

Abstract
Epidermal growth factor receptor (EGFR) is a ubiquitously expressed receptor tyrosine kinase involved in the etiology of several human cancers. Cetuximab is an EGFR blocking-antibody that has been approved for the treatment of patients with cancers of the head and neck (HNSCC) and metastatic colorectal cancer (mCRC). Previous reports have shown that EGFR translocation to the nucleus is associated with cell proliferation. Here we investigated mechanisms of acquired resistance to cetuximab using a model derived from the non-small cell lung cancer line H226. We demonstrated that cetuximab-resistant cells overexpress HER family ligands including epidermal growth factor (EGF), amphiregulin (AR), heparin-binding EGF (HB-EGF) and β-cellulin. Overexpression of these ligands is associated with the nuclear translocation of the EGFR and this process was mediated by the Src family kinases (SFK). Treatment of cetuximab-resistant cells with the SFK inhibitor, dasatinib, resulted in loss of nuclear EGFR, increased membrane expression of the EGFR and re-sensitization to cetuximab. In addition, expression of a nuclear localization sequence tagged EGFR in cetuximab-sensitive cells increased resistance to cetuximab both in vitro and in mouse xenografts. Collectively, these data suggest that nuclear expression of EGFR may be an important molecular determinant of resistance to cetuximab therapy and provides a rationale for investigating nuclear EGFR as a biomarker for cetuximab response. Further, these data suggest a rationale for the design of clinical trials that examine the value of treating patients with cetuximab-resistant tumors with inhibitors of SFKs in combination with cetuximab.
Keywords: EGFR, nuclear, cetuximab, resistance, Src-family kinases, dasatinib
Introduction
The epidermal growth factor receptor (EGFR) is a ubiquitously expressed receptor tyrosine kinase (RTK) that is important in oncogenesis. Ligand activation of the EGFR leads to downstream signaling of several pro-survival cascades, such as the RAS/RAF/MEK/ERK/MAPK and PI(3)K/Akt pathways. Activation of the EGFR ultimately promotes cell proliferation, angiogenesis, invasion, metastasis, and survival making it one of the most promising molecular targets in cancer therapy (Arteaga, 2003; Bianco et al., 2005; Camp et al., 2005; Gupta et al., 2002; Kobayashi et al., 2005; Pao et al., 2005; Viloria-Petit and Kerbel, 2004; Yarden and Sliwkowski, 2001).
EGFR has been an actively investigated target over the last decade with five FDA agents approved in oncology since 2003. Two distinct molecular approaches to EGFR inhibition have emerged: 1) monoclonal antibodies (mAbs) directed against the ligand-binding site of the EGFR and 2) tyrosine kinase inhibitors directed against the intracellular tyrosine kinase domain (TKD). Although both strategies of inhibition have demonstrated promising anti-tumor activity in patients with metastatic colorectal cancer (mCRC), non-small cell lung cancer (NSCLC), breast cancer, and head and neck squamous cell carcinoma (HNSCC), only ∼10 to 20% of patients manifest major clinical responses. Response to these agents appears to be limited by intrinsic as well as acquired resistance (Arteaga, 2003; Bianco et al., 2005; Camp et al., 2005; Gupta et al., 2002; Kobayashi et al., 2005; Pao et al., 2005; Viloria-Petit and Kerbel, 2004). Several mechanisms of resistance to cetuximab have emerged including deregulated ubiquitination, amplified HER3 signals, increased transforming growth factor alpha (TGFα), and altered angiogenesis (Lu et al., 2007; Rajput et al., 2007; Viloria-Petit et al., 2001; Wheeler et al., 2008). Although each of these studies suggests rational mechanisms of intrinsic and/or acquired resistance, our detailed knowledge of EGFR inhibitor resistance remains relatively ill defined (Mukohara et al., 2005).
