

Abstract
Peptide inhibitors of Methuselah (Mth), a G protein-coupled receptor (GPCR), were reported that can extend the lifespan of Drosophila melanogaster. Mth is a class B GPCR, which is characterized by a large, N-terminal ectodomain that is often involved with ligand recognition. The crystal structure of the Mth ectodomain, which binds to the peptide inhibitors with high affinity, was previously determined. Here we report the predicted structures for RWR motif peptides in complex with the Mth ectodomain. We studied representatives of both Pro-class and Arg-class RWR motif peptides and identified ectodomain residues Asp139, Phe130, Asp127, and Asp78 as critical in ligand binding. To validate these structures we predicted the effects of various ligand mutations on the structure and binding to Mth. The binding of five mutant peptides to Mth was characterized experimentally by surface plasmon resonance, revealing measured affinities that are consistent with predictions. The electron density map calculated from our MD structure compares well with the experimental map of a previously determined peptide/Mth crystal structure and could be useful in refining the current low-resolution data. The elucidation of the ligand binding site may be useful in analyzing likely binding sites in other class B GPCRs.
G protein-coupled receptors (GPCRs1) constitute a superfamily of transmembrane proteins that play critical roles in transmitting extracellular signals to the interior of the cell. Methuselah (Mth) is a class B (secretin-like) GPCR that was previously shown to be involved in stress response and biological aging (1). Like other class B GPCRs, Mth has a large (195 residues) amino-terminal extracellular domain essential for ligand-binding (2). However, the sequence similarity of the mth gene to other class B GPCRs is observed solely within the transmembrane regions (1). The crystal structure shows that the Mth ectodomain consists primarily of β-sheets (3), revealing a topology distinct from that of other hormone receptors such as the corticotropin-releasing factor (CRF) or glucose-dependent insulinotropic polypeptide (GIP) receptors (4, 5).
Recently Ja et al. reported a series of peptide antagonists that inhibit Mth signaling and extend Drosophila melanogaster lifespan (6). Since the extracellular N-terminal domains of other hormone GPCRs have been shown to be properly folded and function in ligand-binding (7–9), Ja et al. used mRNA display selection to identify high-affinity peptide ligands targeting the Mth ectodomain (6). The selected peptides contain a highly conserved consensus— [R/P]xxWxxR—which is denoted as the RWR motif. The crystal structure of a selected peptide and the ectodomain complex was reported, but the coordinates of the ligand could not be resolved. Therefore it was not possible to extract the detailed atomistic description of the ligand-receptor interactions that would be critical in understanding the properties of the binding site.
Here we use computational tools to predict atomistic models of the Mth ectodomain complex structure for four high-affinity peptide ligands. We also compute the electron density map with our atomistic structure of the complex for comparison with the experimental map (6). We then use our predicted structure to carry out a computational mutagenesis study that suggests alternative peptide ligands that might improve or diminish the binding affinity. Experimental measurements of binding affinity for five mutant peptides are subsequently performed and found to be consistent with our predictions. Our predicted structures suggest additional experimental validation studies that could be helpful in characterizing the binding of other Mth ligands.
METHODS
Modeling of the Mth ectodomain
Two X-ray crystal structures of the Mth ectodomain (the N-terminal 188 residues of Mth without the signal sequence) were published with and without a peptide inhibitor in complex (PDB ID: 2PZX and 1FJR, respectively) (3, 6). The resolution for the co-crystal structure was not sufficient to determine the coordinates for the ligand. Since the RMSD of Cα atoms between these two X-ray structures is 0.65 Å, we chose the structure with the better resolution (PDB ID: 1FJR). We then refined this crystal structure computationally by equilibrating it in explicit water solvent for 1 ns. Only chain A was extracted from the dimer in the unit cell. Two Pb2+ ions close to Asp or Glu residues were replaced with Zn2+ ions and the water molecules within 5 Å from the protein were retained. The hydrogen atoms were placed using the Whatif program (10). The system was fully solvated into an equilibrated water box of 64×74×70 Å3 using the Visual Molecular Dynamics (VMD) molecular graphics program (11). The VMD autoionize plugin was then used to randomly place the ions necessary to neutralize the system. The resulting system contained 27,643 atoms within the periodic unit cell; 2,993 protein atoms, 24,642 water atoms, 2 Zn2+ and 6 Na+ atoms.
The system was minimized using 5000 conjugate gradient steps and equilibrated subsequently at 310 K for 100 ps while the protein coordinates were kept fixed. Next, the full system was minimized using 5000 conjugate gradient steps with no restraints and then equilibrated at 100 K for 1 ns. This equilibrated system was finally subjected to 5000 steps of conjugate gradient minimization. This system was gradually heated from 0 K to the target temperature using Langevin molecular dynamics with a damping coefficient of 1 ps−1. A constant pressure of 1 atm was maintained using the Langevin piston method.
All simulations used periodic boundary conditions and the electrostatic interactions were computed using the Particle Mesh Ewald (PME) method. The simulations were carried out with the NAMD 2.6 (12) parallel molecular dynamics code using the CHARMM22 forcefield (FF) (13, 14) for proteins and the TIP3P water model (15).
