

Abstract
Oleoylethanolamide (OEA) is a biologically active lipid amide that is released by small-intestinal enterocytes during the absorption of dietary fat and inhibits feeding by engaging the nuclear receptor, peroxisome proliferator-activated receptor-α (PPAR-α). Previous studies have shown that the anorexic effects of systemically administered OEA require the activation of sensory afferents of the vagus nerve. The central circuits involved in mediating OEA-induced hypophagia remain unknown. In the present study, we report the results of in situ hybridization and immunohistochemistry experiments in rats and mice, which show that systemic injections of OEA (5–10 mg kg−1, intraperitoneal) enhance expression of the neuropeptide oxytocin in magnocellular neurons of the paraventricular nucleus (PVN) and supraoptic nucleus (SON) of the hypothalamus. No such effect is observed with other hypothalamic neuropeptides, including vasopressin, thyrotropin-releasing hormone and pro-opiomelanocortin. The increase in oxytocin expression elicited by OEA was absent in mutant PPAR-α-null mice. Pharmacological blockade of oxytocin receptors in the brain by intracerebroventricular infusion of the selective oxytocin antagonist, L-368,899, prevented the anorexic effects of OEA. The results suggest that OEA suppresses feeding by activating central oxytocin transmission.
Introduction
The ingestion of dietary fat stimulates epithelial cells of the small intestine to produce the bioactive lipid amide, OEA (Rodríguez de Fonseca et al., 2001; Fu et al., 2007; Schwartz et al., 2008), an endogenous high-affinity agonist of the nuclear receptor PPAR-α (Fu et al., 2003). Acting within the gut, newly formed OEA engages sensory fibers of the vagus nerve to cause a behaviorally selective suppression of food intake (Rodríguez de Fonseca et al., 2001; Gaetani et al., 2003; Proulx et al., 2005; Fu et al., 2008), and stimulates enterocytes to express genes involved in lipid absorption (Yang et al., 2007). Both responses require the activation of PPAR-α, which is highly expressed in duodenal and jejunal enterocytes (Bünger et al., 2007; Fu et al., 2007). These results suggest that small-intestinal OEA signaling serves as a fat-sensing mechanism that cooperates with premeal insulin release and other cephalic responses to optimize lipid utilization after ingestion of a fat-rich meal (Schwartz et al., 2008).
The central neurotransmitter systems recruited by peripheral OEA to inhibit food intake are still unknown. Identifying them would help, however, to understand how animals monitor the ingestion of fat-containing foods and how such monitoring processes might become dysfunctional in obesity and other eating disorders characterized by excessive fat intake. Previous work has shown that systemic administration of OEA promotes, via afferent vagal fibers, expression of the activity-dependent gene c-fos in the paraventricular (PVN) and supraoptic (SON) nuclei of the hypothalamus (Rodríguez de Fonseca et al., 2001). Neurons in these nuclei express a variety of neuropeptides, including oxytocin and vasopressin, which are involved in the central coordination of metabolic signals originated in peripheral tissues (Broberger and Hökfelt, 2001; Berthoud and Morrison, 2008). In the present study, we examined whether neuropeptide-secreting neurons in the PVN and SON might be implicated in mediating the anorexic effects of OEA.
Materials and Methods
Animals.
We purchased adult male Wistar rats (250–300 g) from Harlan Italy, PPAR-α−/− mice (B6.129S4-PparatmtGonzN12) and wild-type controls (C57BL/6 mice) from Jackson Laboratory (through Charles River Laboratories, Italy). The animals were housed under controlled conditions of temperature and humidity and were kept on a 12 h light/dark cycle, with lights on at 6:30 P.M. Water and standard chow pellets (Prolab RMH 2500, PMI Nutrition International) were available ad libitum. The animals were accustomed to handling and injections for 7 d before experiments. On day 8, they received drug of vehicle injections ∼10 min before dark. At the end of the experiments, the animals were killed with an overdose of chloral hydrate (0.5 g kg−1, i.p.). All procedures met the guidelines from the Italian Ministry of Health, from the United States National Institutes of Health, detailed in the Guide for the Care of Laboratory Animals, and the European Community directives 86/609/EEC regulating animal research.
In situ hybridization.