It has been reported that EGFR family members can be shuttled from the plasma membrane to the nucleus (Giri et al., 2005; Liao and Carpenter, 2007; Lin et al., 2001; Lo et al., 2005a; Lo et al., 2006b; Lo and Hung, 2006; Massie and Mills, 2006; Ni et al., 2001; Offterdinger et al., 2002; Wang et al., 2004; Wang et al., 2006; Xie and Hung, 1994). Nuclear EGFR (nEGFR) has been demonstrated in highly proliferative tissues and linked with poor clinical outcome in breast, oropharyngeal squamous cell carcinoma, and ovarian cancer (Lo and Hung, 2006; Psyrri et al., 2005; Xia et al., 2008). Nuclear EGFR has two identified functions in the nucleus, 1) as a transcription factor and 2) in the direct phosphorylation of proliferating cell nuclear antigen (PCNA). As a transcription factor, EGFR has been shown to interact with STAT3 and E2F1 to mediate transcription of cyclin D1, iNOS, B-Myb and Aurora Kinase A (Cao et al., 1995; Hanada et al., 2006; Hung et al., 2008; Lin et al., 2001; Lo et al., 2005a; Lo et al., 2006b; Lo and Hung, 2006; Marti et al., 1991). In addition to transcription-regulated events, the EGFR has been shown to phosphorylate PCNA on tyrosine 211, thus stabilizing chromatin bound PCNA. This phosphorylation event correlates with pronounced cell proliferation (Wang et al., 2006).
In this study we investigated the role of nuclear EGFR in the resistance to cetuximab therapy. We showed that cells with acquired resistance to cetuximab have increased Src Family Kinase (SFK) activity, and this is linked to the translocation of the EGFR to the nucleus. Collectively, our findings reported herein suggest that nuclear expression of EGFR may be a critical determinant for resistance to cetuximab therapy and provides a rationale for designing clinical trials to examine the value of treating patients with resistance to cetuximab with inhibitors of SFKs in combination with cetuximab.
Results
Cetuximab-resistant cells have increased nuclear EGFR expression
We have previously described the development and characterization of NCI-H226 NSCLC lines with acquired resistance to cetuximab (Wheeler et al., 2008). The development of this model resulted in six stable cetuximab-resistant clones for the H226 NSCLC line: HC1, HC4, HC5, HC6, HC7 and HC8. The sensitive parental line was designated HP. Three of these clones (HC1, HC4 and HC8) were tested for their sensitivity to cetuximab relative to the parental controls (Figure 1A). All clones displayed a robust cetuximab-resistant phenotype when challenged with 100 nM of cetuximab as compared to parental controls.
Figure 1. Cetuximab-resistant cells have increased nuclear EGFR expression.
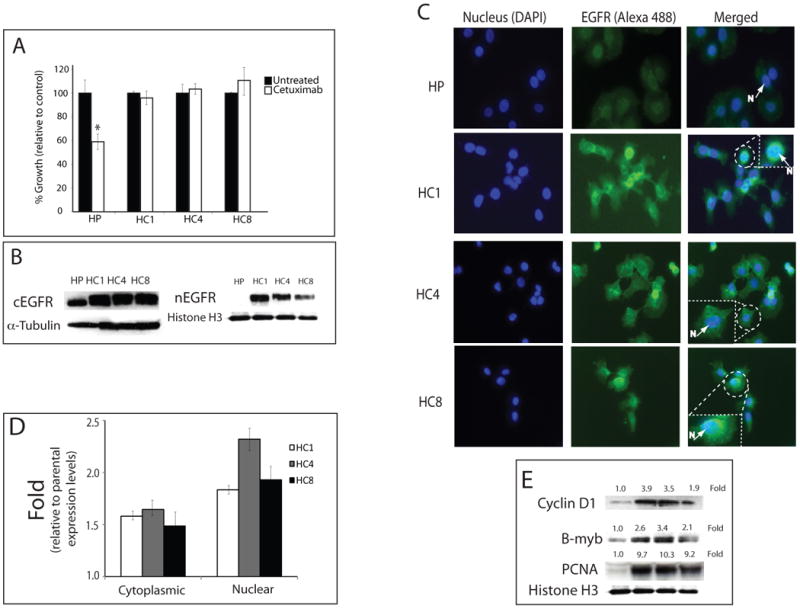
A) Cetuximab growth response using the NSCLC line NCI-H226 cetuximab-resistant clones. Cells were treated with 100 nM of cetuximab and growth was measured using the growth proliferation assay as described in materials and methods. Results are graphed as a percentage of growth relative to the untreated control cells. HP; cetuximab-sensitive parental line, HC1, HC4, HC8; cetuximab-resistant clones. Data points are represented as mean +/- SEM. (n=3). *, P<0.05
B) Nuclear EGFR is increased in cetuximab-resistant cells. After harvesting cetuximab-resistant cells (HC1, HC4 and HC8) and parental control (HP), cytoplasmic and nuclear protein was collected and fractionated by SDS-PAGE followed by immunoblotting for the indicated proteins. α-tubulin and histone H3 were used as loading and purity controls of each cellular fraction.
