860600785080. [DOI] [PubMed] [Google Scholar]
- 15.Matriano JA, Cormier M, Johnson J, Young WA, Buttery M, Nyam K, et al. Macroflux microprojection array patch technology: A new and efficient approach for intracutaneous immunization. Pharm Res. 2002;19:63–70. doi: 10.1023/a:1013607400040. [DOI] [PubMed] [Google Scholar]
- 16.Widera G, Johnson J, Kim L, Libiran L, Nyam K, Daddona PE. Effect of delivery parameters on immunization to ovalbumin following intracutaneous administration by a coated microneedle array patch system. Vaccine. 2006;24:1653–64. doi: 10.1016/j.vaccine.2005.09.049. [DOI] [PubMed] [Google Scholar]
- 17.Mikszta JA, Alarcon JB, Brittingham JM, Sutter DE, Pettis RJ, Harvey NG. Improved genetic immunization via micromechanical disruption of skin-barrier function and targeted epidermal delivery. Nat Med. 2002;8:415–9. doi: 10.1038/nm0402-415. [DOI] [PubMed] [Google Scholar]
- 18.Teo MA, Shearwood C, Ng KC, Lu J, Moochhala S. In vitro and in vivo characterization of MEMS microneedles. Biomed Microdevices. 2005;7:47–52. doi: 10.1007/s10544-005-6171-y. [DOI] [PubMed] [Google Scholar]
- 19.Wang PM, Cornwell M, Hill J, Prausnitz MR. Precise microinjection into skin using hollow microneedles. J Invest Dermatol. 2006;126:1080–7. doi: 10.1038/sj.jid.5700150. [DOI] [PubMed] [Google Scholar]
- 20.Davis SP, Martanto W, Allen MG, Prausnitz MR. Hollow metal microneedles for insulin delivery to diabetic rats. IEEE Trans Biomed Eng. 2005;52:909–15. doi: 10.1109/TBME.2005.845240. [DOI] [PubMed] [Google Scholar]
- 21.Schuetz YB, Naik A, Guy RH. Emerging strategies for the transdermal delivery of peptide and protein drugs. Expert Opin Drug Deliv. 2005;2:533–48. doi: 10.1517/17425247.2.3.533. [DOI] [PubMed] [Google Scholar]
- 22.Miyano T, Tobinaga Y, Kanno T, Matsuzaki Y, Takeda H, Wakui M, et al. Sugar micro needles as transdermic drug delivery system. Biomed Microdevices. 2005;7:185–8. doi: 10.1007/s10544-005-3024-7. [DOI] [PubMed] [Google Scholar]
- 23.Miyano T, Miyachi T, Okanishi T, Todo H, Sugibayashi K, Uemura T, et al. Hydrolytic microneedles as transdermal drug delivery system. Transducers; Proceedings of the 14th International Conference on Solid-State Senors, Actuators and Microsystems; Lyon, France. 2007; June 10-14.pp. 355–8. [Google Scholar]
- 24.Kolli CS, Banga AK. Characterization of solid maltose microneedles and their use for transdermal delivery. Pharm Res. 2008;25:104–13. doi: 10.1007/s11095-007-9350-0. [DOI] [PubMed] [Google Scholar]
- 25.Wilke N, Mulcahy A, Ye SR, Morrissey A. Process optimization and characterization of silicon microneedles fabricated by wet etch technology. Microelectronics J. 2005;36:650–6. [Google Scholar]
- 26.Johnson ME, Berk DA, Blankschtein D, Golan DE, Rakesh KJ, Langer RS. Lateral diffusion in human stratum corneum and model lipid bilayer system. Biophys J. 1996;71:2656–68. doi: 10.1016/S0006-3495(96)79457-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Donnelly RF, Morrow DIJ, McCarron PA, Juzenas P, Woolfson AD. Pharmaceutical analysis of 5-aminolevulinic acid in solution and in tissues. J Photochem Photobiol B. 2006;82:59–71. doi: 10.1016/j.jphotobiol.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 28.Shabir GA. Validation of high-performance liquid chromatography methods for pharmaceutical analysis. Understanding the differences and similarities between validation requirements of the US food and drug administration, the US Pharmacopoeia and the International Conference on Harmonization. J Chromatogr A. 2003;987:57–66. doi: 10.1016/s0021-9673(02)01536-4. [DOI] [PubMed] [Google Scholar]
- 29.ICH-Topic Q2B: Validation of Analytical Procedures: Methodology; International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use; Geneva, Switzerland. 1996. [Google Scholar]
- 30.McCarron PA, Woolfson AD, Donnelly RF, Andrews GP, Zawislak A, Price JH. Influence of plasticiser type and storage conditions on the properties of poly(methyl vinyl ether-co-maleic anhydride) bioadhesive films. J Appl Polym Sci. 2004;91:1576–89. [Google Scholar]
- 31.Seo J, Oh J, Kim DJ, Kim HK, Hwang Y. Making monosaccharide and disaccharide sugar glasses by using microwave oven. J Non-Cryst Solids. 2004;333:111–4. [Google Scholar]
- 32.Ito Y, Hagiwara E, Saeki A, Sugioka N, Takada K. Sustained-release self-dissolving micropiles for percutaneous absorption of insulin in mice. J Drug Target. 2007;15:323–6. doi: 10.1080/10611860701349794. [DOI] [PubMed] [Google Scholar]
- 33.Yaylayan AV. Classification of the Millard reaction: A conceptual approach. Trends Food Sci Technol. 1997;8:13–8. [Google Scholar]
- 34.Cilurzo F, Minghetti P, Sinico C. Newborn pig skin as model membrane in in vitro drug permeation studies: A technical note. AAPS PharmSciTech. 2007;8:E1–3. doi: 10.1208/pt0804094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Simon GA, Maibach HI. The pig as an experimental animal model of percutaneous permeation in man: Qualitative and quantitative observations—An overview. Skin Pharmacol Appl Skin Physiol. 2000;13(5):229–34. doi: 10.1159/000029928. [DOI] [PubMed] [Google Scholar]