Building the peptide ligands
Two peptides representing the Pro- and Arg-classes of RWR motif peptides (LP1 and LR1 in Table 1) were built as canonical α-helices using the Biograf program. Based on the spacing of the critical residues in the RWR motif, the ligand regions contacting the binding site are likely to be helical. The side chains of the peptide were replaced using the SCREAM side chain optimization program (V. W. T. Kam and W. A. Goddard III, to be published ). These side chain conformations were further optimized with three cycles of annealing molecular dynamics using the SGB implicit solvent protocol (16). The isolated helix was heated from 50 K to 600 K and cooled down to 50 K in 50 K temperature steps while the coordinates of the backbone atoms were fixed. At each temperature the equilibration was carried out for 300 fs. The annealing MD was performed using the DREIDING FF (17) with the charges from CHARMM22. MPSim was used for all energy and force calculations (18). The cell multipole method was used for the calculation of non-bonded interactions (19).
Table 1.
Peptide ligands for the Mth ectodomain. Consensus residues from the RWR motif ([R/P]xxWxxR) are in bold (6).
The optimized helix structure was then equilibrated in explicit water solvent as described previously. The equilibration was performed at 100 K for 1 ns, then 200 K for 1 ns and 310 K for 1 ns. The equilibrated structure was minimized with a conjugate gradient of 5000 steps and used for the docking study.
Docking of the LP1 ligand to the Mth ectodomain
To search for the LP1 peptide binding site on the Mth ectodomain, rigid docking was carried out using ZDOCK (20). The search area on Mth was limited to residues 126–144, 148–159, and 164 –188, based on the approximate binding regions provided by the electron density map from the experimental crystal structure of the LP1–Mth ectodomain complex (6). From the 2000 configurations generated, we selected the 80 configurations in which the N…O distance of the salt-bridge between R8 and Asp139 was within 4 Å.2 Here we assumed a close interaction between R8 in LP1 (which is one of the critical residues identified in the previous study) and Asp139. Asp139 is a negatively charged residue present in the putative binding crevice. The side chain conformations of the peptide ligands were replaced using SCREAM and then the ligands were minimized with 50 conjugate gradient steps. These calculations, including the following optimization steps, were carried out using the DREIDING FF and CHARMM22 charges with the MPSim MD code (18).
Among the top 20 configurations ranked by the interaction energy (sum of intermolecular Coulombic, van der Waals and hydrogen bond energies), we chose two configurations in which W5, another residue critical for receptor binding, had favorable interactions with Mth. These two ligand-protein complexes were further optimized as follows: the ligand was subjected to conjugate gradient minimization to an RMS force of 0.5 kcal/mol/Å followed by 10 ps NVT dynamics at 50 K using SGB implicit solvent; then we minimized it to an RMS force of 0.3 kcal/mol/Å. We next used SCREAM to replace side chain conformations of the peptide ligand and of Mth residues within 3 Å from the ligand. Finally, the entire complex structure was minimized to 0.3 kcal/mol/Å of RMS force with conjugate gradient. The structures for both complexes were subsequently equilibrated in an explicit water box as described previously. The water box was chosen to extend by ~8 Å from all atoms of the complex. This equilibration was carried out for 3 ns (100K for 1 ns, then 200K for 1 ns and 310 K for 1 ns).
Docking of the LR1 ligand to the Mth ectodomain
The LR1 peptide was docked to the receptor by matching the Cα atoms of residues 5–8 (which include the key W5 and R8 residues of the RWR motif) to those of the LP1 ligand in the equilibrated structure of the complex. From the last 500 ps of the 1 ns MD trajectory for the LR1 ligand we selected 4 snapshots out of 50 (the snapshots were taken every 10 ps) that had no clashes with the receptor after being matched and minimized them further. Side chains of the ligand and of receptor residues within 3.5 Å of the ligand were replaced using SCREAM and the complex structure was minimized to an RMS force of 0.5 kcal/mol/Å. The final best complex structure was chosen based on the FF energy and subsequently equilibrated in the explicit water solvent by following the same procedures as for the LP1 ligand complex. Here we extended the equilibration time to 3 ns at 310 K to allow for full conformational relaxation of the docked ligand since the LR1 ligand has an internal proline residue that induced a kink in the helix of the equilibrated free ligand. To decrease the equilibration time scale and assist conformational change, the side chain conformations in the cavity around R2 of the ligand (which was expected to be critical in interacting with the receptor (6)) were re-assigned using SCREAM. After replacement, the complex was further equilibrated for 1 ns. This newly assigned complex structure showed better interaction energy (−407.77±25.88kcal/mol) than the original complex structure (−389.61±38.18kcal/mol). Here the interaction energy includes electrostatic and van der Waals interactions and is averaged over the last 1 ns trajectory.
Computational mutagenesis study
Starting from the 1 ns-equilibrated Mth/LP1 complex structure, five residues (W5, R8, Y9, L11 and R15) of the LP1 ligand were selected for computational mutagenesis. For each of these 5 residues we considered the 19 natural amino acids excluding proline. We selected 10 sidechain conformations using SCREAM with the 0.1 Å rotamer library, then we conjugate gradient minimized for 10 steps with MPSim and selected the rotamer with the best energy. To reduce the bias in interaction energies due to electrostatic interactions with charged groups, each charged residue was neutralized by protonating or de-protonating before calculating the differential interaction energy. The structure of the ligand protein complex was minimized to 0.25 kcal/mol/Å of the RMS force after the neutralization step. The energies from these calculations are listed in Table 2.
Table 2.