C) Expression of nEGFR in cetuximab-resistant cells by immunofluorescence microscopy. EGFR is shown in the middle column (green), the left column shows DNA (blue) and the right column is a merged image of EGFR and DNA. Box represents enlarged area of insert. N; Nucleus, HP; cetuximab-sensitive parental line, HC1, HC4, HC8; cetuximab-resistant clones. 200× magnification.
D) Fold change of EGFR intensity levels in cetuximab-resistant clones relative to parental controls. Intensity of EGFR in each cetuximab-resistant clones (HC1, HC4, HC8) were measured by BD Pathway 855 (BD sciences, San Jose, CA) and graphed as fold change relative to the parental cell lines (HP). Data points are presented as mean +/- SEM. (n=10).
E) Relative amounts of cyclin D1, B-myb and PCNA in cetuximab-resistant clones. (HC1, HC4, HC8) were quantified by ImageJ software and normalized against the parental, cetuximab-sensitive line (HP). histone H3 was used as a loading and nuclear fraction purity control.
Nuclear expression of EGFR has been inversely correlated with survival in breast, oropharyngeal and ovarian cancer (Lo et al., 2005b; Psyrri et al., 2005). To build upon these published results, we investigated nEGFR expression in cetuximab-resistant cells relative to the parental controls to determine if nEGFR can function as a mechanism of escape to cetuximab therapy. We analyzed the cytoplasmic and nuclear distribution of EGFR in cetuximab-resistant cells. Figure 1B shows all three cetuximab-resistant clones had a dramatic increase in nuclear expression of the EGFR relative to the parental controls. We next utilized confocal microscopy to confirm nEGFR localization in cetuximab-resistant clones. These findings showed robust EGFR expression in all three clones in the nucleus, perinuclear membranes and cytosol (Figure 1C). Quantification of the cytoplasmic and nuclear fractions of each cetuximab-resistant clone, relative to parental controls, is represented in Figure 1D and indicated a ∼1.5-fold increase in cytosolic expression and a ∼2 to 2.5-fold increase in nuclear expression.
Nuclear EGFR has been shown to be associated with the transcription of cell cycle genes (cyclin D1, B-myb and Aurora kinase A), DNA repair, and DNA synthesis activity (PCNA phosphorylation and stability) (Bandyopadhyay et al., 1998; Dittmann et al., 2005; Friedmann et al., 2006; Hung et al., 2008; Lin et al., 2001; Lo et al., 2005a; Lo et al., 2006b; Wang et al., 2006). To confirm that the nEGFR observed in the cetuximab-resistant clones had nuclear function, we investigated the level of expression of these proteins in cetuximab-resistant clones relative to the parental controls. These experiments revealed that Cyclin D1, B-myb and PCNA were increased in their expression (Figure 1E). Collectively, these data indicate that cells with acquired resistance to cetuximab have increased expression of nEGFR, Cyclin D1, B-myb and PCNA.
