

Abstract
The majority of human gastrointestinal stromal tumours (GIST) are driven by activating mutations in the proto-oncogene KIT, a tyrosine kinase receptor. Clinical treatment with imatinib targets the kinase domain of KIT, but tumour regrowth occurs as a result of the development of resistant mutations in the kinase active site. An alternative small-molecule approach to GIST therapy is described, in which the KIT gene is directly targeted, and thus kinase resistance may be circumvented. A naphthalene dimiide derivative has been used to demonstrate the concept of dual quadruplex targeting. This compound strongly stabilises both telomeric quadruplex DNA and quadruplex sites in the KIT promoter in vitro. It is shown here that the compound is a potent inducer of growth arrest in a patient-derived GIST cell line at a concentration (ca 1μM) that also results in effective inhibition of telomerase activity and almost complete suppression of KIT mRNA and KIT protein expression. Molecular modelling studies with a telomeric quadruplex have been used to rationalise aspects of the experimental quadruplex melting data.
Keywords: gastrointestinal cancer, drug, quadruplex, telomerase
Introduction
The proto-oncogene KIT encodes for a 145-160 kDa tyrosine kinase receptor (1), which regulates several key signal transduction cascades in order to control cell growth and proliferation (2). As the KIT protein plays a critical role in establishing normal cell growth, mutations in functionally-important regions, or its over-expression, result in aberrant function and can contribute to oncogenic cellular transformation in melanocyte, mast cell, myeloid, germ-cell, and interstitial cell of Cajal lineages. As one example, aberrant function can result from constitutive activation of the KIT protein, where mutations result in constant kinase activity, rather than activity only in response to the external stimulus of binding of its natural cytokine ligand, stem cell factor (2).
Such gain–of–function mutations are found in several highly malignant human cancers, including gastrointestinal stromal tumours (GIST). >80% of GIST tumour cells contain activating mutations in KIT extracellular, cytoplasmic juxtamembrane, or kinase domains (3-5). These oncogenic mutations result in increased dimer formation by bringing together two KIT kinase molecules into close proximity and facilitating autophosphorylation, and consequently disregulated cell growth. Current GIST therapy uses the drug imatinib to inhibit the phosphoactivation of the KIT enzyme (6, 7); use of imatinib is directly linked to tumour regression, but the onset of resistance occurs, sometimes after several years of therapy, in most patients (8, 9). Second-generation KIT kinase inhibitors have been more recently developed (10), notably the multi-kinase targeting compound sunitinib (11, 12); however the heterogeneity of patterns of mutations in the KIT protein and in resistance mechanisms suggests that the clinical challenges of resistance still remain to be overcome (13).
We describe here an alternative approach to inhibiting GIST cell growth by using a small molecule (the naphthalene diimide derivative 1: Figure 1a). We suggest that this targets two distinct categories of quadruplex DNA structures in the KIT gene itself: (i) induction of a quadruplex arrangement and displacement of bound telomeric proteins at the single-stranded 3′ ends of telomeric DNA results in inhibition of telomerase, disruption of telomere maintenance, and selective inhibition of cancer cell growth, and (ii) stabilization of guanine-rich sequences as quadruplex structures in the promoter region of the KIT gene, resulting in down-regulation of KIT expression. A wide range of small molecules have been developed to target telomeric quadruplexes (14, 15). The targeting of promoter quadruplexes with small molecules has been less studied to date, although several reports (16-19) have shown the feasibility of the approach in principle. The promoter region of the KIT gene contains sequences (20) with quadruplex features, and the dependence of GIST on abnormal KIT expression suggests that this cancer can be a paradigm for evaluating this approach. We have previously reported on a series of naphthalene diimide derivatives with potent telomeric quadruplex affinity and telomerase inhibitory activity (21) in standard laboratory cancer cell lines. A compound from this series (Figure 1a) is examined in depth in the present study, with the aim of ascertaining if there is activity in a clinically-relevant GIST cell line, and if so, whether this can be correlated with quadruplex binding and their subsequent molecular consequences.
Figure 1.

Structures of (a) compound 1, (b) BRACO-19 and (c) TMPyP4
Results
The KIT quadruplexes
We have previously identified two sequences (KIT1 and KIT2) in the KIT promoter upstream of the transcription start site that are capable of forming quadruplex structures (22, 23), and the 3-D structure of the KIT1 quadruplex has been determined in detail (24, 25). Both sequences are in nuclease hypersensitive regions. Since the stability of these particular quadruplexes is highly sensitive to mutations in the sequences involved, a 690 nucleotide region in the KIT promoter that contains these two sites, taken from the DNA of the GIST882 cell line, was sequenced. The sequence data in Table 1 shows that both sites are intact, with no mutations in or adjacent to them.
Table 1.
Part of the 690 nucleotide region immediately upstream of the transcription start site of the KIT gene sequenced in this study, together with part of exon 1 (in mauve), using DNA extracted from GIST882 cells. The sequence highlighted in cyan corresponds to the quadruplex sequence KIT1, and that in yellow to the quadruplex sequence KIT2
| KIT | GCCCCCCGGCATTAACACGTCGAAAGAGCAGGGGCCAGACGCCGCCGGGAAGAAGCGAGA | -107 |
| PCR_A_1 | GCCCCCCGGCATTAACACGTCGAAAGAGCAGGGGCCAGACGCCGCCGGGAAGAAGCGAGA | -107 |
| PCR_A_2 | GCCCCCCGGCATTAACACGTCGAAAGAGCAGGGGCCAGACGCCGCCGGGAAGAAGCGAGA | -107 |
| PCR_A_3 | GCCCCCCGGCATTAACACGTCGAAAGAGCAGGGGCCAGACGCCGCCGGGAAGAAGCGAGA | -107 |
| PCR_B_1 | GCCCCCCGGCATTAACACGTCGAAAGAGCAGGGGCCAGACGCCGCCGGGAAGAAGCGAGA | -107 |
| PCR_B_2 | GCCCCCCGGCATTAACACGTCGAAAGAGCAGGGGCCAGACGCCGCCGGGAAGAAGCGAGA | -107 |
| PCR_B_3 | GCCCCCCGGCATTAACACGTCGAAAGAGCAGGGGCCAGACGCCGCCGGGAAGAAGCGAGA | -107 |
| KIT | CC AGGCGAGGAGGGGCGTGGCCGGCGCGCAG AGGCGAGGAGGGGCGTGGCCGGCGCGCAG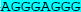
|
-48 |
| PCR_A_1 | CCAGGCGAGGAGGGGCGTGGCCGGCGCGCAG
|
-48 |
| PCR_A_2 | CCAGGCGAGGAGGGGCGTGGCCGGCGCGCAG
|
-48 |
| PCR_A_3 | CCAGGCGAGGAGGGGCGTGGCCGGCGCGCAG
|
-48 |
| PCR_B_1 | CCAGGCGAGGAGGGGCGTGGCCGGCGCGCAG
|
-48 |
| PCR_B_2 | CCAGGCGAGGAGGGGCGTGGCCGGCGCGCAG
|
-48 |
| PCR_B_3 | CCAGGCGAGGAGGGGCGTGGCCGGCGCGCAG
|
-48 |
| KIT |
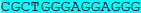 GCTGCTGCTCGCCGCTCGCGGCTCTGGGGGCTC GCTGCTGCTCGCCGCTCGCGGCTCTGGGGGCTC
|
13 |
| PCR_A_1 |
GCTGCTGCTCGCCGCTCGCGGCTCTGGGGGCTC
|
13 |
| PCR_A_2 |
GCTGCTGCTCGCCGCTCGCGGCTCTGGGGGCTC
|
13 |
| PCR_A_3 |
GCTGCTGCTCGCCGCTCGCGGCTCTGGGGGCTC
|
13 |
| PCR_B_1 |
GCTGCTGCTCGCCGCTCGCGGCTCTGGGGGCTC
|
13 |
| PCR_B_2 |
GCTGCTGCTCGCCGCTCGCGGCTCTGGGGGCTC
|
13 |
| PCR_B_3 |
GCTGCTGCTCGCCGCTCGCGGCTCTGGGGGCTC
|
13 |
The ability of compound 1 and two other established quadruplex-binding ligands ligands, the trisubstituted acridine experimental drug molecule BRACO-19 (26-31) and the N-methylpyridylylporphyrin compound TMPyP4 (32) (Figure 1a-c), to stabilise these quadruplexes as well as a human telomeric quadruplex and a duplex sequence, all as isolated sequences in vitro, was evaluated by Fluorescence Resonance Energy Transfer (FRET) methods (Figure 2a). The data in Table 2 shows that of the three compounds compound 1 has the greatest overall ability to stabilise the human telomeric and the KIT2 quadruplexes, with very similar ΔTm values for each, and a significantly greater ΔTm value for the KIT2 quadruplex compared to either BRACO-19 or TMPyP4. It is notable that all three compounds have significantly lower ΔTm values for the KIT1 quadruplex compared to KIT2. Control experiments with imatinib show no significant interactions with any of these DNAs. Selectivity for quadruplex vs duplex DNA was evaluated with a competition assay, using varying ratios of calf thymus DNA (Figure 2b). This suggests a similar pattern of selectivity for all three compounds. For all three the ΔTm values for the telomeric quadruplex are not significantly affected by the presence of calf thymus DNA at a low duplex:quadruplex ratio, but are diminished at higher ratios. The pattern of melting behaviour with the KIT2 quadruplex shows some differences at the lowest duplex:quadruplex ratio, with the ΔTm value for compound 1 unaffected compared to BRACO-19 and TMPyP4. The appearance of a second transition in the melting curve (Figure 2a) at low ligand concentrations is suggestive of possible folding reorganisation induced by ligand binding, although slow exchange cannot be ruled out in the absence of further studies.