We used RNA polymerases (Roche Diagnostics) in the presence of both [35S]CTP and [35S]UTP, to prepare radioactively labeled riboprobes ([35S]cRNA) of c-fos (coding region 583-1250 of rat c-fos), oxytocin (coding region 1-378 of rat oxytocin), vasopressin (coding region 5-461 of rat vasopressin), thyrotropin-releasing hormone (TRH, coding region 1-768 of rat TRH), proopiomelanocortin (POMC, coding region 68-666 of rat POMC). We prepared the digoxigenin-labeled (DIG) oxytocin riboprobe using a DIG RNA labeling kit (Roche Diagnostics) following the manufacturer's instructions. We cut serial coronal brain sections (20-μm-thick) on a cryostat (Microm) and hybridized them at 60°C for 16 h in a buffer containing [35S]cRNA (75 × 106 dpm ml−1), 10% dextran sulfate, 50% formamide, 1 × Denhardt's solution, 100 μg ml−1 denatured salmon sperm DNA, 0.15 mg ml−1 tRNA and 40 mm dithiothreitol. For in situ double hybridization experiments, we prepared a mixture of DIG-oxytocin riboprobe (200 mg ml−1) and radiolabeled c-fos riboprobe (75 × 106 dpm ml−1) in the same hybridization buffer. After hybridization, the sections were exposed to Kodak Biomax film (Sigma-Aldrich) for 8–72 h. The specificity of the hybridization signal was ascertained by hybridization of same sections labeled with corresponding sense probes. The colocalization of signals after in situ double hybridization was evaluated by immunocytochemical staining of the DIG-labeled probe using an anti-DIG antibody conjugated to alkaline phosphatase (Roche Diagnostics) and successive exposure to Kodak autoradiography emulsion (Integra Biosciences) for 18 d at 4°C. We examined the slices using a Nikon 80i Eclipse microscope, and captured images in bright field for oxytocin hybridization and dark-field for c-fos hybridization. We evaluated probes colocalization by analyzing the 2 merged images, using the Scion Image software (National Institutes of Health, Bethesda, MD).
Densitometric analyses.
We scanned autoradiography films at high resolution (1200 dpi) and used a brain atlas (Paxinos and Watson, 1997) to define localization of brain structures. Quantitative analyses of hybridized signals were performed using the Scion Image software. Optical density (OD) values were converted into radioactivity concentrations by densitometric analysis of 14C-microscale standards (American Radiolabeled Chemicals), so as to create for each film a calibration curve with a linear coefficient r2 > 0.9. In every brain section, the OD of the corpus callosum was used as background and an integrated OD value was calculated as radioactivity per extension of hybridized area. Measurements were obtained in at least 4 consecutive tissue sections containing the desired structure.
Immunohistochemistry.
We perfused rats, through their left heart ventricle, with a saline solution and then with a fixation solution containing 4% paraformaldehyde in 0.1 m phosphate buffer (PB, pH 7.2). Brains were collected, postfixed for 1 d, and cut in coronal sections with a vibratome (40 μm thickness). Sections were incubated for 24 h at 4°C with anti-c-fos antibody (1:500 dilution, Santa Cruz Biotechnology) and anti-oxytocin antibody (1:1000 dilution, Millipore Bioscience Research Reagents), then rinsed in 0.1 m PB and exposed to anti-mouse Alexa Fluor 488 and anti-rabbit Alexa Fluor 546 (1:1000 dilution, Invitrogen) for 2 h. After an additional 10 min rinse, the sections were observed under a Nikon Eclipse C1 confocal microscope (at 0.90 μm optical thickness). Sagittal sections of the pituitary gland (15-μm-thick) were cut on a cryostat and mounted on Superfrost plus slides. The sections were treated with 0.3% H2O2 in methanol for 20 min to inactivate endogenous peroxidase, exposed to blocking serum (2% normal goat serum) for 1 h at room temperature, and incubated overnight at room temperature with a biotinylated anti-oxytocin antibody (1:10,000 dilution, Millipore Bioscience Research Reagents). Sections were washed and then incubated in biotinylated goat anti-mouse serum (1:500 dilution, Jackson ImmunoResearch) for 60 min at room temperature. After additional 10 min rinsing with 0.1 m PB, sections were treated with avidin–biotin–peroxidase complex (ABC, 1:200 dilution, Vector Laboratories), and developed with diaminobenzidine (DAB) with metal enhancer (Sigma-Aldrich). Slides were rinsed, dehydrated in ascending ethanol concentrations, cleared in xylene and mounted with Eukitt mounting medium (Sigma-Aldrich). Slice images were captured using a Nikon 80i Eclipse microscope and oxytocin expression was evaluated by reading OD values in the neurohypophysis, taking the OD values of the adenohypophysis as background.
Drugs and treatments.