Predicted changes in binding energy (kcal/mol) from combinatorial mutation calculations of the LP1 Mth peptide inhibitor. a The shading indicates cases selected for experimental validations.
| L11 | Y9 | W5 | R8 | R15 | |
|---|---|---|---|---|---|
| A | −0.84 | 4.86 | 19.46 | 45.23 | 21.40 |
| C | −1.21 | 2.87 | 17.39 | 45.18 | 22.20 |
| D | −2.25 | 4.05 | 11.79 | 48.48 | 19.24 |
| E | −1.42 | 5.17 | 12.31 | 46.07 | 18.91 |
| F | −0.40 | −0.18 | 8.81 | 42.25 | 14.86 |
| G | −0.91 | 5.21 | 20.90 | 44.40 | 15.75 |
| H | −0.72 | 1.62 | 8.71 | 40.69 | 18.83 |
| I | −1.04 | 1.70 | 15.15 | 40.03 | 21.47 |
| K | −8.21 | 2.76 | −4.62 | 20.28 | 11.17 |
| L | 0.00 | 2.83 | 13.92 | 41.99 | 21.83 |
| M | −1.86 | 2.42 | 11.48 | 39.68 | 16.96 |
| N | −1.15 | 8.70 | 13.18 | 46.92 | 22.37 |
| Q | −5.63 | 3.77 | 13.17 | 47.28 | 12.92 |
| R | −11.38 | −0.54 | 4.19 | 0.00 | 0.00 |
| S | −0.86 | 5.29 | 12.99 | 46.72 | 22.82 |
| T | −1.12 | 4.35 | 19.25 | 44.79 | 22.39 |
| V | −0.97 | 3.80 | 16.99 | 41.85 | 20.85 |
| W | 0.80 | −0.61 | 0.00 | 37.67 | 14.25 |
| Y | 1.03 | 0.00 | 10.64 | 43.54 | 16.34 |
 | |||||
Delphi solvation energy is included; Relative to the bind energy in WT, B.E. (WT) = −81.79 kcal/mol; Negative means increase of binding
Based on these calculations we selected five mutations of the LP1 ligand (W5K, W5F, Y9F, L11Q and R15Q) as good tests of our predicted ligand-protein structure (the reason why they were chosen is explained in the Results and Discussion section). Each of these was equilibrated in explicit water solvent to allow for conformational relaxation due to perturbation from mutation. To minimize the effect of the initial solvent and ion configurations on conformational change, we started with the fully solvated wild-type (WT) complex structure and mutated the corresponding residue with the same mutant rotamer previously optimized. Any water molecules that clashed were removed. The system was re-neutralized by adding or deleting Cl− counter-ions as necessary. Each system was first subjected to a minimization of 1000 steps and then the solvent molecules were equilibrated for 100 ps at 310 K while the coordinates of Mth/ligand complex structure were fixed. Finally, the entire system was equilibrated for 2 ns at 310 K. The MD simulation was carried out with the NAMD 2.6 program as described previously.
Experimental binding analysis of peptides using surface plasmon resonance (SPR)
LP1 (FPSSWLQRYYLAKRR) and mutant peptides (W5K, W5F, Y9F, L11Q and R15Q) were synthesized and purified (>95%) by Genscript Corp. (Piscataway, NJ) with N-terminal acetylation and C-terminal amidation. Lyophilized peptides were dissolved in ddH2O and quantitated by measuring absorbance at 280 nm. C-terminal biotinylated Mth ectodomain was prepared as described previously (6). SPR measurements were performed at 25 °C on a Biacore T100 instrument (Biacore, Piscataway, NJ). A CM5-streptavidin chip was prepared in-house by standard NHS/EDC amine coupling (Biacore) and achieved ~6000 RU of immobilized streptavidin per flow cell. Biotinylated Mth ectodomain was immobilized to different flow cells of the CM5-streptavidin chip to surface densities of approximately 500, 600, and 800 RU. HBS-EP+ (Biacore, 10 mM HEPES at pH 7.4, 150 mM NaCl, 3 mM EDTA, and 0.05% v/v Surfactant P20) was used as the running buffer and Flow cell 1 was left as a streptavidin negative control for all experiments. To collect kinetics data, a concentration series for each peptide was injected for 90 s at a flow rate of 100 μL/min. Numerous buffer blank injections were also included for double referencing with the streptavidin negative control surface. Peptides were allowed to dissociate for >5 min, which allowed the signal to return to baseline before injection of the next sample. Raw data were processed with Biacore T100 Evaluation Software using a 1:1 bimolecular interaction model and KD values were calculated (kd/ka) from the determined on and off rates. Rate constants were determined from each Mth-containing flow cell and are reported as averages (± s.d.) except for the W5F mutant, which yielded measurable data from only the flow cell with the highest density of immobilized Mth ectodomain.
RESULTS AND DISCUSSION
In silico equilibration of apo receptor and free peptide ligands
After the Mth ectodomain was equilibrated in the explicit water box for 1 ns, we found that the overall folding topology remained the same as the initial X-ray crystal structure (RMSD of Cα atoms = 0.47 Å). This indicates that the force field parameters describe our current system well. The LP1 ligand remained in the initial α-helical conformation during the 1 ns equilibration and for the LR1 ligand we observed the proline kink in the middle of the helix, as expected. The bend angle was 27.5° for the final equilibrated structure after 1 ns and 17.5° for the conformation showing the best FF energy after docking (snapshot at 940 ps).
Characterization of the predicted binding site of the LP1 peptide ligand
After equilibration of two candidate ligand-protein complexes at 310 K for 1 ns, the interactions between the LP1 ligand and the receptor were identified. We then selected the putative ligand-protein structure (1 ns snapshot of Fig. 1) based on the nature of the important non-bonding intermolecular interactions and the interaction energy (−347.68±27.23 kcal/mol vs. −213.31±24.44 kcal/mol, averaged over 1 ns). This final complex structure was further equilibrated for 5 ns. Three representative snapshots at 1 ns, 3 ns and 5 ns are shown in Fig. 1, along with the initial minimized structure. Fig. 2 presents the changes in distances over the 5 ns equilibration for several key residue pairs involved in the intermolecular interactions.