Nuclear EGFR in cetuximab-resistant cells is driven by EGFR ligands
It has been clearly demonstrated that EGF stimulates translocation of the EGFR to the nucleus (Hsu and Hung, 2007). We therefore, hypothesized that cetuximab-resistant cells may have increased expression of EGFR ligands leading to chronic EGFR translocation to the nucleus. We performed DNA microarray analysis comparing HP parental cells versus the HC4 cetuximab-resistant clone. Upregulation of several HER family ligands including EGF (∼20-fold), heparin-binding EGF (HB-EGF, ∼12-fold), amphiregulin (AR, ∼20-fold) and β-cellulin (∼3-fold) were identified in this microarray analysis (data not shown). To extend these findings to all resistant clones we performed real-time quantitative PCR to validate EGF, HB-EGF, AR and β-cellulin gene expression in passage controlled cetuximab resistant clones HC1, HC4 and HC8 relative to parental control (Figure 2A). Figures 2A-2D represent several functional assays to confirm the importance of EGFR ligands in the translocation of EGFR to the nucleus. To determine which of these ligands had the potential to drive the EGFR into the nucleus, we treated HP parental cells (low nEGFR expression) with purified ligands (200 ng/mL) and measured nEGFR. Each ligand (EGF, HB-EGF, AR, β-cellulin) induced nuclear translocation of the EGFR to varying degrees in HP parental cells (Figure 2A).
Figure 2. Nuclear EGFR in cetuximab-resistant cells is driven by EGFR ligands.
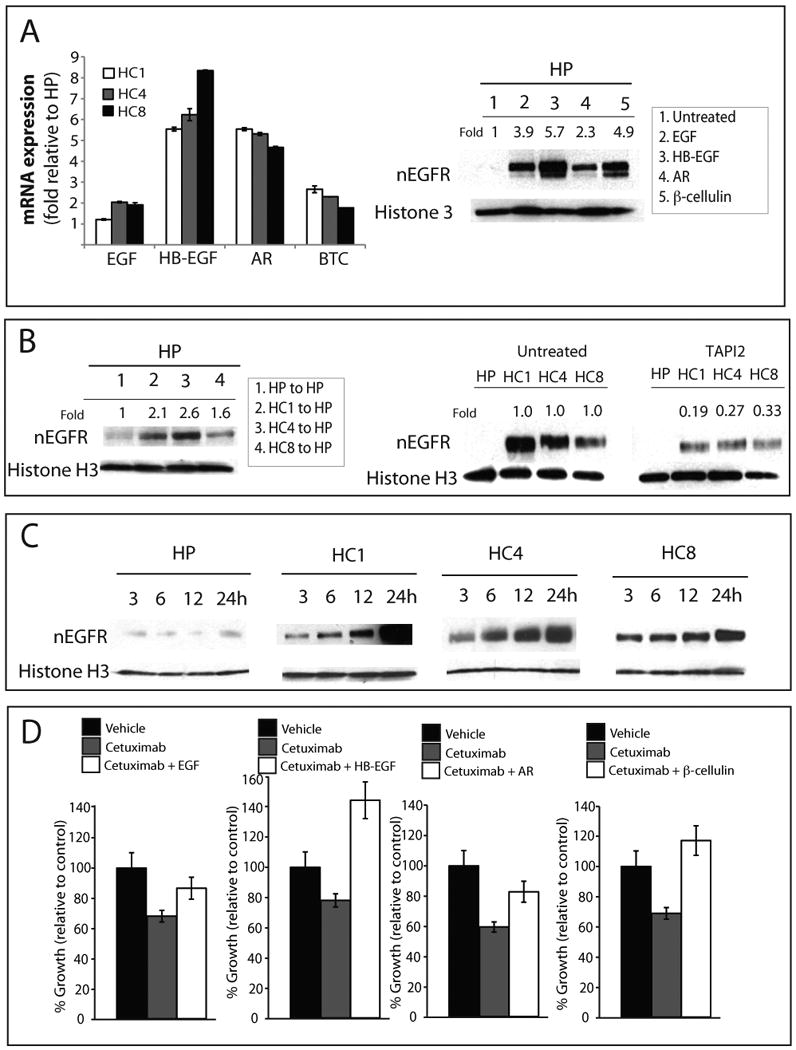
A) Real-time quantitative PCR analysis of HER family ligands in cetuximab-resistant clones HC1, HC4 and HC8. Data is represented as fold increase relative to the HP parental control. Data points are represented as mean +/- SEM. (n=3). Parental cells (HP) were treated with 200 ng/mL of each HER family ligand for 1 hour after 24 hours of serum starvation. Cells were harvested and nuclear protein was fractionated by SDS-PAGE followed by immunoblotting for indicated proteins. histone H3 was used as a loading control. Expression of nuclear EGFR (nEGFR) after treatment with each ligand was quantitated using ImageJ software and normalized against untreated cells.