We prepared OEA in the laboratory (Giuffrida et al., 2000) and administered it by intraperitoneal injection in a vehicle of saline/polyethylene glycol/Tween 80 (90/5/5, v/v). We purchased L-368,899 from Tocris Bioscience and administered into the brain third ventricle in 1 μl of saline 10 min before OEA or vehicle administration.
Oxytocin measurements.
We used an enzyme immunoassay kit (Assay Designs) to measure oxytocin levels in plasma samples following the manufacturer's instruction.
Intracerebroventricular infusions.
We anesthetized rats with equithesin and stereotaxically implanted a 26-gauge guide cannula (18 mm) positioned 1 mm dorsal to the third ventricle (Paxinos and Watson, 1997): AP −2.2, L 0.05, and DV −7.6 from bregma). Infusions were made through a 30-gauge infusion cannula that extended 2 mm beyond the end of the guide cannula and that was connected to a 10 μl Hamilton syringe by a PE-20 polyethylene tubing. The syringe was driven by an automated pump (CMA 400) at the rate of 1 μl min−1 to provide an infusion volume of 1 μl. Cannula placements were validated pharmacologically and verified histologically at end of experiments. Briefly, 1 week after surgery, we infused angiotensin II (10 ng in 1 μl of saline) into the cannula and used in subsequent experiments only those animals that drank ≥5 ml of water in the 30 min following angiotensin II infusion. Experiments were conducted at least 2 d after angiotensin II infusions.
Feeding behavior.
Food intake was recorded with automated cages (PRS Italia) equipped with lickometers (to monitor water consumption as number of licks) and food trays continuously accessible to the rats. The food trays contained standard chow pellets and were connected to weight sensors. The rats were habituated to test cages for 2 d before trials, and had ad libitum access to food and water. Experiments began 30 min before dark and lasted 18 h.
Statistical analyses.
Statistical analyses were performed using the Prism software (GraphPad Software). Statistical significance for mRNA expression and data from behavioral experiments was determined by two-way or one-way ANOVA, depending on experimental setting, and multiple comparisons were performed by Tukey's post hoc test. Microdialysis results were analyzed by two-way ANOVA for repeated measures with treatment (OEA, or vehicle) as between-subject factor and time as within-subject factor. In all instances, statistical significance threshold was set at p < 0.05.
Results
Peripheral OEA induces c-fos mRNA expression in oxytocin neurons of PVN and SON
Systemically administered OEA does not readily enter the CNS (Campolongo et al., 2009), presumably because of the high expression of its degrading enzyme, fatty acid amide hydrolase, in the blood–brain barrier (Egertová et al., 2000). We administered OEA to rats by intraperitoneal injection, at a dosage that does not allow penetration into the brain (10 mg kg−1) (Campolongo et al., 2009), and measured c-fos mRNA expression in the CNS by in situ hybridization. As previously reported (Rodríguez de Fonseca et al., 2001), OEA administration produced a localized increase of c-fos mRNA in PVN and SON, but not other regions of the forebrain (Fig. 1 A,B). Densitometric analyses illustrated in Figure 1, C and D, show that this effect was statistically detectable 30–60 min after OEA injection. In situ double-hybridization experiments revealed that ∼70% of PVN neurons, which responded to OEA with an elevation in c-fos expression, also contained oxytocin mRNA (Fig. 1 E,F). Other neuronal populations activated by OEA administration were not identified in the present study. Immunohistochemical experiments confirmed that OEA-induced c-fos expression occurred in oxytocin-expressing magnocellular neurons (Fig. 1 G). The results suggest that systemic administration of OEA activates prevalently, albeit not exclusively, oxytocin neurons of the hypothalamus.
Figure 1.