FIGURE 1.

Binding of the LP1 ligand to the Mth ectodomain after 0 ns, 1 ns, 3 ns and 5 ns equilibration. The residues from the receptor (green) are in three-letter code and those from the ligand (magenta) are in single-letter code. Hydrogen bonds and aromatic interactions are specified with black dotted lines and red arrows, respectively. The HBPLUS program was used to calculate hydrogen bonds (maximum D-A distance = 3.9 Å, minimum D-H-A angle = 90.0°).
FIGURE 2.

Changes during 5 ns MD simulation of key intermolecular interactions in the Mth/LP1 complex. Shown are distances (in Å) between side chain heavy atoms. (a) N…O between R15 and Asp127 (gray solid) and between R15 and Asp78 (black dotted), (b) two N…O pairs between R8 and Asp139, (c) centroid-to-centroid between W5 and Phe130 (gray solid) and W5 and Phe153 (black dotted), (d) O…O between S4 and Glu136 (gray solid) and N…O between W5 and Asp139 (black dotted).
Two defining residues of the RWR motif, W5 and R8, were previously determined to be critical in binding to Mth (6). The initial predicted structure has W5 making favorable hydrophobic interactions with the aromatic residues Phe130 (5.0 Å at 1 ns, the centroid-to-centroid distance) and Phe153 (6.1 Å). However, the contact with Phe153 loosens after 1.5 ns (~8.0 Å). The indole nitrogen of W5 also forms a hydrogen bond with Asp139, which is quite stable during the 5 ns (remaining at ~3 Å).
The other key residue, R8, interacts via salt-bridge with Asp139 (~2.6 Å), which remains buried and constant during the dynamics (Fig. 2-(b)). R8 also forms a salt-bridge with Glu136 at early times (2.77 Å at 1 ns), but this becomes weaker later (4.46 Å at 5 ns). R8 also forms a hydrogen bond (~2.7 Å) with the backbone carbonyl oxygen of Tyr131.
At the C-terminus of the ligand, R15 makes a salt-bridge with Asp127 (2.67 Å at 1 ns). Since R15 is exposed to solvent, we find fluctuations in the distance between R15 and Asp127, from 2.7 and 15 Å, as water molecules move in and out. Interestingly, R15 switches its interaction to Asp78 after 2 ns and then back to Asp127 after 4 ns (Fig. 2-(a)). R15 is located at the terminus of the ligand, which allows for this conformational flexibility. These two Asp residues present in different loop regions enable R15 to maintain receptor interactions while remaining free to sweep through space. Hence, this binding interaction is likely to have some favorable aspects of both enthalpy and entropy. Arginine residues C-terminal to the RWR motif are weakly conserved in the previously identified peptide ligands (6), with the location of the Arg residues varying by several residues between sequences. The presence of these Asp residues in the loop regions of Mth could explain this variation.
We identified two other favorable contacts: F1 initially interacts closely with Phe153 and later the backbone nitrogen forms a salt-bridge with Glu137 (at 3 ns) and then Glu136 (at 5 ns). We find that P2, which is one of the consensus residues of the RWR motif, does not interact directly with the receptor. However, the flexibility in the conformation induced by Pro likely plays a role in permitting F1 to make contacts with the receptor. In addition, S4 contacts Glu136 through hydrogen bonding, but with fluctuations.
Characterization of the predicted binding site of the LR1 peptide ligand
Binding of the LR1 ligand to the Mth ectodomain reduces the kink induced by the proline residue (from a bend angle of 17.5° to <10°). The intermolecular contacts between the LR1 ligand and the ectodomain are shown in Fig. 3 after equilibration for 5 ns. W5 and R8 show the same interactions with the ectodomain as for the LP1 ligand, except that Glu136 no longer interacts with the peptide. S4 of LP1, which interacts with Mth Glu136, is replaced with V4 in the LR1 ligand. This leaves Glu136 in the LR1/Mth complex exposed and solvated with water molecules. Therefore, we would expect that mutation of Glu136 might reduce the binding affinity for the LP1 ligand preferentially and assaying this mutation could provide validation for our prediction.
FIGURE 3.

Binding of the LR1 ligand to the Mth ectodomain after 0 ns, 1 ns, 3 ns and 5 ns equilibration. The receptor is colored green and the ligand magenta. Hydrogen bonds and the aromatic interactions are specified with dotted lines and arrows, respectively.
R2, a critical residue of the conserved RWR motif, shows favorable electrostatic interaction with Asp154. This contact is fairly stable throughout the equilibration as shown in Fig. 4-(c). R2 also contacts the side chain of Gln138 through a hydrogen bond, which is formed after 1 ns and preserved thereafter.
FIGURE 4.

Changes during the 5 ns MD simulation of key intermolecular interactions in the Mth/LR1 complex. Shown are distances (in Å) between side chain heavy atoms. (a) N…O between R15 and Asp78 (gray solid) and between R17 and Asp127 (black dotted), (b) N…O between R8 and Asp139 (gray solid) and between W5 and Asp139 (black dotted), (c) N…O between R2 and Asp154 (gray solid) and N…O between R2 and Gln138 (black dotted), (d) centroid-to-centroid distance between F12 and Tyr131 (gray solid) and W5 and Phe130 (black dotted).