B) Conditioned media from cetuximab-resistant cells can lead to increased nuclear translocation of the EGFR. Conditioned media from cetuximab-resistant clones (HC1, HC4, HC8) or parental cells (HP) was placed on HP cells and incubated for 1 hours; cetuximab-resistant cells and parental control were treated with 100 μM TAPI2 for 24 hours. Cells were harvested and nuclear protein fractionated on SDS-PAGE followed by immunoblotting for EGFR. histone H3 was used as a loading control. Expression of nEGFR was quantitated using ImageJ software; the untreated cells were compared to TAPI2 treated cells.
C) Serum starvation leads to increased nEGFR in cetuximab-resistant clones, but not parental cell lines. Cetuximab-resistant cells and parental control were placed in serum-free medium and cells were harvested at 3, 6, 12 and 24 hours. Nuclear protein was fractionated by SDS-PAGE followed by immunoblotting for anti-EGFR antibody. histone H3 was used as loading control.
D) HER family ligands can enhance cetuximab resistance in cetuximab-sensitive cells. HP cells were treated with cetuximab or the combination of cetuximab and 200 ng/ml of ligand (EGF, HB-EGF, AR or β-cellulin) for 72 hours. Growth was measured at 72 hours after treatment using proliferation assays and plotted as a percentage of growth relative to the untreated control cells. Data points are represented as mean +/- SEM. (n=3).
We expanded on these findings to determine if conditioned media from the cetuximab-resistant clones (HC1, HC4, and HC8) could induce nuclear translocation of the EGFR in HP parental cells (low nEGFR expression). Conditioned media from HP, HC1, HC4 and HC8 were collected after 72 hours and were incubated with HP cells that have been serum starved for 24 hours. Nuclear proteins were isolated 1 hour after the addition of the conditioned media and fractionated by SDS-PAGE. These results showed that conditioned media from the cetuximab-resistant clones, but not from the HP parental cells, could induce EGFR translocation to the nucleus (Figure 2B).
EGFR ligands are released from the cytoplasmic membrane via activation of tumor necrosis factor converting enzyme, TACE (Borrell-Pagès et al., 2003; Sunnarborg et al., 2002). We used the TACE inhibitor, TAPI2, to block the release of EGFR ligands in cetuximab-resistant cells as well as the parental control and measured the effect on nuclear translocation of the EGFR (Figure 2B). The results of these experiments indicated an approximate 3 to 5-fold decrease in nEGFR expression in cetuximab-resistant clones in the TAPI2 treated groups. These data suggest that blockade of TACE and thus release of growth factors results in decreased nuclear translocation of the EGFR.
In Figure 2C HP, HC1, HC4, and HC8 were serum starved for 3, 6, 12, and 24 hours and nEGFR translocation was then assessed. All cetuximab-resistant clones (HC1, HC4 and HC8), but not the parental HP, had increased nuclear translocation of the EGFR as time increased. These results suggest that factors from the cetuximab-resistant cells are released and lead to nuclear translocation of the EGFR over time.
If cetuximab-resistant cells have increased EGFR ligands and these ligands appear to drive nEGFR, we investigated whether or not this process would result in resistance to cetuximab. Growth proliferation assays indicated that the addition of ligand results in robust resistance to cetuximab (Figure 2D). Interestingly, HB-EGF and β-cellulin, which induced the most pronounced nEGFR translocation, resulted in the largest increase in resistance to cetuximab (Figure 2A and 2D).
Src family Kinases mediate ligand-induced EGFR translocation to the nucleus
We have previously shown that cells with acquired resistance to cetuximab have increased SFK activity relative to parental control (Wheeler et al., 2009). We hypothesized that ligand-induced EGFR translocation to the nucleus may be mediated by SFK. Pretreatment of the HP parental cells with the SFK inhibitor, dasatinib for 24 hours prior to ligand stimulation completely blocked EGFR translocation to the nucleus by these ligands (Figure 3A).