Figure 2.

FRET melting curves. (a) shows the change in normalized fluorescent intensity with respect to temperature for the G4-Tel sequence 5′-FAM-d[G3(T2AG3)3]-TAMRA-3′ at a concentration of 200mM in potassium chloride/ cacodylate buffer. The curves at four different concentrations of compound 1 show in an increase in melting temperature (ΔTm) with respect to increasing ligand concentration. (b) shows the results of competition experiments for the G4-Tel, KIT1 and KIT2 quadruplexes and compound 1, in which increasing concentrations of calf thymus DNA are added to each melting experiment. The decrease in ΔTm for each quadruplex is a qualitative indication of relative selectivity at that quadruplex:duplex DNA ratio.
Table 2.
Thermal stabilization of various DNAs, expressed as ΔTm values in °, for ligand concentrations of 0.5μM, using a FRET method (21). Sequences are given in the Methods section. The average esd is ± 0.2°
| G4-tel | Duplex DNA | G4-KIT1 | G4-KIT2 | |
|---|---|---|---|---|
| Imatinib | 0.2 | 0.1 | 0.2 | 0.2 |
| Compound 1 | 28.7 | 5.7 | 11.2 | 29.0 |
| BRACO-19 | 21.6 | 7.2 | 6.5 | 18.5 |
| TMPyP4 | 27.8 | 8.7 | 11.1 | 21.6 |
Molecular modelling studies
Several distinct native telomeric quadruplex structures are now available (33), whose diversity demonstrates the polymorphism produced by closely-related human quadruplex sequences. We have not used any of these structures as starting-points for the modeling, since relevant crystal structures of quadruplex-ligand complexes are now available. The quadruplex binding of ligands 1 and BRACO-19 has been modeled by docking them in to an end G-quartet face of the co-crystal structures of BRACO-19 and a naphthalene diimide compound in complexes with human telomeric quadruplexes (34, 35). The low-energy docked positions for the planar chromophore of compound 1 are in the centre of a terminal G-quartet, with the charged side chains extending into the four grooves of the quadruplex (Figure 3a). The final docked and optimized position of the chromophore is closely similar to that observed in the crystal structure of the naphthalene diimide complex (34). The acridine moiety of the BRACO-19 molecule is asymmetrically stacked on one half of the terminal G-quartet (Figure 3b). In this arrangement the charged nitrogen atom of the chromophore is situated above the potassium channel at the centre of the quadruplex, in accord with the crystal structure (35). The BRACO-19 side-chains can only access three grooves, with the anilino group at the 9-position being extremely restricted in its orientation, similarly in accord with the crystal structure. The four side chains in compound 1, each with a +1 charge, presumably enables it to participate in more electrostatic interactions and thereby contributes to more effective binding than does BRACO-19.
Figure 3.
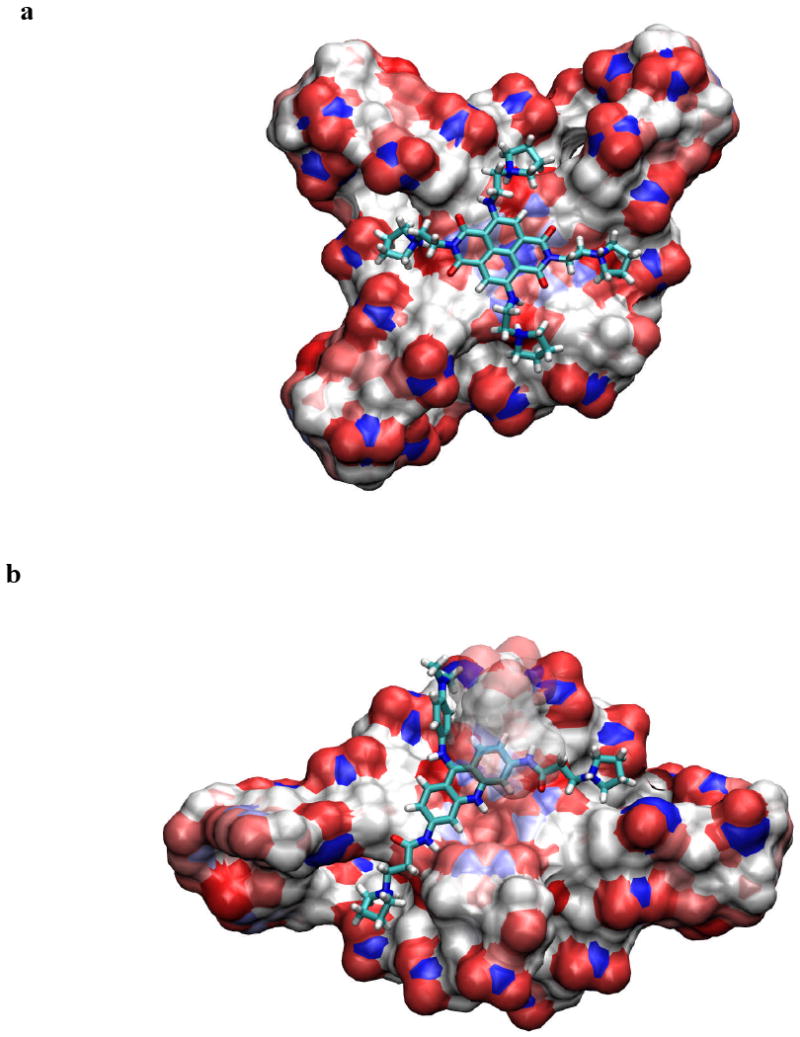
Results of the docking and simulated annealing/molecular dynamics calculations, showing in each case the low-energy position of the ligand (shown in stick representation) stacked onto a terminal G-quartet, shown in solvent-accessible surface representation mode (47). (a), (b) show compound 1 and BRACO-19 respectively bound to human telomeric quadruplexes. Structures were drawn using the VMD program (48).
A continuum model for both complexes has been computed that describes electrostatic interactions between each ligand and a quadruplex by numerically solving the Poisson-Boltzmann equation using the program APBS (36). This has enabled relative binding energies of the ligand-quadruplex complexes to be calculated (Table 3) and compared. These energies indicate that BRACO-19 binds with substantially less affinity to a human telomeric quadruplex compared to compound 1; this ranking is in accord with the order of ΔTm values (Table 2).
Table 3.