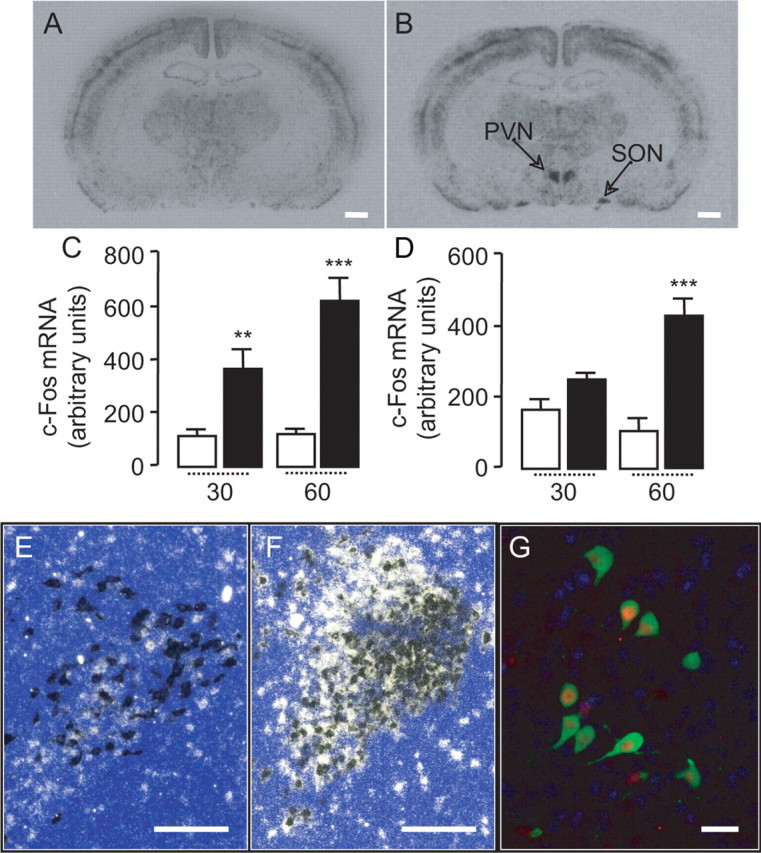
OEA stimulates c-fos mRNA expression in oxytocin neurons of the hypothalamus. A, B, Rat brain coronal sections showing the effects of vehicle (A) or OEA (10 mg kg−1, i.p.) (B) on c-fos mRNA levels in the paraventricular (PVN) and supraoptic (SON) nuclei of the hypothalamus. Calibration bar, 1 mm. C, D, Time course (in minutes) of OEA-induced c-fos mRNA expression in PVN (C) and SON (D). Open bar, vehicle; closed bar, OEA (10 mg kg−1, i.p.). E, F, In situ double hybridization images showing the effects of vehicle (E) or OEA (F) on mRNA levels of c-fos (white spots) and oxytocin (dark spots) in the PVN. Calibration bar, 0.25 mm. G, Immunohistochemical detection of c-fos protein (red) and oxytocin (green) in neurons of the PVN. Calibration bar, 50 μm. **p < 0.01, ***p < 0.001, versus respective control; n = 5–6.
Peripheral OEA induces oxytocin mRNA expression in PVN and SON
To further test this idea, we quantified oxytocin mRNA in the brain following OEA injection (5–10 mg kg−1, i.p.). The treatment stimulated, in a dose- and time-dependent manner, oxytocin expression in the PVN and SON (Fig. 2 A,B; supplemental Fig. 1, available at www.jneurosci.org as supplemental material). By contrast, OEA injection did not change the expression of three additional hypothalamic peptides that are implicated in energy balance: vasopressin and TRH, which are highly expressed in the PVN and SON, and POMC, which is highly expressed in the arcuate nucleus and generates peptide signals that control PVN and SON activity (Morton et al., 2006) (Fig. 2 A,B; supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Notably, OEA (10 mg kg−1) did not alter oxytocin mRNA levels in mutant mice lacking PPAR-α (Fig. 2 C,D), indicating that the same receptor system that mediates the satiety-inducing effects of OEA (Fu et al., 2003) is also involved in regulating oxytocin expression. As expected from our localization studies, which pointed to magnocellular neurons as a primary site of OEA-induced oxytocin expression (Fig. 1 G), we found that systemic OEA administration increased oxytocin immunoreactivity in the neurohypophysis (Fig. 3 A–C) and oxytocin levels in plasma (Fig. 3 D).
Figure 2.

OEA induces oxytocin mRNA expression in PVN and SON. A, B, Effects of vehicle (open bars) or OEA (closed bars, 5 and 10 mg kg−1, i.p.) on oxytocin and vasopressin mRNA levels in the rat PVN (A) and SON (B). C, D, Effects of vehicle or OEA (10 mg kg−1, i.p.) on oxytocin mRNA expression in wild-type C57BL/6 mice (Wt, open bars) and PPAR-α−/− mice (closed bars) in the PVN (C) and SON (D). Animals were killed 1 h after OEA administration. *p < 0.05; **p < 0.01, versus respective control; n = 5–6.
Figure 3.

OEA increases oxytocin expression in the neurohypophysis and plasma. A, B, Effect of vehicle (A) or OEA (B) (10 mg kg−1, i.p) on oxytocin Immunoreactivity in rat neurohypophysis. Calibration bar, 0.25 mm. C, Quantitative oxytocin expression, assessed by immunohistochemistry, in rat neurohypophysis. D, Time course (in minutes) of oxytocin release into plasma. Open bars, Vehicle; closed bars, OEA (10 mg kg−1, i.p.).*p < 0.05, versus respective control; n = 5–6.