R15 forms a salt-bridge with Asp78 as observed in the LP1 case. However in the LR1 case this interaction remains tight throughout the equilibration since the C-terminus of the LR1 ligand is extended from R15 by additional residues. R17 is in the proximity of Asp127, but appears to interact weakly. During the first 1 ns equilibration, the N…O distance between them fluctuates between 2.5 and 10.5 Å. After ~1.7 ns R17 gets closer to Asp127, but a water molecule still intervenes between them occasionally.
F12 has a strong aromatic interaction with Tyr131 and its backbone carbonyl group forms a hydrogen bond with Asn79 at earlier times. The carbonyl group of M1 also contacts Gln138.
The interaction energy between the ligand and the ectodomain was averaged over the last 4 ns to compare the energetics between the LP1 and LR1 ligand cases. The LR1 ligand showed better interaction energy than the LP1 ligand (−437.34±44.58 kcal/mol vs. −310.97±78.33 kcal/mol), in qualitative agreement with experimental binding affinities (6). The relatively large standard deviation for the LP1 case reflects greater conformational fluctuations, indicating that the binding of the LP1 ligand would be entropically more favorable as previously described.
Characterization of the predicted binding sites of the LP2 and LR2 ligands
The LP2 ligand has seven additional amino acids extended from the N-terminus of LP1 (Table 1). Starting with the 1 ns-equilibrated Mth/LP1 complex structure in Fig. 1, we added these seven residues in the α-helical conformation. The torsion angles for the unraveled amino-terminal F-P in the LP1 ligand were modified to be α-helical. However this full α-helix conformation caused a clash with the receptor and therefore these additional seven residues likely form a random coil. In any case, they would not have much contact with the receptor since they are hung peripherally. This may explain the similar binding affinities between the LP1 and LP2 peptides, measured previously (6).
The structure of the Mth/LR2 complex was built from the Mth/LR1 structure by using SCREAM to mutate the corresponding residues. Since the LR2 ligand does not have the proline that caused the kink in LR1 (it was replaced with phenylalanine), the torsion angle was modified to assume a straight α-helix conformation after replacement of the amino acids. The two additional amino-terminal residues in LR2 were built in the extended conformation first and then optimized with annealing molecular dynamics. The final equilibrated complex structure is shown in Fig. S1. During the entire trajectory of the 1 ns equilibration, three arginine residues common with the LR1 ligand formed salt-bridges with Asp residues of the receptor. The aromatic interactions of W7 from the LR2 ligand (corresponding to W5 of the LR1 peptide) also remain stable.
Comparison of predicted and experimental electron density maps
We computed the electron density map from our Mth/LP1 complex structure using the CCP4 program suite (21) for comparison with the experimental density map published in the previous study (6). The unit cell for the Mth/LP1 complex structure was built with the same space group as the crystal structure (PDB ID: 2PZX). Starting with the structure factor file deposited in the Protein Data Bank, we recalculated the structure factors and phases for our complex structure and computed the electron density map. The density map covering the peptide ligand for the 5 ns-equilibrated structure is shown in Fig. 5-(a). The density maps also overlapped well with the experimental density map of the ligand (Fig. 5-(b)), supporting our prediction of the LP1 peptide binding mode. These computational results could be useful in refining the relatively low-resolution crystal structure.
FIGURE 5.

Calculated electron density maps (EDM). (a) The calculated EDM from the predicted Mth/LP1 complex structure after 5 ns equilibration. The Mth ectodomain is in green, LP1 peptide in magenta, and EDM in blue. For clarity, only the part of map covering the ligand is shown. (b) Comparison with the experimental map (ref. 6). The Cα trace of the crystal structure is in cyan and the experimental map in orange. The calculated maps from equilibrated structures of 1 ns, 3 ns and 5 ns are colored magenta, yellow and blue, respectively.
Computational mutagenesis study
We carried out a computational mutagenesis study for the LP1 ligand to design better alternative peptide ligands and also to provide candidates for mutagenesis experiments. Five residues were considered for mutation: L11, Y9, W5, R8 and R15.
We selected L11 because it is located on the boundary of the ligand-receptor contact and is therefore exposed to solvent even though it is nonpolar. Moreover, the nearby Asn79 has no interaction partner. When replacing this L11 with 18 other amino acids (see Table 2 for the energies), we found three mutations with dramatically enhanced binding energy, K (by 8.2 kcal/mol), R (by 11.4) and Q (by 5.6), with all of them forming a hydrogen bond with Asn79. We selected L11Q for experimental measurements since the other cases would introduce charges and hence might affect the ligand structure.
We chose Y9 as a control mutation candidate since we expected that mutations of Y9 would have little effect on binding. Although it is close to Phe130 and W5, Y9 does not interact tightly with the receptor. Most predicted mutations made the interaction worse (Table 2), and the changes were small as expected. However, mutation to R enhanced the affinity due to hydrogen bonding with the backbone carbonyl group. A conserved mutation to either F or W barely affected binding, thus we decided to assay Y9F experimentally.
Since W5, R8 and R15 all strongly interact with the receptor, mutation of these residues generally leads to a dramatic decrease in the calculated binding energy. One exception is the W5K mutation, which the calculations suggested would improve binding (by ~5 kcal/mol) by forming a salt-bridge with Asp139 (which previously made a hydrogen bond with the side chain nitrogen of W5). In contrast, mutation of either R8 or R15 to K was predicted to not improve binding even though these mutations preserve the positive charge. For the R8K mutation, the K residue can no longer reach Glu136 leading to loss of this contact. For R15K, Asp127 formed a salt-bridge, but the binding energy was 11 kcal/mol smaller than the WT. These results are compatible with the previous observation that Arg and not Lys is prevalent in this region of the peptide ligands (6). To avoid inducing large conformational instability in the peptide ligand itself, we chose to experimentally assay W5F and R15Q, in addition to W5K.