Computed free energies of binding for two ligand-quadruplex complexes
| System | Computed Energies (kcal/mol) | |||
|---|---|---|---|---|
| Complex | Quadruplex | Ligand | Binding | |
| G4-tel + BRACO-19 | 2949.2 | 2861.1 | 74.2 | 13.9 |
| G4-tel + compound 1 | 2578.1 | 2465.4 | 91.4 | 21.6 |
Short term growth inhibition
Table 4 shows that compound 1 has potent growth inhibitory activity against a range of cancer cell lines, representing human gastric (HGC-27) and colorectal carcinomas (HT-29) as well as the primary KIT-dependent gastrointestinal tumour cell line GIST882. It also shows high activity against the human breast carcinoma cell line MCF-7. The activity of compound 1 is closely comparable with that of imatinib in the GIST882 line, although the former is much more active in the laboratory lines. The established telomeric quadruplex agent BRACO-19 (26-31) on the other hand, while showing activity in the HGC-27, HT-29 and MCF-7 cell lines, has no activity against GIST882 up to at least a 25μM ligand concentration. The porphyrin molecule TMPyP4 shows a similar profile, although its activity against GIST882 cells is at least 2-fold greater than that of BRACO-19.
Table 4.
Short-term antiproliferative activity evaluated by the sulforhodamine B assay in several cancer cell lines, given as IC50 values in μM, for 96hr exposure. Values are means of experiments in triplicate, and esds are shown in parentheses
| GIST882 | HGC-27 | HT-29 | MCF7 | |
|---|---|---|---|---|
| Imatinib | 1.7(2) | 2.4(2) | 0.06(3) | 11.5(3) |
| Compound 1 | 1.6(2) | 0.04(3) | 0.03(3) | 0.02(3) |
| BRACO-19 | >25.0(3) | 2.6(2) | 2.7(2) | 2.2(2) |
| TMPyP4 | 13.2(3) | 2.7(2) | 4.5(2) | 6.1(3) |
The luminescence-based cell viability assay was performed on a larger panel of cell lines for compound 1, including several non-KIT lines (Figure 4). This confirmed the high level of potency of the compound in the GIST882 cell line, with yet higher potency in the GISTT1 line. In contrast there is little activity in the KIT-negative lines GIST48B and LMS04.
Figure 4.

Effects of varying concentrations (shown on a logarithmic scale) of compound 1 on the viability of a panel of KIT-related cancer cell lines, shown as normalized cell counts.
Effects on KIT transcription and expression in GIST cells
This study has focussed on the effects of the ligands on the primary GIST cell line GIST882. Figure 5a-c shows that KIT mRNA is reduced in a dose-dependent manner following 24 hr exposure to compound 1. At approximately the IC50 dose (1μM) KIT mRNA is reduced by ∼90% relative to a GAPDH control. No significant changes in KIT transcription levels were found with imatinib treatment (Figure 5d, e) or BRACO-19 and TMPyP4 (data not shown). The effects of compound 1 on the transcription of two other proto-oncogenes containing putative quadruplex sequences MYC (16, 17, 37) and N-RAS (38) as well as on hTERT (39), the catalytic domain of human telomerase, were also examined. A 25-30% change in MYC expression was observed, together with a smaller change in hTERT and no significant change in N-RAS expression (Figures 6a-h).
Figure 5.
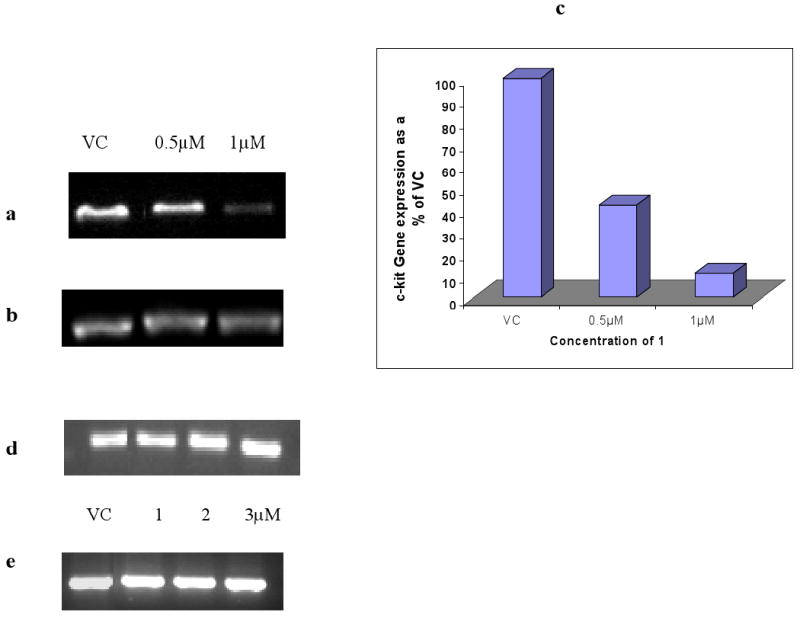
(a), (b) Changes in KIT expression in GIST882 cells following 24 hr treatment with compound 1. (c) shows the quantification of these changes in KIT expression normalised against untreated control with GAPDH expression.
(d), (e) Changes in KIT and GAPDH expression in GIST882 cells following 24hr treatment with imatinib.
Figure 6.
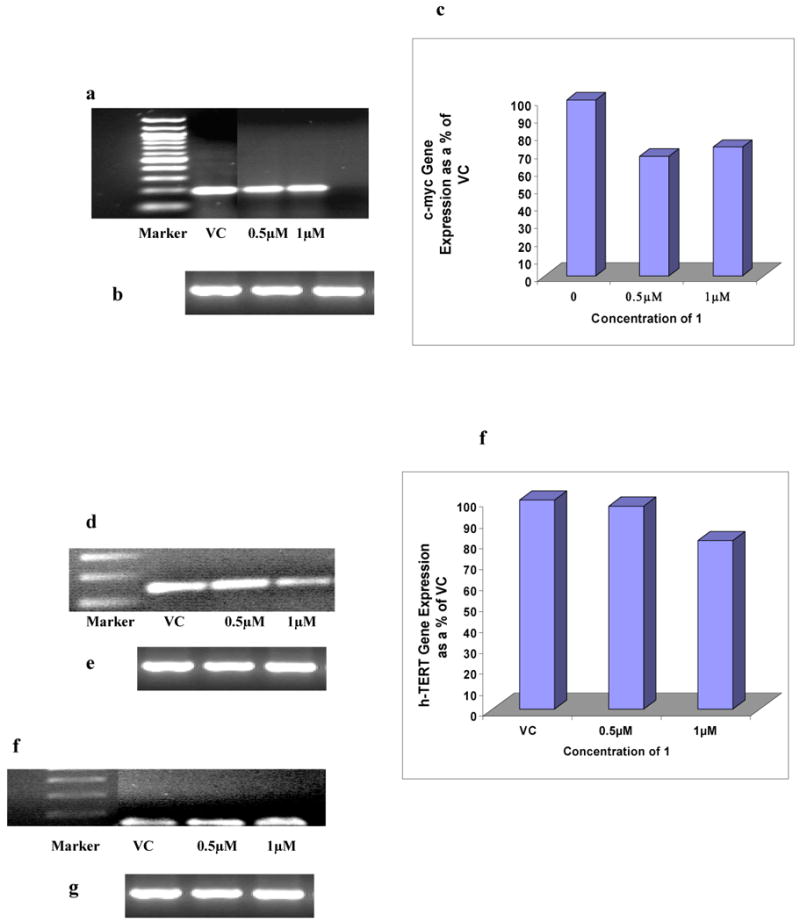
Changes in MYC expression in GIST882 cells following 24 hr treatment with compound 1. (a) and (b) represent MYC and GAPDH expression respectively. (c) shows the quantitation of changes in MYC expression normalised against untreated control GAPDH expression.
(d) and (e) Changes in hTERT expression in GIST882 cells following 24 hr treatment with compound 1, showing hTERT and GAPDH expression levels respectively. (f) shows the change in hTERT gene expression normalised against untreated control GAPDH expression.
The changes in N-RAS expression in GIST882 cells following 24 hr treatment with compound 1 are shown in gels (g) N-RAS and (h) GAPDH expression respectively.
Immunoblots of KIT protein (Figure 7a, b) show that 24hr exposure of GIST882 cells to a 1 μM concentration of compound 1 results in complete inhibition of KIT protein expression. By contrast KIT protein expression levels are unimpaired by imatinib treatment (Figure 7c, d), or by BRACO-19 or TMPyP4 (data not shown).
Figure 7.