Central oxytocin receptor blockade prevents OEA-induced hypophagia
Multiple lines of evidence indicate that central oxytocin transmission is implicated in the modulation of feeding behavior (Verbalis et al., 1986; Kirchgessner et al., 1988; Arletti et al., 1990; Swaab et al., 1995; Douglas et al., 2007; Amico et al., 2008; Kublaoui et al., 2008). We examined, therefore, whether oxytocin may contribute to the anorexic effects of peripheral OEA. Thirty min before dark, we infused two doses of the selective oxytocin receptor antagonist, L-368,899 (0.05 and 0.5 nmol) (Thompson et al., 1997), into the third ventricle of free-feeding rats, which then received systemic injections of either vehicle or OEA (10 mg kg−1, i.p.). The selectivity of this agent for oxytocin receptors has been previously documented both in vitro and in vivo (Williams et al., 1994; Chiu et al., 1995; Thompson et al., 1997; Gupta et al., 2008). We used an automated system to monitor the amount of food and water consumed by the animals for 18 h following drug administration. Systemic injections of OEA caused a profound reduction in total food intake (Rodríguez de Fonseca et al., 2001; Gaetani et al., 2003), an effect that was dose-dependently inhibited by L-368,899 (Fig. 4 A). Underscoring the selectivity of this response, the oxytocin antagonist did not alter food intake when infused alone (Fig. 4 A) and did not influence OEA-induced c-fos expression in the nucleus of the solitary tract (NST) (Rodríguez de Fonseca et al., 2001) (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). None of the drugs had any significant influence on water intake (Fig. 4 B). The finding that intracerebroventricular infusion of a selective oxytocin antagonist prevents the hypophagia evoked by OEA indicates that central oxytocin receptors play an obligatory role in this response.
Figure 4.

OEA anorexic effect is prevented by oxytocin receptor antagonist. A, B, Effects of L-368,899 (0.05 nmol of and 0.5 nmol) and/or OEA (10 mg kg−1, i.p.) on food intake (A) and water intake (B) in rats. **p < 0.01, versus respective control; ## p < 0.01 versus OEA group; n = 5–6.
Discussion
Fat is an essential component of the mammalian diet, but its availability in nature is often unpredictable. Mammals have adapted to this environmental challenge by developing a network of neural and hormonal signals that align food intake and lipid metabolism to the variable accessibility of fat-rich foods (Gillum et al., 2008; Schwartz et al., 2008). The lipid mediator OEA appears to play an important role in this network. Activation of small-intestinal OEA signaling by dietary fat engages the sensory vagus to prolong the interval between meals (Gaetani et al., 2003; Schwartz et al., 2008) and concomitantly enhances lipid uptake by enterocytes (Fu et al., 2003; Yang et al., 2007), two effects that are expected to optimize fat utilization. PPAR-α localized to enterocytes and, possibly, sensory terminals is thought to mediate this response (Fu et al., 2003). In the present report, we show that systemic administration of OEA stimulates, at doses that cause a behaviorally selective inhibition of food intake (Gaetani et al., 2003; Proulx et al., 2005), the expression of oxytocin in magnocellular neurons of the PVN and SON. We further demonstrate that central oxytocin receptor blockade by the selective antagonist L-368,899 abrogates the ability of systemic OEA to alter feeding behavior. Together, these results suggest that peripheral OEA signaling suppresses food intake by activating oxytocin transmission in the hypothalamus.
Several signals of peripheral origin, such as the gut peptide cholecystokinin, control feeding by engaging vagal sensory fibers that converge on the NST of the brainstem. Higher-order brain regions are engaged to process the information coming from that nucleus: pivotal in this regard is the PVN of the hypothalamus, which integrates central and peripheral satiety signals and orchestrates autonomic responses by adjusting the balance between energy intake and energy expenditure (for review, see Schwartz et al., 2000; Berthoud and Morrison, 2008). Several lines of evidence suggest that OEA may act like cholecystokinin to suppress feeding. First, OEA fails to reduce food intake in rats in which the subdiaphragmatic vagus nerve has been severed (Rodríguez de Fonseca et al., 2001). Second, rats deprived of peripheral sensory fibers by treatment with the neurotoxin capsaicin fail to respond to OEA or other PPAR-α agonists, but retain their ability to respond to centrally acting anorexic drugs, such as the serotonergic agonist CP-93129 (Rodríguez de Fonseca et al., 2001). Third, although potent when administered peripherally, OEA produces no change in feeding after injection into the rat brain ventricles (Rodríguez de Fonseca et al., 2001). Fourth, in situ hybridization experiments show that systemically administered OEA is selective at activating expression of c-fos in the NST, PVN and SON (Rodríguez de Fonseca et al., 2001 and present study). The latter result, in particular, is consistent with an involvement of hypothalamic oxytocin in mediating the anorexic effects of OEA. It is noteworthy, in this regard, that oxytocin receptor blockade in the brain does not influence the ability of OEA to engage the NTS, even though it prevents OEA-induced hypophagia. This suggests that activation of the NTS precedes that of oxytocinergic neurons in the PVN and SON.