Experimental binding study of mutant ligands
Based on our computational mutagenesis predictions, we measured the binding kinetics of five mutant ligands (L11Q, Y9F, W5F, W5K and R15Q) by SPR. The biotinylated Mth ectodomain was immobilized on the surface of a sensor chip while the peptides were in solution. Binding of the ligands to the Mth ectodomain was observed as a refractive index change on the sensor chip surface and was measured in real-time in resonance units (RU).
The results with the LP1 wild-type (WT) peptide compare well with previous measurements and may reflect slight differences in the synthetic peptides tested (peptides in this study had N- and C-terminal acetylation and amidation, respectively) (6). The L11Q, Y9F, and R15Q mutants exhibited similar rate constants to the WT peptide. Mutations to the strongly conserved W5 residue drastically reduced binding, with W5F exhibiting a KD of 12 μM and W5K showing no binding to the Mth ectodomain at the highest concentrations tested (1 μM).
Molecular dynamics simulations for mutant complex structures
In the computational mutagenesis study described above, we did not allow conformational changes from the single-residue mutations. For optimal comparison to the experimental results, we carried out MD simulations in explicit water solvent for the complex structures of the five experimentally tested peptide mutants, allowing for full conformational relaxation. Indeed we see that some initial contacts change within the 2 ns equilibration. The interactions in the core regions composed of W5, R8, Asp139 and Phe130 remain fairly strong for all five cases, as in the WT. We can observe some changes in contacts between the receptor and the ligand, which vary case by case. The final equilibrated structures are shown in Fig. 6. The binding energies for these mutant complex structures were computed in the same way as shown in Table 2 of the computational mutagenesis study. We also calculated the non-bond interaction energy between the ligand and the receptor residues within 5 Å from the ligand (Table 3).
FIGURE 6.

Five mutant complex structures after 2 ns equilibrations in the fully solvated water box (~23000 atoms, 54×86×48 Å3).
Table 3.
Comparison of experimental KD values (nM) of the LP1 peptide and mutant ligands with the energies (kcal/mol) calculated both from the combinatorial mutation calculations and from the MD simulationsa.
| KD(exp’t) | ΔB.E.(calcComb)b | ΔB.E.(calcMD)c | ΔEint(calc)d | |
|---|---|---|---|---|
| WT | 18.5 | 0.00 | 0.00 | 0.00 |
| L11Q | 24.5 | −5.63 | −13.49 | −22.66 |
| Y9F | 49.5 | −0.18 | −11.00 | −12.88 |
| W5F | 12000 | 8.81 | 5.66 | 10.78 |
| W5K | No binding | −4.62 | 6.30 | 8.02 |
| R15Q | 37.7 | 12.92 | −8.63 | 0.91 |
Relative to WT, for the 2 ns-equilibrated complex structures after mutation
Calculated binding energy from combinatorial mutation calculation (from Table 2)
Calculated binding energy after MD; delphi solvation energy is included; B.E. (WT) = −68.25 kcal/mol
Intermolecular interaction energies, calculated for the residues within 5 Å of the ligand; Eint (WT) = −61.62 kcal/mol
-
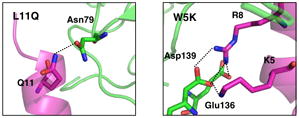
L11Q
We made this experimental mutation because the combinatorial mutation calculations suggested that it might increase binding by ~6 kcal/mol. However the full solvent MD found that the initial hydrogen bond between Q11 and Asn79 is unstable, breaking after 0.5 ns of equilibration. At 2 ns, the side chain of Q11 forms a hydrogen bond with the hydroxyl group of Tyr131. However, this contact is water-mediated with an average distance of ~6 Å after equilibration. Indeed the L11Q mutation caused the salt-bridge of R15 with Asp127 to break, becoming water-mediated. However, L11Q leads to improved hydrogen bonding of Glu136 with R8 and S4. The net result from the MD is that L11Q improves the binding (by ~13–23 kcal/mol, as expected from the combinatorial mutation calculations). The experiments find a slightly decreased binding affinity (18 to 24 nM), indicating very similar binding.
-
W5F
We expected W5F to decrease binding (by ~9 kcal/mol). Obviously, the hydrogen bond with Asp139 shown for the indole nitrogen of W5 is no longer available for W5F. Indeed the MD shows much decreased binding (by ~6–11 kcal/mol). The salt-bridge interaction between R15 and Asp127 on the C-terminus of the ligand is preserved in the MD. However, this mutation leads to a loss of the hydrogen bonding of F1 to the Glu residue, indicating a loosening of the N-terminus of the ligand. The experiments find a dramatic decrease in binding (18 nM to 12 μM) as expected from the prediction.