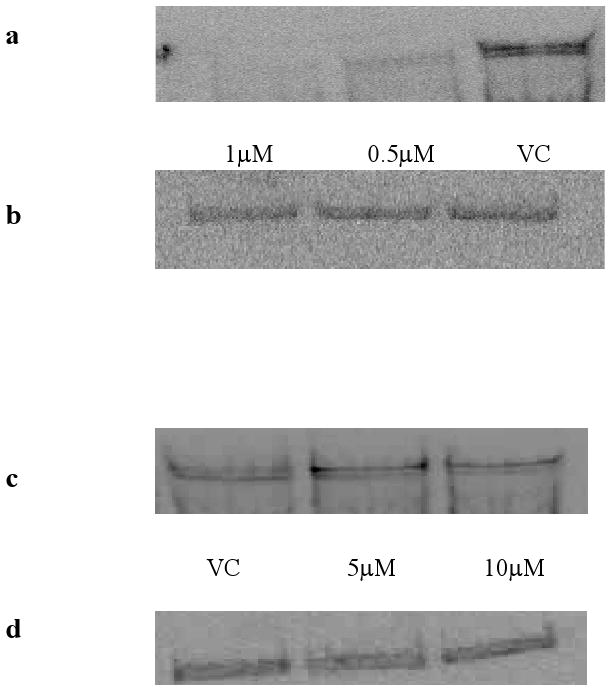
Immunoblotting of total KIT protein in GIST882 cells. Panel (a) shows total KIT after treatment with compound 1 for 24 hrs and panel (b) represents an actin control blot. Panel (c) shows total KIT protein in GIST882 cells treated with Imatinib for 24hrs, and panel (d) shows an actin control.
Effects on telomerase activity
Telomerase activity was assessed following exposure of GIST882 cells to compound 1 at a concentration of 0.5 μM (ie significantly below the IC50 value). Data obtained using the modified TRAP-LIG assay (28) shows that telomerase activity was absent after this exposure (Figure 8a), indicating potent telomerase inhibition. A subsequent series of long-term cell proliferation studies were performed for three weeks exposure at several sub-toxic concentrations. At the lowest, 0.1 μM, some modest effects on proliferation were observed after two weeks, when growth started to decrease relative to the control arm of the experiment (Figure 8b). At slightly higher concentrations (0.25, 0.5 μM), growth was more rapidly and profoundly affected, and cell growth and proliferation was completely arrested after week one of exposure.
Figure 8.
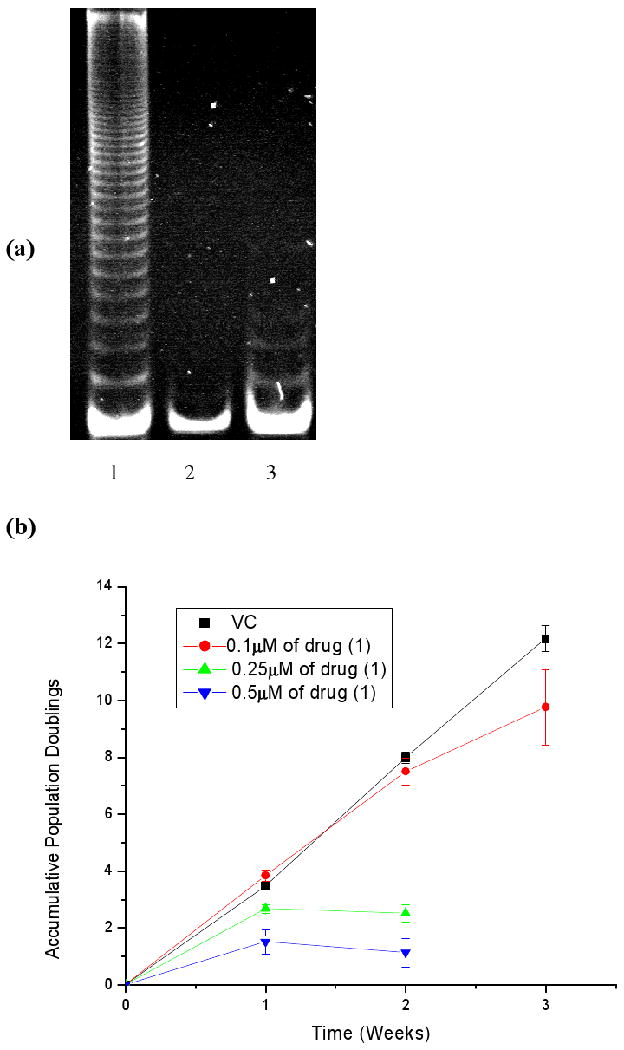
(a) Polyacrylamide gel showing telomerase activity in GIST882 cells treated with compound 1 for one week. Lane 1; Vehicle Control, Lane 2; 0.5μM of compound 1, Lane 3; Negative control. (b) Long term accumulative population doublings of GIST 882 cells treated with increasing concentrations of compound 1.
Discussion
We have shown here that the naphthalene diimide derivative 1 has a high level of potency against primary tumour GIST cell lines, whose viability has been established to be KIT-dependent (6). Moreover we have shown that this molecule produces 90% down-regulation of KIT transcription and complete inhibition of KIT protein expression at the same dosage levels as are required for growth inhibition in one of these lines. This is strongly suggestive of the compound acting at the KIT gene level; although at this stage it has not been unequivocally demonstrated in cells that compound 1 stabilises one or both of the two KIT promoter quadruplex sequences, it is our current working hypothesis that this is a likely significant contributor to its cellular effects. The hypothesis is also supported by the finding (Figure 4) that compound 1 is significantly less potent in other KIT-independent GIST lines We have also found that compound 1 has selectivity for down-regulating KIT expression and does not have a comparable effect on several other cancer-associated genes for which promoter quadruplexes have been postulated, although a more definitive conclusion must await a genome-wide study of the effects of compound 1. A small effect on MYC expression is apparent, which may be related to the amplification of this gene in GIST (3), as well as a small effect on hTERT expression, which may be related to the presence of an extended set of G-tracts in the upstream promoter region of this gene.
Surface plasmon resonance studies have previously shown that the naphthalene diimides display exceptionally strong binding to human telomeric quadruplexes (21), with Ka values of > 107 M-1. The binding constants of BRACO-19 to a human telomeric quadruplex and a DNA duplex have been estimated, again by surface plasmon resonance, to be 3.1 × 107 and 5 × 105 M-1 respectively (30b). By contrast, TMPyP4, as judged by competition dialysis experiments (32b), has only modest selectivity for quadruplex over duplex DNA, contrary to data from the melting experiments (Table 2 and Figure 2), Thus, measures of thermal stabilisation should only be taken as indicative measures of ligand selectivity and affinity for quadruplex vs duplex DNAs. The molecular modelling studies concur with the trends found in the melting studies for telomeric quadruplexes. We have not undertaken any modelling for the KIT1 quadruplex drug complexes since even though a structure is available for this native quadruplex (24), no structural information is available on any ligand complexes. The molecular structure of the KIT2 quadruplex has not as yet been disclosed.
Compound 1 is the most effective of the three ligands reported here, in stabilising the two KIT quadruplexes (Table 2), although the stabilization is relatively slight for the KIT1 quadruplex compared to the KIT2 one. The relatively low ability of BRACO-19 to stabilise these quadruplexes appears to be reflected in the low potency of this compound in the GIST882 cell line (Table 4) and inability to affect KIT expression, and suggests that there is a threshold level of quadruplex stabilisation required for a ligand to show these effects. Uptake effects cannot be discounted though, and compound 1 has been shown to rapidly taken up into cancer cells in culture (21). This factor combined with its high quadruplex affinity suggests that there would be sufficient of compound 1 available to saturate the very small number of telomeric and KIT quadruplex sites even though very many more duplex sites would be available. Future analogues with enhanced quadruplex selectivity are clearly desirable and are the subject of current studies in our laboratories.