Figure 3. Src family kinases mediate ligand-induced EGFR translocation to the nucleus.
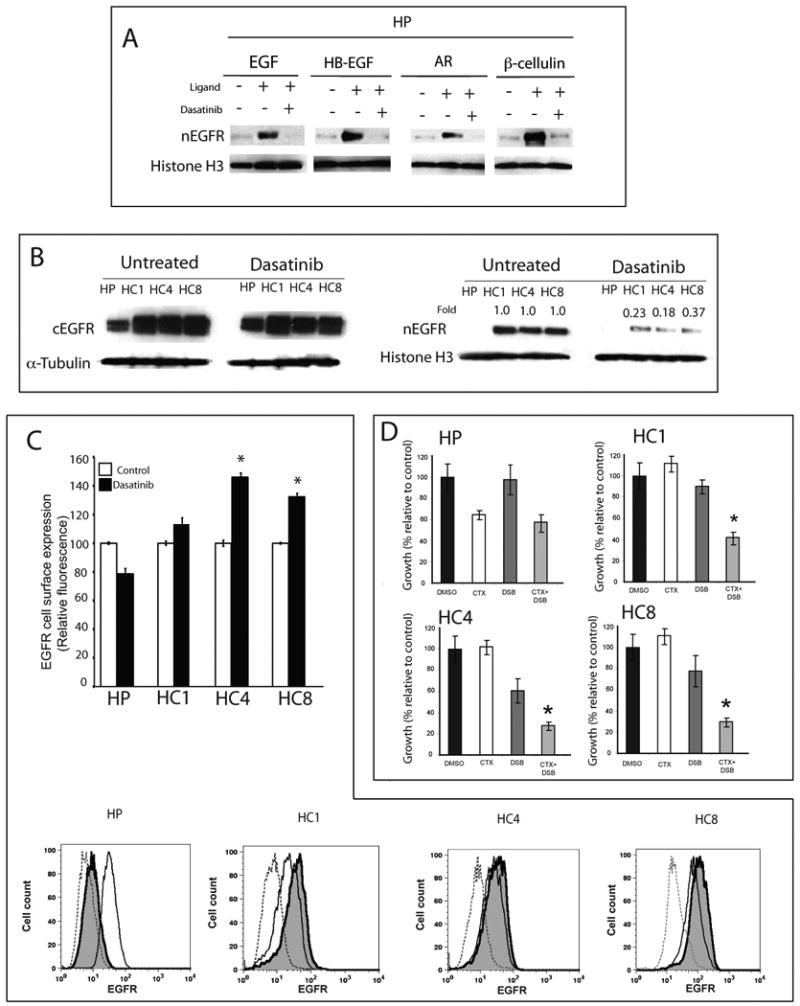
A) Dasatinib inhibits HER family ligands signaling in parental cells (HP). HP cells were untreated, treated for 24 hours with 50 nM of dasatinib alone, or followed by 200 ng/ml of indicated ligand for 1 hour prior to harvesting. Nuclear protein was collected and fractionated by SDS-PAGE followed by immunoblotting for EGFR. histone H3 was used as loading control.
B) Dasatinib inhibits nuclear expression of EGFR in cetuximab-resistant cell lines. Parental cells (HP) and cetuximab-resistant cell lines (HC1, HC4, HC8) were treated with 50 nM of dasatinib for 24 hours. After cells were harvested, cytoplasmic and nuclear protein was fractionated by SDS-PAGE followed by immunoblotting for EGFR. α-tubulin and histone H3 were used as loading controls and purity controls of each cellular fraction. Expression of nEGFR after dasatinib treatment in cetuximab-resistant clones was quantitated using ImageJ software and normalized against the amounts of untreated cells.