W5K
-
We expected from the combinatorial mutation calculations that W5K might improve the binding (by ~5 kcal/mol). This mutation of the aromatic residue to a non-aromatic results in the loss of hydrophobic contacts with Phe130 and Phe153. However, the initial hydrophilic interactions remain during the 2 ns equilibration, including the contact of K5 with Asp139. The net result from the MD is much worse binding (by 6 to 8 kcal/mol). The experiments observed no binding at the highest peptide concentrations tested (KD >15 μM). To explore the origin of the lack of measureable binding we carried out MD calculations for isolated W5K in water solvent (Fig. 7), starting with the α-helical docked conformation, and found that the W5K peptide unraveled in ~2 ns. Hence, the W5K peptide likely suffers a higher entropic cost than that of the WT peptide upon binding to Mth. In silico, this would lead to a low probability of observing the predicted W5K complex where the helical conformation of the ligand was retained.R15Q
We expected from the combinatorial mutation calculations that this mutant would have dramatically decreased binding (by ~13 kcal/mol). Indeed the MD simulation finds that the initial hydrogen bond of Q15 with Asp127 breaks, making a salt-bridge to R14. However, the R15Q preserves the hydrophobic interaction between W5 and Phe153 in the MD, whereas this interaction is not preserved in the MD for the WT. Moreover the hydrogen bond of Glu136 to S4 is lost during the MD of the R15Q mutant. The net results from the MD is that the binding for R15Q is essentially the same (~1 kcal/mol worse) as for the WT. The experiments find a slightly decreased binding affinity (18 to 38 nM), indicating very similar binding.
-
Y9F
Compared with Y9 in the WT, the mutated F shows more conformational fluctuation. The hydroxyl group of Y9 in WT holds it closer to the receptor through water-mediated hydrogen bond with the backbone of the receptor. The centroid-to-centroid distance between F9 and Phe130 represents this behavior (initially ~5 Å and occasionally ~8Å (Fig. S2-(b))). W5 has the original hydrophobic contact with Phe153, which becomes loose after further equilibration in WT. The experiments find a slightly decreased binding affinity (18 to 49 nM) indicating very similar bonding.
FIGURE 7.

The structures of WT and W5K mutant of LP1 peptide ligands after 5 ns equilibration (red) in the fully solvated water box (4500 atoms, 36×36×44 Å3). The initial docked conformation is colored blue.
To summarize, the experiments and predictions indicated that L11Q, Y9F and R15Q mutant ligands exhibit comparable interactions to the WT LP1 peptide. We expected that W5F would lead to worse binding, which was confirmed by the MD simulation and experimental results. For the W5K complex, we thought that an additional hydrophilic interaction from mutation of W5 to K might compensate for the missing aromatic interactions. However, MD simulations of the W5K peptide alone and in complex with Mth suggested a more severe decrease in binding, which was experimentally verified as W5K showed no measureable affinity to Mth.
The changes in binding energy are not in quantitative agreement with experiments. Several possibilities for this include: 1) the energies in the dynamics fluctuate greatly as various charged side groups and counter-ions move about, 2) no account is taken of the entropic effects upon binding, and 3) the length of the dynamics might not sample all significant binding modes. Regardless, the predictions on how the binding changes from the combinatorial mutation calculations and the MD simulations are qualitatively consistent with experiments, indicating which ones should lead to comparable binding. Thus such calculations are likely to distinguish bad binders from good binders.
CONCLUSION
The docking study of the Mth ectodomain with the peptide ligands was carried out using rigid docking methods followed by side chain replacement and force-field based scoring. The RWR motif of the ligands showed favorable aromatic and electrostatic interactions with the ectodomain, demonstrating the importance of these residues on binding as suggested from the conservation seen in the previous experimental study (6). Indeed these studies show that the critical residues of the protein are: Asp139 interacting with W5 and R8, Phe130 with W5, either Asp78 or Asp127 with R15 and Asp154 with R2 in the LR ligands. This provides a number of predictions that can be subjected to experimental tests.
Using the predicted structures, we illustrate the combinatorial mutation calculation strategy to predict interesting mutations for experimental study. This is a complement to experimental screening or selection studies. Based on the computational mutagenesis results, we measured the kinetics of binding of the mutant peptide ligands with the Mth ectodomain. Here we found that conformational relaxation of the mutant complex structures from MD simulations is essential to obtain qualitative correlations with the experiments. The computational predictions are consistent with experiments but clearly an improvement in binding scoring will be needed to use such methods for optimizing ligands.
It is encouraging that the current method of docking such a large peptide ligand to a receptor protein leads to results in apparently excellent agreement with experiments. We expect that the binding characteristics examined in this study for the Mth ectodomain will provide insight helpful in investigating interactions between the N-terminal domains of other class B GPCRs and their peptide ligands. To understand the overall process of GPCR function, it will be necessary to model ligand binding to the entire receptor structure, including the transmembrane domains. Our studies of the ectodomain-ligand binding are the first step for elucidating this process.
Supplementary Material
Acknowledgments
We thank J. Kaiser for help in calculating the density map, A.P. West, Jr. for providing the experimental density map, and J. Vielmetter and the Protein Expression Center of the Beckman Institute for technical assistance on the Biacore T100.
Footnotes
Abbreviations: GPCR, G protein-coupled receptor; Mth, Methuselah; CRF, corticotropin-releasing factor; GIP, glucose-dependent insulinotropic polypeptide; RMSD, root mean square deviation; VMD, Visual Molecular Dynamics; PME, Particle Mesh Ewald; FF, forcefield; SPR, surface plasmon resonance; MD, molecular dynamics
Throughout the text, the Mth ectodomain residues are denoted by their three-letter abbreviations while peptide ligand residues are denoted by their one-letter codes.
This work was supported by funding from the NIH (K99AG030493 to W.W.J.; AG16630 to S.B.). The computational parts of this study were supported in part by NSF (CTS-0608889) and NIH (1 RO1 CA 112293-01).