Several GIST cell lines have been previously characterised as telomerase-positive (see for example, ref. 40). We have recently shown that compound 1 is an effective inhibitor of telomerase in a small panel of other cancer cell lines (21). We report here that the GIST882 line strongly expresses catalytically-active telomerase, and find that compound 1 produces profound growth arrest after several days of treatment at sub-toxic concentrations (Figure 8b), characteristic of quadruplex-mediated inhibition of telomerase (27, 41-42). This compound is the most potent telomerase inhibitor in the library of naphthalene diimides described previously (21), which may explain in part why it is active in patient-derived GIST882 cells. In addition, preliminary studies (Figure 4) have demonstrated similar potency effects in GIST48 cells, which were derived from a patient with GIST progressing on imatinib therapy. GIST48 contains kinase-domain imatinib resistance mutations, and is resistant to imatinib in vitro (IC50 of 18.7 μM) whereas the activity of compound 1 remains high (IC50 of 0.5 μM). We suggest that compound 1 and analogues may therefore be useful in the treatment of human GIST (and perhaps other cancers such as leiomyosarcoma and mesothelioma) which is clinically resistant to imatinib or other KIT-kinase inhibitor drugs.
The data presented here is consistent with the hypothesis that compound 1 has dual action both on telomerase activity and expression of the KIT gene, which plays a key role in the growth of GIST cell lines, and that the compound is targeting and stabilising one or both quadruplexes in the KIT promoter region, although further studies, beyond the scope of the present paper, will be required to prove that this is the case. This dual action is independent of and unaffected by mutational changes in the KIT protein that are consequential to inhibitors acting on the nucleotide-binding domain - we show here that the GIST882 cell line does not contain any mutations in either KIT quadruplex sequence, and the same has been found for the GIST48 imatinib-resistant line (data not shown). The availability of co-crystal structures of a naphthalene diimide compound (35) with human telomeric quadruplexes may be useful in the rational design of further analogues with improved KIT quadruplex specificity, as well as optimized inhibitory and pharmacological properties. These may find use in the therapy of human GIST that is resistant to conventional kinase inhibitors.
Experimental
Cell Lines
GIST882 and GISTT1 are human cell lines established from imatinib-sensitive GISTs. GIST882 contains a homozygous K642E KIT exon 13 mutation, and GISTT1 has a heterozygous 57-base pair deletion in KIT exon 11 (43, 44). GIST48 and GIST430 are human cell lines established from progressing imatinib-resistant GISTs. GIST48 contains a homozygous V560D KIT exon 11 primary mutation and a heterozygous D820A KIT exon 17 secondary mutation. GIST430 has a heterozygous KIT exon 11 in-frame deletion primary mutation and a heterozygous V654A KIT exon 13 secondary mutation. GIST48B is a stable sub-cell line developed from the GIST48 parental cell line. GIST48B was established after 17-AAG drug pressure which resulted in nearly undetectable KIT transcript and protein. EWS502 is a human cell line established from a patient with Ewing's sarcoma. EWS502 contains wild-type KIT which is over-expressed but not activated. LMS04 and MESO924 are human cell lines established from patients with leiomyosarcoma and mesothelioma, respectively, and contain wild-type KIT. GIST882, GISTT1, EWS502, LMS04 and MESO428 were maintained in RPMI 1640 supplemented with 15% fetal bovine serum. GIST430, GIST48 and GIST48B were maintained in F10 supplemented with 15% fetal bovine serum, mito + serum extender and bovine pituitary extract. HGC-27, HT-29 and MCF-7 cells were purchased from ATCC. MCF-7 and HT-29 cells were both maintained in DMEM. HGC-27 was maintained in MEM. Both DMEM and MEM were supplemented with 10% FBS.
Small molecule inhibitors
Imatinib mesylate (Glivec®) was purchased from ACC Corporation. A 10mM stock solution was prepared in 100% DMSO and kept at -20°C in 10μl aliquots. Stock solution of compound 1 as free base were prepared in 100% DMSO and kept at -20°C. Two established quadruplex-binding ligands, the acridine BRACO-19 (synthesised in-house) and the porphyrin TMPyP4 (purchased from Calbiochem Inc), were also used in this study. A 1mM working solution of each compound was freshly prepared in filter-sterilised distilled water with 1% HCl, on the day of an experiment.
DNA melting studies
FRET DNA melting assays were performed as described previously (22), using 96-well plates and an Opticon DNA Engine instrument (MJ Research). Oligonucleotides (Eurogentec Ltd., U.K.) used were:
G4-Tel: 5′-FAM-d[G3(T2AG3)3]-TAMRA-3′
G4-KIT1: 5′-FAM-d[AGAG3AG2GCGCTG3AG2AG3GCT]-TAMRA-3′
G4-KIT2: 5′-FAM-d[C3G3CG3CGCGAG3AG4AG2]-TAMRA-3′
Duplex DNA: 5′-FAM-d[(TA)2GC(TA)2T6(TA)2GC(TA)2]-TAMRA-3′
where FAM is 6-carboxyfluorescein and TAMRA 6-carboxytetramethylrhodamine.
FRET-labelled oligonucleotides were annealed as 400 nM solutions in 60mM potassium chloride/ cacodylate buffer (50 mM potassium chloride + 10 mM potassium hydroxide adjusted to pH 7.4 with cacodylic acid) by heating to 90°C and allowing to cool slowly to ambient temperature. Into each well of a 96 well plate was transferred 50 μl of the above solution and, in the case of control wells, 50 μl of buffer added to give a final oligonucleotide concentration of 200 nM concentration. For wells in which the Tm shifts were to be determined, 25 μl of 2 mM ligand stock in buffer were added together with 25 μl of buffer to give a final ligand concentration of 500 nM. In the case of wells where competition binding was to be measured the 25 μl of buffer was substituted with 25 μl of calf thymus DNA stock solutions in buffer at base pair concentrations of 2.4 μM, 24 μM, 240 μM, or 480 μM, leading to the final concentrations shown in the legend to Figure 2b. The 96-well plates were measured with a DNA engine Opticon 1 (MJ Research) with excitation at 450–495 nm and detection at 515–545 nm. Fluorescence readings were taken at intervals of 0.5°C over the range 30–100°C, a ramp rate of 1°C min-1 being used. Final analysis of the data was carried out using a script written in the program Origin 7.0 (Origin Lab Corp., Northampton, MA). Tm values were determined as the maximum of the derivative of the fluorescence vs. temperature plots except for the oligonucleotide KIT1 where the plots were fitted to a sigmoid curve.
Molecular modeling studies
The coordinates from the crystal structures of human telomeric quadruplexes complexed with a naphthalene diimide compound similar to ND1 (PDB id 3CDM) and with BRACO-19 (PDB id 3CE5) were obtained from the Protein Data Bank (PDB). The ligands were built using the Builder module in the Insight II suite of programs (www.accelrys.com). The partial charges for the ligands were derived by fitting the HF/6-31G* electrostatic potential, calculated with the GAMESS ab initio software package (45).
Docking was performed with the Affinity Docking module of the Insight II package. The ligand binding sites were based on the relative positions of the ligands bound in the crystal structures. This included the terminal G-quartet in the 3CDM structure. The side chains of the closely-related naphthalene diimide compound in the 3CDM crystal structure were modified to become compound 1 and were minimized and docked into the structure, while keeping the position of the central chromophore fixed. This protocol enables the positioning of the ligand to be close to that observed in the crystal structure.
The next phase of modeling involved simulated annealing. The van der Waals radii were adjusted to their full values, charges included with a distance-dependent dielectric of 4rij and the non-bonded cutoff set to 12 Å. The system was again minimized for 300 steps of conjugate gradient followed by molecular dynamics simulations. The initial starting temperature was set to 500 K and gradually cooled down to 300 K over 10ps. The resulting structures were again subjected to 500 steps of conjugate gradient minimization. The final docked model in each case was chosen on the basis of two criteria: total energy of the system and the total number of hydrogen bonds that each ligand made with the quadruplex DNA. The positively-charged ions were retained in their crystallographic positions or were assigned between the quartets along the central axis of the structures and were assumed to be integral parts of the systems.