The integrative function served by oxytocinergic transmission in social behaviors is well documented (Insel and Young, 2001; Insel, 2003; Lee et al., 2009), but there is also a growing appreciation for the role played by this peptide in the central regulation of energy balance (Amico et al., 2005). For example, electrophysiological studies have shown that magnocellular oxytocin-secreting neurons in the hypothalamus become strongly activated during feeding, which is suggestive of a role for such neurons in the regulation of satiety (Douglas et al., 2007). Consistent with this view, mutant mice that do not express oxytocin show an enhanced intake of sweet and nonsweet carbohydrate solutions (Sclafani et al., 2007) and develop late-onset obesity (Camerino, 2009). Similarly, mice with heterozygous inactivation of the transcription factor single-minded 1 (SIM1), which reduces oxytocin expression in the hypothalamus, develop hyperphagic obesity (Kublaoui et al., 2008). Mutations affecting SIM1 also lead to profound hyperphagia and obesity in humans (Faivre et al., 2002). Our findings support a role for oxytocin as a central regulator of satiety and provide the first evidence of a link between fat-dependent OEA signaling in the gut and oxytocin transmission in the brain. Future studies should examine whether such a link extends to other physiological and behavioral processes that are influenced by central and peripheral oxytocin.
Footnotes
This study was supported by two national grants (PRIN) from the Italian Ministry of Education to S.G. (2007R2WFBZ) and to V.C. (2007WRJNMX_003), and by a National Institute of Diabetes and Digestive and Kidney Diseases grant (DK073955) to D.P. We thank Dr. Mario Falchi for use of the confocal microscope.
References
- Amico JA, Vollmer RR, Cai HM, Miedlar JA, Rinaman L. Enhanced initial and sustained intake of sucrose solution in mice with an oxytocin gene deletion. Am J Physiol Regul Integr Comp Physiol. 2005;289:R1798–R1806. doi: 10.1152/ajpregu.00558.2005. [DOI] [PubMed] [Google Scholar]
- Amico JA, Miedlar JA, Cai HM, Vollmer RR. Oxytocin knockout mice: a model for studying stress-related and ingestive behaviours. Prog Brain Res. 2008;170:53–64. doi: 10.1016/S0079-6123(08)00405-6. [DOI] [PubMed] [Google Scholar]
- Arletti R, Benelli A, Bertolini A. Oxytocin inhibits food and fluid intake in rats. Physiol Behav. 1990;48:825–830. doi: 10.1016/0031-9384(90)90234-u. [DOI] [PubMed] [Google Scholar]
- Berthoud HR, Morrison C. The brain, appetite, and obesity. Annu Rev Psychol. 2008;59:55–92. doi: 10.1146/annurev.psych.59.103006.093551. [DOI] [PubMed] [Google Scholar]
- Broberger C, Hökfelt T. Hypothalamic and vagal neuropeptide circuitries regulating food intake. Physiol Behav. 2001;74:669–682. doi: 10.1016/s0031-9384(01)00611-4. [DOI] [PubMed] [Google Scholar]
- Bünger M, van den Bosch HM, van der Meijde J, Kersten S, Hooiveld GJ, Müller M. Genome-wide analysis of PPAR-alpha activation in murine small intestine. Physiol Genomics. 2007;30:192–204. doi: 10.1152/physiolgenomics.00198.2006. [DOI] [PubMed] [Google Scholar]
- Camerino C. Low sympathetic tone and obese phenotype in oxytocin-deficient mice. Obesity. 2009;17:980–984. doi: 10.1038/oby.2009.12. [DOI] [PubMed] [Google Scholar]
- Campolongo P, Roozendaal B, Trezza V, Cuomo V, Astarita G, Fu J, McGaugh JL, Piomelli D. Fat-induced satiety factor oleoylethanolamide enhances memory consolidation. Proc Natl Acad Sci U S A. 2009;106:8027–8031. doi: 10.1073/pnas.0903038106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu SH, Thompson KA, Vincent SH, Alvaro RF, Huskey SW, Stearns RA, Pettibone DJ. The role of drug metabolism in drug discovery: a case study in the selection of an oxytocin receptor antagonist for development. Toxicol Pathol. 1995;23:124–130. doi: 10.1177/019262339502300204. [DOI] [PubMed] [Google Scholar]