SUPPORTING INFORMATION AVAILABLE
Kinetics analysis of peptide binding by surface plasmon resonance (Table S1), binding charateristics of the LR2 ligand into the Mth ectodomain (Figure S1), changes in distance during the 2 ns MD simulation of key intermolecular interactions in mutant complexes (Figure S2) and SPR sensorgrams of LP1 wild-type (WT) and mutant peptides with the Mth ectodomain (Figure S3). The material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Lin YJ, Seroude L, Benzer S. Extended life-span and stress resistance in the Drosophila mutant methuselah. Science. 1998;282:943–946. doi: 10.1126/science.282.5390.943. [DOI] [PubMed] [Google Scholar]
- 2.Harmar AJ. Family-B G-protein-coupled receptors. Genome Biol. 2001;2:reviews3013.3011–reviews3013.3010. doi: 10.1186/gb-2001-2-12-reviews3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.West AP, Llamas LL, Snow PM, Benzer S, Bjorkman PJ. Crystal structure of the ectodomain of Methuselah, a Drosophila G protein-coupled receptor associated with extended lifespan. Proc Natl Acad Sci USA. 2001;98:3744–3749. doi: 10.1073/pnas.051625298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grace CRR, Perrin MH, Gulyas J, DiGruccio MR, Cantle JP, Rivier JE, Vale WW, Riek R. Structure of the N-terminal domain of a type B1 G protein-coupled receptor in complex with a peptide ligand. Proc Natl Acad Sci USA. 2007;104:4858–4863. doi: 10.1073/pnas.0700682104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Parthier C, Kleinschmidt M, Neumann P, Rudolph R, Manhart S, Schlenzig D, Fanghanel J, Rahfeld JU, Demuth HU, Stubbs MT. Crystal structure of the incretin-bound extracellular domain of a G protein-coupled receptor. Proc Natl Acad Sci USA. 2007;104:13942–13947. doi: 10.1073/pnas.0706404104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ja WW, West AP, Delker SL, Bjorkman PJ, Benzer S, Roberts RW. Extension of Drosophila melanogaster life span with a GPCR peptide inhibitor. Nat Chem Biol. 2007;3:415–419. doi: 10.1038/nchembio.2007.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cao YJ, Gimpl G, Fahrenholz F. The Amino-Terminal Fragment of the Adenylate-Cyclase Activating Polypeptide (Pacap) Receptor Functions as a High-Affinity Pacap Finding Domain. Biochem Bioph Res Co. 1995;212:673–680. doi: 10.1006/bbrc.1995.2021. [DOI] [PubMed] [Google Scholar]
- 8.Chow BKC. Functional antagonism of the human secretin receptor by a recombinant protein encoding the N-terminal ectodomain of the receptor. Recept Signal Trans. 1997;7:143–150. [PubMed] [Google Scholar]
- 9.Runge S, Schimmer S, Oschmann J, Schiodt CB, Knudsen SM, Jeppesen CB, Madsen K, Lau J, Thogersen H, Rudolph R. Differential structural properties of GLP-1 and exendin-4 determine their relative affinity for the GLP-1 receptor N-terminal extracellular domain. Biochemistry. 2007;46:5830–5840. doi: 10.1021/bi062309m. [DOI] [PubMed] [Google Scholar]
- 10.Vriend G. What If - a Molecular Modeling and Drug Design Program. J Mol Graphics. 1990;8:52–56. doi: 10.1016/0263-7855(90)80070-v. [DOI] [PubMed] [Google Scholar]
- 11.Humphrey W, Dalke A, Schulten K. VMD: Visual molecular dynamics. J Mol Graphics. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 12.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mackerell AD, Bashford D, Bellott M, Dunbrack RL, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph D, Kuchnir L, Kuczera K, Lau FTK, Mattos C, Michnick S, Nguyen DT, Ngo T, Prodhom B, Roux B, Schlenkrich M, Smith J, Stote R, Straub J, Wiorkiewiczkuczera J, Karplus M. Self-Consistent Parameterization of Biomolecules for Molecular Modeling and Condensed Phase Simulations. Faseb J. 1992;6:A143–A143. [Google Scholar]
- 14.MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FTK, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiorkiewicz-Kuczera J, Yin D, Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 15.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of Simple Potential Functions for Simulating Liquid Water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 16.Ghosh A, Rapp CS, Friesner RA. Generalized born model based on a surface integral formulation. J Phys Chem B. 1998;102:10983–10990. [Google Scholar]
- 17.Mayo SL, Olafson BD, Goddard WA. Dreiding - a Generic Force-Field for Molecular Simulations. J Phys Chem. 1990;94:8897–8909. [Google Scholar]
- 18.Lim KT, Brunett S, Iotov M, McClurg RB, Vaidehi N, Dasgupta S, Taylor S, Goddard WA. Molecular dynamics for very large systems on massively parallel computers: The MPSim program. J Comput Chem. 1997;18:501–521. [Google Scholar]
- 19.Ding HQ, Karasawa N, Goddard WA. Atomic Level Simulations on a Million Particles - the Cell Multipole Method for Coulomb and London Nonbond Interactions. J Chem Phys. 1992;97:4309–4315. [Google Scholar]
- 20.Chen R, Li L, Weng ZP. ZDOCK: An initial-stage protein-docking algorithm. Proteins. 2003;52:80–87. doi: 10.1002/prot.10389. [DOI] [PubMed] [Google Scholar]
- 21.Bailey S. The Ccp4 Suite - Programs for Protein Crystallography. Acta Crystallogr D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.