Electrostatics contributions to the overall energy for the implicit models were calculated using the APBS electrostatics package (36). The online PDB2PQR server (http://agave.wustl.edu/pdb2pqr/) was used to prepare the docked complexes for electrostatic calculations by optimizing hydrogen-bonding networks (46). Charges, radii and atom types were assigned to each ligand atoms in accordance with the AMBER forcefield. The systems were then subjected to APBS electrostatic calculations. A dielectric constant of 2, a solvent dielectric of 80 and grid spacing were all optimally chosen such that the grid was always finer than 0.5 Å. The remaining parameters were kept at default values. The binding free energies were calculated based on the simple thermodynamics cycle:
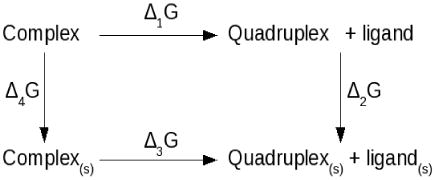
where [complex, quadruplex, ligand] and [complex(s), quadruplex(s), ligand(s)] represent the non-solvated and solvated states of the system respectively. The program APBS calculates binding free energy using the equation:
This binding free-energy cycle illustrates binding in terms of the transfer of free energies from a homogeneous dielectric environment to an inhomogeneous dielectric environment with differing internal and external dielectric constants (36). The contributors to the binding energies are detailed in Table 3.
Gene expression using RT-PCR
Changes in the expression of KIT, MYC, hTERT, N-RAS and GAPDH genes were measured following exposure to imatinib and compound 1 at sub-cytotoxic concentrations for 24hr in GIST882 cells. Gene expression changes were monitored using the One-step Superscript RT-PCR system (Invitrogen). 1×106 cells were seeded in T25cm2 flasks with appropriate concentrations of drug and incubated as above. At the end of 24hr cells were washed with 1XPBS, trypsinised and centrifuged at 8000g for 3mins. Total RNA was extracted from the pellets using the Absolutely RNA® Miniprep Kit from Stratagene. 250ng of RNA was used to amplify expression of these genes. PCR products were resolved in 2% Agarose gel stained with ethidium bromide. The intensity of each PCR product was quantified using a gel scanner and gene tool software (Sygene, Cambridge, UK) and was normalized for each gene against appropriate GAPDH expression. Primers used for the amplification of each gene are given below.
| Gene | Primer sequences |
| KIT | Forward: TGA CTT ACG ACA GGC TCG TG |
| Reverse: AAG GAG TGA ACA GGG TGT GG | |
| MYC | Forward: TGG TGC TCC ATG AGG AGA CA |
| Reverse: GTG GCA CCT CTT GAG GAC CT | |
| h-TERT | Forward: CGG AAG AGT GTC TGG AGC AA |
| Reverse: GGA TGA AGC GGA GTC TGG A | |
| N-RAS | Forward: CAG GTT CTT GCT GGT GTG AA |
| Reverse: CTC TAT GGT GGG ATC ATA TT | |
| GAPDH | Forward: GGC AGT GAT GGC ATG GAC TG |
| Reverse: CGG GAA GCT TGT GAT CAA TGG |
Western blotting analysis of total KIT expression
To assess changes in KIT protein expression protein lysates were prepared from GIST882 cells treated with imatinib at 5 and 10μM concentrations and compound 1 at 0.5 and 1 μM concentrations for 24hrs. Protein concentrations were determined by the Bradford assay followed by electrophoresis and immunoblotting.
Determination of cellular telomerase activity
Inhibition of telomerase activity following long-term exposure to compound 1 was measured using the TRAP–LIG modified telomere repeat amplification protocol assay (28), which is designed to ensure that ligand is not carried over into the second PCR step of the assay. Protein (1000 ng) from GIST882 cells treated for one week with compound 1 was incubated with master mix containing the TS forward primer (0.1μg 5′-AAT CCG TCG AGC AGA GTT-3′), TRAP buffer (20mM Tris-HCl [pH 8.3], 68mM KCl, 1.5mM MgCl2, 1mM EGTA, 0.05% v/v Tween-20), bovine serum albumin (0.05μg), and dNTPs (125μM each), protein extract (500ng/sample) diluted in lysis buffer (10 mM Tris-HCl, pH 7.5, 1 mM MgCl2, 1 mM EGTA, 0.5% CHAPS, 10% glycerol, 5mM β-mercaptoethanol, 0.1mM AEBSF). Telomerase elongation step was carried out for 10 min at 30°C, followed by 94°C for 5 min and a final maintenance of the mixture at 20°C. Elongated products were purified using QIA quick nucleotide purification kit (Qiagen) according to the manufacturer's instructions. The purified samples were freeze-dried and then re-dissolved in PCR-grade water at room temperature prior to the amplification step. Purified telomerase extended samples were then subject to PCR amplification. For this, a second PCR master mix was prepared consisting of ACX reverse primer (1μM; 5′-GCG CGG [CTTACC]3 CTA ACC-3′), TS forward primer (0.1μg; 5′-AAT CCG TCG AGC AGA GTT-3′), TRAP buffer, BSA (5μg), 0.5mM dNTPs and 2U of TAQ polymerase (RedHot, ABgene, Surrey, UK). An aliquot of 10μl of the master mix was added to the purified telomerase extended samples and amplified for 35 cycles of 94°C for 30 sec, at 61°C for 1 min and 72°C for 1 min. Samples were separated on a 12% PAGE and visualised with SYBR green (Aldrich) staining. Gels were quantified using a gel scanner and gene tool software (Sygene, Cambridge, UK). Intensity data were obtained by scanning and integrating the total intensity of each PCR product ladder in the denaturing gels. Drug treated samples were normalised against positive control containing untreated protein only. All samples were corrected for background by subtracting the fluorescence reading of the negative control.
Sequencing the c-kit promoter region
A 943 base pair long KIT gene fragment was amplified by two PCR reactions using genomic DNA isolated from GIST882 cells. This comprises 690 nucleotides of the KIT promoter region, exon 1 and the initial 100 nucleotides of intron 1. PCR was carried out in a final volume of 50 μl containing 50 ng of genomic DNA, 50 pmol of each primer (5′-NNNGAATTCACTTCGCCACGCGCC-3′ and 5′-NNNNAAGCTTCGCAGTCCTCTCTCCGGATG -3′), 200 μM of each dNTP, 2.5 U Herculase polymerase (Stratagene) and 10× PCR buffer. After heating for 5 minutes at 94°C, the reaction was incubated for 30 cycles with denaturation (90 s, 94°C) annealing (60 s, 55°C) and elongation (120 s, 72°C) steps followed by 10 min extension at 72°C. The correct-size fragment was separated on agarose gel and ligated into pUC19. DNA sequences of the PCR fragment contained in the vector were determined by Eurofins MWG Operon. Three clones were sequenced from each strand by two independent amplification reactions (PCR_A, PCR_B) and aligned with the sequence taken from the Ensembl website (http://www.ensembl.org).
In Vitro Assays
Short-term antiproliferative activity in several cancer cell lines was evaluated by the sulforhodamine B assay, and calculated as IC50 values in μM, for 96 hr exposure (23, 30). Viability studies were also conducted using the CellTiter-Glo luminescent assay (Promega, Madison, WI, USA) which utilizes a luciferase-catalyzed luciferin/ATP reaction to provide a qualitative measure of metabolically active cells. Cell lines were plated at 10,000 to 30,000 cells per well in a 96-well flat bottom plate (Falcon, Lincoln, NJ, USA), cultured in serum-containing media overnight, and treated with compound 1 for 3 to 6 days, including water-solvent control. The luminescence signal was quantitated using a Veritas Microplate Luminometer (Turner Biosystems, Sunnyvale, CA, USA) and normalized to the control. All experimental points were measured in triplicate.
Acknowledgments
Grant support: Cancer Research UK Programme Grant and European Union FP6 Molecular Cancer Medicine Grant (to S.N.) and by an anonymous donor, GI SPORE 1P50CA127003-02, the Life Raft Group, the Daniel K. Ludwig Trust for Cancer Research, and the Stutman GIST Cancer Research Fund (to J.A.F.).