- Douglas AJ, Johnstone LE, Leng G. Neuroendocrine mechanisms of change in food intake during pregnancy: a potential role for brain oxytocin. Physiol Behav. 2007;91:352–365. doi: 10.1016/j.physbeh.2007.04.012. [DOI] [PubMed] [Google Scholar]
- Egertová M, Cravatt BF, Elphick MR. Fatty acid amide hydrolase expression in rat choroid plexus: possible role in regulation of the sleep-inducing action of oleamide. Neurosci Lett. 2000;282:13–16. doi: 10.1016/s0304-3940(00)00841-7. [DOI] [PubMed] [Google Scholar]
- Faivre L, Cormier-Daire V, Lapierre JM, Colleaux L, Jacquemont S, Geneviéve D, Saunier P, Munnich A, Turleau C, Romana S, Prieur M, De Blois MC, Vekemans M. Deletion of the SIM1 gene (6q16.2) in a patient with a Prader-Willi-like phenotype. J Med Genet. 2002;39:594–596. doi: 10.1136/jmg.39.8.594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu J, Gaetani S, Oveisi F, Lo Verme J, Serrano A, Rodríguez de Fonseca F, Rosengarth A, Luecke H, Di Giacomo B, Tarzia G, Piomelli D. Oleylethanolamide regulates feeding and body weight through activation of the nuclear receptor PPAR-alpha. Nature. 2003;425:90–93. doi: 10.1038/nature01921. [DOI] [PubMed] [Google Scholar]
- Fu J, Astarita G, Gaetani S, Kim J, Cravatt BF, Mackie K, Piomelli D. Food intake regulates oleoylethanolamide formation and degradation in the proximal small intestine. J Biol Chem. 2007;282:1518–1528. doi: 10.1074/jbc.M607809200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu J, Kim J, Oveisi F, Astarita G, Piomelli D. Targeted enhancement of oleoylethanolamide production in proximal small intestine induces across-meal satiety in rats. Am J Physiol Regul Integr Comp Physiol. 2008;295:R45–R50. doi: 10.1152/ajpregu.00126.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaetani S, Oveisi F, Piomelli D. Modulation of meal pattern in the rat by the anorexic lipid mediator oleoylethanolamide. Neuropsychopharmacology. 2003;28:1311–1316. doi: 10.1038/sj.npp.1300166. [DOI] [PubMed] [Google Scholar]
- Gillum MP, Zhang D, Zhang XM, Erion DM, Jamison RA, Choi C, Dong J, Shanabrough M, Duenas HR, Frederick DW, Hsiao JJ, Horvath TL, Lo CM, Tso P, Cline GW, Shulman GI. N-acylphosphatidylethanolamine, a gut-derived circulating factor induced by fat ingestion, inhibits food intake. Cell. 2008;135:813–824. doi: 10.1016/j.cell.2008.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giuffrida A, Rodríguez de Fonseca F, Piomelli D. Quantification of bioactive acylethanolamides in rat plasma by electrospray mass spectrometry. Anal Biochem. 2000;280:87–93. doi: 10.1006/abio.2000.4509. [DOI] [PubMed] [Google Scholar]
- Gupta J, Russell R, Wayman C, Hurley D, Jackson V. Oxytocin-induced contractions within rat and rabbit ejaculatory tissues are mediated by vasopressin V1A receptors and not oxytocin receptors. Br J Pharmacol. 2008;155:118–126. doi: 10.1038/bjp.2008.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Insel TR. Is social attachment an addictive disorder? Physiol Behav. 2003;79:351–357. doi: 10.1016/s0031-9384(03)00148-3. [DOI] [PubMed] [Google Scholar]
- Insel TR, Young LJ. The neurobiology of attachment. Nat Rev Neurosci. 2001;2:129–136. doi: 10.1038/35053579. [DOI] [PubMed] [Google Scholar]
- Kirchgessner AL, Sclafani A, Nilaver G. Histochemical identification of a PVN-hindbrain feeding pathway. Physiol Behav. 1988;42:529–543. doi: 10.1016/0031-9384(88)90154-0. [DOI] [PubMed] [Google Scholar]