Abbreviations
- GIST
gastrointestinal stromal tumours
- FRET
Fluorescence Resonance Energy Transfer
References
- 1.Yarden Y, Kuang WJ, Yang-Feng T, Coussens L, Munemitsu S, Dull TJ, Chen E, Schlessinger J, Francke U, Ullrich A. Human proto-oncogene C-Kit - A new cell-surface receptor tyrosine kinase for an unidentified ligand. EMBO J. 1987;6:3341–3351. doi: 10.1002/j.1460-2075.1987.tb02655.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roskoski R., Jr Structure and regulation of Kit protein-tyrosine kinase - the stem cell factor receptor. Biochem Biophys Res Commun. 2005;37:307–1315. doi: 10.1016/j.bbrc.2005.09.150. [DOI] [PubMed] [Google Scholar]
- 3.Yang J, Du X, Lazar AJ, Pollock R, Hunt K, Chen K, Hao X, Trent J, Zhang W. Genetic aberrations of gastrointestinal stromal tumors. Cancer. 2008;113:1532–1543. doi: 10.1002/cncr.23778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fletcher JA, Rubin BP. KIT mutations in GIST. Curr Opin Genet Dev. 2007;7:3–7. doi: 10.1016/j.gde.2006.12.010. [DOI] [PubMed] [Google Scholar]
- 5.Hoeben A, Schoffski P, Debiec-Rychter M. Clinical implications of mutational analysis in gastrointestinal stromal tumours. Brit J Cancer. 2008;98:684–688. doi: 10.1038/sj.bjc.6604217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tuveson DA, Willis NA, Jacks T, Griffin JD, Singer S, Fletcher CD, Fletcher JA, Demetri GD. STI571 inactivation of the gastrointestinal stromal tumor c-KIT oncoprotein: biological and clinical implications. Oncogene. 2001;20:5054–5058. doi: 10.1038/sj.onc.1204704. [DOI] [PubMed] [Google Scholar]
- 7.Zhu MJ, Ou WB, Fletcher CD, Cohen PS, Demetri GD, Fletcher JA. KIT oncoprotein interactions in gastrointestinal stromal tumors: therapeutic relevance. Oncogene. 2007;26:6386–6395. doi: 10.1038/sj.onc.1210464. [DOI] [PubMed] [Google Scholar]
- 8.Heinrich MC, Corless CL, Demetri GD, Blanke CD, von Mehren M, Joensuu H, McGreevey LS, Chen CJ, Van den Abbeele AD, Druker BJ, Kiese B, Eisenberg B, Roberts PJ, Singer S, Fletcher CD, Silberman S, Dimitrijevic S, Fletcher JA. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol. 2003;21:4342–4349. doi: 10.1200/JCO.2003.04.190. [DOI] [PubMed] [Google Scholar]
- 9.Heinrich MC, Corless CL, Blanke CD, Demetri GD, Joensuu H, Roberts PJ, Eisenberg BL, von Mehren M, Fletcher CD, Sandau K, McDougall K, Ou WB, Chen CJ, Fletcher JA. Molecular correlates of imatinib resistance in gastrointestinal stromal tumors. J Clin Oncol. 2006;24:4764–4774. doi: 10.1200/JCO.2006.06.2265. [DOI] [PubMed] [Google Scholar]
- 10.von Mehren M. Beyond imatinib: second generation c-KIT inhibitors for the management of gastrointestinal stromal tumors. Clin Colorectal Cancer. 2006;6:S30–34. doi: 10.3816/ccc.2006.s.005. [DOI] [PubMed] [Google Scholar]
- 11.Abrams TJ, Lee LB, Murray LJ, Pryer NK, Cherringham JM. SU11248 inhibits KIT and platelet-derived growth factor receptor beta in preclinical models of human small cell lung cancer. Mol Cancer Ther. 2003;2:471–478. [PubMed] [Google Scholar]
- 12.Le Tourneau C, Raymond E, Faivre S. Sunitinib: a novel tyrosine kinase inhibitor. A brief review of its therapeutic potential in the treatment of renal carcinoma and gastrointestinal stromal tumors (GIST) Ther Clin Risk Manag. 2007;3:341–348. doi: 10.2147/tcrm.2007.3.2.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liegl B, Kepten I, Le C, Zhu M, Demetri GD, Heinrich MC, Fletcher CD, Corless CL, Fletcher JA. Heterogeneity of kinase inhibitor resistance mechanisms in GIST. J Pathol. 2008;216:64–74. doi: 10.1002/path. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Monchaud D, Teulade-Fichou MP. A hitchhiker's guide to G-quadruplex ligands. Org Biomol Chem. 2008;6:627–636. doi: 10.1039/b714772b. [DOI] [PubMed] [Google Scholar]
- 15.Tan JH, Gu LQ, Wu JY. Design of selective G-quadruplex ligands as potential anticancer agents. Mini Rev Med Chem. 2008;8:1163–1178. doi: 10.2174/138955708785909880. [DOI] [PubMed] [Google Scholar]
- 16.Siddiqui-Jain A, Grand CL, Bearss DJ, Hurley LH. Direct evidence for a G-quadruplex in a promoter region and its targeting with a small molecule to repress c-MYC transcription. Proc Natl Acad Sci USA. 2002;99:11593–11598. doi: 10.1073/pnas.182256799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hurley LH, Von Hoff DD, Siddiqui-Jain A, Yang D. Drug targeting of the c-MYC promoter to repress gene expression via a G-quadruplex silencer element. Seminars Oncol. 2006;33:498–512. doi: 10.1053/j.seminoncol.2006.04.012. [DOI] [PubMed] [Google Scholar]
- 18.Liu JN, Deng R, Guo JF, Zhou JM, Feng GK, Huang ZS, Gu LQ, Zeng YX, Zhu XF. Inhibition of myc promoter and telomerase activity and induction of delayed apoptosis by SYUIQ-5, a novel G-quadruplex interactive agent in leukemia cells. Leukemia. 2007;21:1300–1302. doi: 10.1038/sj.leu.2404652. [DOI] [PubMed] [Google Scholar]
- 19.Bejugam M, Sewitz S, Shirude PS, Rodriguez R, Shahid R, Balasubramanian S. Trisubstituted isoalloxazines as a new class of G-quadruplex binding ligands: small molecule regulation of c-kit oncogene expression. J Am Chem Soc. 2007;129:12926–12927. doi: 10.1021/ja075881p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yamamoto K, Tojo A, Aoki N, Shibuya M. Characterization of the promoter region of the human c-kit proto-oncogene. Jpn J Cancer Res. 1993;84:1136–1144. doi: 10.1111/j.1349-7006.1993.tb02813.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cuenca F, Greciano O, Gunaratnam M, Haider S, Munnar D, Nanjunda R, Wilson WD, Neidle S. Tri- and tetra-substituted naphthalene diimides as potent G-quadruplex ligands. Bioorg Med Chem Lett. 2008;18:1668–1673. doi: 10.1016/j.bmcl.2008.01.050. [DOI] [PubMed] [Google Scholar]
- 22.Rankin S, Reszka AP, Huppert J, Zloh M, Parkinson GN, Todd AK, Ladame S, Balasubramanian S, Neidle S. Putative DNA quadruplex formation within the human c-kit oncogene. J Am Chem Soc. 2005;127:10584–10589. doi: 10.1021/ja050823u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fernando H, Reszka AP, Huppert J, Ladame S, Rankin S, Venkitaraman AR, Neidle S, Balasubramanian S. A conserved quadruplex motif located in a transcription activation site of the human c-kit oncogene. Biochemistry. 2006;45:7854–7860. doi: 10.1021/bi0601510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Phan AT, Kuryavyi V, Burge S, Neidle S, Patel DJ. Structure of an unprecedented G-quadruplex scaffold in the human c-kit promoter. J Am Chem Soc. 2007;129:4386–4392. doi: 10.1021/ja068739h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Todd AK, Haider SM, Parkinson GN, Neidle S. Sequence occurrence and structural uniqueness of a G-quadruplex in the human c-kit promoter. Nucleic Acids Res. 2007;35:5799–808. doi: 10.1093/nar/gkm609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schultes CM, Guyen B, Cuesta J, Neidle S. Synthesis, biophysical and biological evaluation of 3,6-bis-amidoacridines with extended 9-anilino substituents as potent G-quadruplex-binding telomerase inhibitors. Bioorg Med Chem Lett. 2004;14:4347–4351. doi: 10.1016/j.bmcl.2004.05.090. [DOI] [PubMed] [Google Scholar]
- 27.Gunaratnam M, Greciano O, Martins C, Reszka AP, Schultes CM, Morjani H, Riou JF, Neidle S. Mechanism of acridine-based telomerase inhibition and telomere shortening. Biochem Pharmacol. 2007;74:679–689. doi: 10.1016/j.bcp.2007.06.011. [DOI] [PubMed] [Google Scholar]
- 28.Reed JE, Gunaratnam M, Beltran M, Reszka AP, Vilar R, Neidle S. TRAP-LIG, a modified TRAP assay to quantitate telomerase inhibition by small molecules. Anal Biochem. 2008;380:99–105. doi: 10.1016/j.ab.2008.05.013. [DOI] [PubMed] [Google Scholar]
- 29.Read M, Harrison RJ, Romagnoli B, Tanious FA, Gowan SM, Reszka AP, Wilson WD, Kelland LR, Neidle S. Structure-based design of selective and potent G quadruplex-mediated telomerase inhibitors. Proc Natl Acad Sci USA. 2001;98:4844–4849. doi: 10.1073/pnas.081560598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.(a) Harrison RJ, Cuesta J, Chessari G, Read MA, Basra SK, Reszka AP, Morrell J, Gowan SM, Incles CM, Tanious FA, Wilson WD, Kelland LR, Neidle S. Trisubstituted acridine derivatives as potent and selective telomerase inhibitors. J Med Chem. 2003;46:4463–4476. doi: 10.1021/jm0308693. [DOI] [PubMed] [Google Scholar]; (b) White EW, Tanious F, Ismail MA, Reszka AP, Neidle S, Boykin DW, Wilson WD. Structure-specific recognition of quadruplex DNA by organic cations: influence of shape, substituents and charge. Biophys Chem. 2007;126:140–153. doi: 10.1016/j.bpc.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 31.Burger AM, Dai F, Schultes CM, Reszka AP, Moore MJ, Double JA, Neidle S. The G-quadruplex-interactive molecule BRACO-19 inhibits tumor growth, consistent with telomere targeting and interference with telomerase function. Cancer Res. 2005;65:1489–1496. doi: 10.1158/0008-5472.CAN-04-2910. [DOI] [PubMed] [Google Scholar]
- 32.(a) Wheelhouse RT, Sun DK, Han HY, Han FXG, Hurley LH. Cationic porphyrins as telomerase inhibitors: the interaction of tetra-(N-methyl-4-pyridyl)porphine with quadruplex DNA. J Am Chem Soc. 1998;120:3261–3262. [Google Scholar]; (b) Ren J, Chaires JB. Sequence and structural selectivity of nucleic acid binding ligands. Biochemistry. 1999;38:16067–16075. doi: 10.1021/bi992070s. [DOI] [PubMed] [Google Scholar]
- 33.Parkinson GN, Lee MP, Neidle S. Crystal structure of parallel quadruplexes from human telomeric DNA. Nature. 2002;417:876–880. doi: 10.1038/nature755. [DOI] [PubMed] [Google Scholar]; Phan AT, Kuryavyi V, Luu KN, Patel DJ. Structure of two intramolecular G-quadruplexes formed by natural human telomere sequences in K+ solution. Nucleic Acids Res. 2007;35:6517–6525. doi: 10.1093/nar/gkm706. [DOI] [PMC free article] [PubMed] [Google Scholar]; Ambrus A, Chen D, Dai J, Bialis T, Jones RA, Yang D. Human telomeric sequence forms a hybrid-type intramolecular G-quadruplex structure with mixed parallel/antiparallel strands in potassium solution. Nucleic Acids Res. 2006;34:2723–2735. doi: 10.1093/nar/gkl348. [DOI] [PMC free article] [PubMed] [Google Scholar]; Dai J, Carver M, Punchihewa C, Jones RA, Yang D. Structure of the hybrid-2 type intramolecular human telomeric G-quadruplex in K+ solution: insights into structure polymorphism of the human telomeric sequence. Nucleic Acids Res. 2007;35:4927–4740. doi: 10.1093/nar/gkm522. [DOI] [PMC free article] [PubMed] [Google Scholar]; Lim KW, Amrane S, Bouaziz S, Xu W, Mu Y, Patel DJ, Luu KN, Phan AT. Structure of the human telomere in K(+) solution: a stable basket-type G-Quadruplex with only two G-tetrad layers. J Am Chem Soc. 2009;131:4301–4309. doi: 10.1021/ja807503g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Parkinson GN, Cuenca F, Neidle S. Topology conservation and loop flexibility in quadruplex-drug recognition: crystal structures of inter- and intramolecular telomeric DNA quadruplex-drug complexes. J Mol Biol. 2008;381:1145–1156. doi: 10.1016/j.jmb.2008.06.022. [DOI] [PubMed] [Google Scholar]
- 35.Campbell NH, Parkinson GN, Reszka AP, Neidle S. Structural basis of DNA quadruplex recognition by an acridine drug. J Am Chem Soc. 2008;130:6722–6724. doi: 10.1021/ja8016973. [DOI] [PubMed] [Google Scholar]
- 36.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci USA. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. Programme available at http://apbs.sf.net. [DOI] [PMC free article] [PubMed]
- 37.Simonsson T, Pecinka P, Kubista M. DNA tetraplex formation in the control region of c-myc. Nucleic Acids Res. 1998;26:1167–1172. doi: 10.1093/nar/26.5.1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kumari S, Bugaut A, Huppert JL, Balasubramanian S. An RNA G-quadruplex in the 5′ UTR of the NRAS proto-oncogene modulates translation. Nature Chem Biol. 2007;3:218–221. doi: 10.1038/nchembio864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gomez D, Lemarteleur T, Lacroix L, Mailliet P, Mergny JL, Riou JF. Telomerase downregulation induced by the G-quadruplex ligand 12459 in A549 cells is mediated by hTERT RNA alternative splicing. Nucleic Acids Res. 2004;32:371–379. doi: 10.1093/nar/gkh181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sakurai S, Fukayama M, Kaizaki Y, Saito K, Kanazawa K, Kitamura M, Iwasaki Y, Hishima T, Hayashi Y, Koike M. Telomerase activity in gastrointestinal stromal tumors. Cancer. 1998;83:2060–2066. doi: 10.1002/(sici)1097-0142(19981115)83:10<2060::aid-cncr3>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 41.Hahn WC, Stewart SA, Brooks MW, York SG, Eaton E, Kurachi A, Beijersbergen RL, Knoll JH, Meyerson M, Weinberg RA. Inhibition of telomerase limits the growth of human cancer cells. Nature Med. 1999;5:1164–1170. doi: 10.1038/13495. [DOI] [PubMed] [Google Scholar]
- 42.Mergny JL, Lacroix L, Teulade-Fichou MP, Hounsou C, Guittat L, Hoarau M, Arimondo PB, Vigneron JP, Lehn JM, Riou JF, Garestier T, Helene C. Telomerase inhibitors based on quadruplex ligands selected by a fluorescence assay. Proc Natl Acad Sci USA. 2001;98:3062–3067. doi: 10.1073/pnas.051620698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lux ML, Rubin BP, Biase TL, Chen CJ, Maclure T, Demetri G, Xiao S, Singer S, Fletcher CD, Fletcher JA. KIT extracellular and kinase domain mutations in gastrointestinal stromal tumors. Am J Pathol. 2000;156:791–795. doi: 10.1016/S0002-9440(10)64946-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Taguchi T, Sonobe H, Toyonaga S, Yamasaki I, Shuin T, Takano A, Araki K, Akimaru K, Yuri K. Conventional and molecular cytogenetic characterization of a new human cell line, GIST-T1, established from gastrointestinal stromal tumor. Lab Invest. 2002;82:663–665. doi: 10.1038/labinvest.3780461. [DOI] [PubMed] [Google Scholar]
- 45.Guest MF, Bush IJ, van Dam HJJ, Sherwood P, Thomas JMH, van Lenthe JH, Havenith RWA, Kendrick J. The GAMESS-UK electronic structure package: algorithms, developments and applications. Mol Physics. 2005;103:719–747. [Google Scholar]
- 46.Dolinsky TJ, Nielsen JE, McCammon JA, Baker NA. PDB2PQR: an automated pipeline for the setup, execution, and analysis of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004;32:W665–W667. doi: 10.1093/nar/gkh381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Connolly ML. Analytical molecular surface calculation. J Appl Cryst. 1983;16:548–558. [Google Scholar]
- 48.Humphrey W, Dalke A, Schulten K. VMD - Visual Molecular Dynamics. J Molec Graphics. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]