- Kublaoui BM, Gemelli T, Tolson KP, Wang Y, Zinn AR. Oxytocin deficiency mediates hyperphagic obesity of Sim1 haploinsufficient mice. Mol Endocrinol. 2008;22:1723–1734. doi: 10.1210/me.2008-0067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, Macbeth AH, Pagani JH, Young WS., 3rd Oxytocin: the great facilitator of life. Prog Neurobiol. 2009;88:127–151. doi: 10.1016/j.pneurobio.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton GJ, Cummings DE, Baskin DG, Barsh GS, Schwartz MW. Central nervous system control of food intake and body weight. Nature. 2006;443:289–295. doi: 10.1038/nature05026. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. San Diego: Academic; 1997. [DOI] [PubMed] [Google Scholar]
- Proulx K, Cota D, Castañeda TR, Tschöp MH, D'Alessio DA, Tso P, Woods SC, Seeley RJ. Mechanisms of oleoylethanolamide-induced changes in feeding behavior and motor activity. Am J Physiol Regul Integr Comp Physiol. 2005;289:R729–R737. doi: 10.1152/ajpregu.00029.2005. [DOI] [PubMed] [Google Scholar]
- Rodríguez de Fonseca F, Navarro M, Gómez R, Escuredo L, Nava F, Fu J, Murillo-Rodríguez E, Giuffrida A, LoVerme J, Gaetani S, Kathuria S, Gall C, Piomelli D. An anorexic lipid mediator regulated by feeding. Nature. 2001;414:209–212. doi: 10.1038/35102582. [DOI] [PubMed] [Google Scholar]
- Schwartz GJ, Fu J, Astarita G, Li X, Gaetani S, Campolongo P, Cuomo V, Piomelli D. The lipid messenger OEA links dietary fat intake to satiety. Cell Metab. 2008;8:281–288. doi: 10.1016/j.cmet.2008.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz MW, Woods SC, Porte D, Jr, Seeley RJ, Baskin DG. Central nervous system control of food intake. Nature. 2000;404:661–671. doi: 10.1038/35007534. [DOI] [PubMed] [Google Scholar]
- Sclafani A, Rinaman L, Vollmer RR, Amico JA. Oxytocin knockout mice demonstrate enhanced intake of sweet and nonsweet carbohydrate solutions. Am J Physiol Regul Integr Comp Physiol. 2007;292:R1828–R1833. doi: 10.1152/ajpregu.00826.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaab DF, Purba JS, Hofman MA. Alterations in the hypothalamic paraventricular nucleus and its oxytocin neurons (putative satiety cells) in Prader-Willi syndrome: a study of five cases. J Clin Endocrinol Metab. 1995;80:573–579. doi: 10.1210/jcem.80.2.7852523. [DOI] [PubMed] [Google Scholar]
- Thompson KL, Vincent SH, Miller RR, Colletti AE, Alvaro RF, Wallace MA, Feeney WP, Chiu SH. Pharmacokinetics and disposition of the oxytocin receptor antagonist L-368,899 in rats and dogs. Drug Metab Dispos. 1997;25:1113–1118. [PubMed] [Google Scholar]
- Verbalis JG, McCann MJ, McHale CM, Stricker EM. Oxytocin secretion in response to cholecystokinin and food: differentiation of nausea from satiety. Science. 1986;232:1417–1419. doi: 10.1126/science.3715453. [DOI] [PubMed] [Google Scholar]
- Williams PD, Anderson PS, Ball RG, Bock MG, Carroll L, Chiu SH, Clineschmidt BV, Culberson JC, Erb JM, Evans BE, Fitzpatrick SL, Freidinger RM, Kaufman MJ, Lundell GF, Murphy JS, Pawluczyk JM, Perlow DS, Pettibone DJ, Pitzenberger SM, Thompson KL, Veber DF. 1-((7,7-Dimethyl-2(S)-(2(S)-amino-4-(methylsulfonyl)butyramido)bicyclo [2.2.1]-heptan-1(S)-yl)methyl)sulfonyl)-4-(2-methylphenyl)piperazine (L-368,899): an orally bioavailable, non-peptide oxytocin antagonist with potential utility for managing preterm labor. J Med Chem. 1994;37:565–571. doi: 10.1021/jm00031a004. [DOI] [PubMed] [Google Scholar]
- Yang Y, Chen M, Georgeson KE, Harmon CM. Mechanism of oleoylethanolamide on fatty acid uptake in small intestine after food intake and body weight reduction. Am J Physiol Regul Integr Comp Physiol. 2007;292:R235–R241. doi: 10.1152/ajpregu.00270.2006. [DOI] [PubMed] [Google Scholar]