C) Dasatinib treatment lead to increased membrane-bound EGFR in cetuximab-resistant cells by flow cytometry analysis. Parental cells (HP) and cetuximab-resistant cells (HC1, HC4 and HC8) were treated with DMSO or 50 nM of dasatinib for 24 hours and membrane expression is represented relative to untreated controls. Mean surface expression of EGFR is represented +/- SEM (n=3). Flow cytometric plots of representative experiments are presented. Shaded histograms represent dasatinib treatment. Controls (dotted line) represent cells labeled with FITC-conjugated normal mouse IgG *, P<0.05
D) Dasatinib re-sensitizes cetuximab-resistant cells to cetuximab growth inhibition. Cetuximab-resistant cells (HC1, HC4 and HC8) and parental controls (HP) were treated with DMSO, 100 nM cetuximab (CTX), 25 nM dasatinib (DSB) or the combination for 72 hours. Growth was measured using the proliferation assay and plotted as a percentage of growth relative to the untreated control cells. Data points are represented as mean +/- SEM. (n=6). *, P<0.05
Since cetuximab-resistant cells have increased expression of EGFR ligands, SFK activity and nEGFR, we designed experiments to determine if these were linked. We investigated whether or not SFK blockade using dasatinib could reduce nEGFR expression in the cetuximab-resistant clones HC1, HC4 and HC8. Cetuximab-resistant clones were treated with vehicle or dasatinib for 24 hours; the cells were then fractionated for the cEGFR or nEGFR. Dasatinib treatments lead to a 2.7 to 5.5-fold decrease in nEGFR expression in the cetuximab-resistant clones (Figure 3B). The results of this experiment indicated that cetuximab-resistant cells have increased release of EGFR ligands, which then induce the translocation of the EGFR to the nucleus and this process is dependent upon SFK.
It has been well characterized that membrane-bound EGFR translocates from the membrane to the nucleus (Hsu and Hung, 2007; Lin et al., 2001; Lo et al., 2006a). We postulated that SFK blockade inhibited this process and would result with higher membrane levels in cetuximab-resistant clones after dasatinib treatment. To test this hypothesis, we treated HP, HC1, HC4 and HC8 with 50 nM dasatinib for 24 hours and performed flow cytometric analysis on membrane EGFR expression (Figure 3C). HP showed a decrease in membrane EGFR levels whereas all three cetuximab-resistant clones showed an increase in membrane expression of the EGFR after dasatinib treatment.
With an increase in EGFR membrane expression and a decrease in nuclear expression after dasatinib treatment, we asked the question of whether or not cetuximab-resistant clones would be more sensitive to cetuximab therapy. We performed proliferation analysis assays using DMSO control, 100 nM cetuximab, 25 nM dasatinib or the combination on HP, HC1, HC4 and HC8. The results of these experiments showed that dasatinib induced mild growth inhibition on cetuximab-resistant cells but the combination of the two drugs showed augmentation of growth inhibition (Figure 3D). These results suggest that blockade of SFK can block nuclear translocation of the EGFR, increase membrane EGFR and thus make cells more sensitive to cetuximab therapy.
Nuclear localization sequence tagged EGFR leads to increased resistance to cetuximab
To isolate the effects of nEGFR from ligand-induced nuclear translocation of EGFR, we designed a retrovirus expressing wild-type human EGFR fused to a nuclear localization sequence tag (NLS), followed by a myc tag for identification of the transgene (EGFR-NLS/Myc, Figure 4A). We infected cetuximab-sensitive parental line, HP, with EGFR-NLS/Myc or vector only. From these experiments we obtained three EGFR-NLS/Myc-expressing clones designated C4, C5, C10 and one vector only clone, V0. Figure 4A shows the characterization of these three clones. The nuclear fractions were purified and immunoprecipitated with a myc antibody. All clones were positive for the myc tag, EGFR and phospho-tyrosine (p-Tyr) of the EGFR, indicating active EGFR by Western blot analysis. These results were followed by immunofluorescence (IF) analysis of EGFR expression in the vector only and three EGFR-NLS/Myc-expressing clones. Each EGFR-NLS/Myc-expressing clone had robust EGFR expression in the nucleus and peri-nuclear membrane as well as cytosolic expression. These results indicated that our transgene was expressed in the nuclear fraction and active as demonstrated by total phosphorylation of tyrosine residues on the EGFR.
Figure 4. EGFR tagged with nuclear localization sequence confers resistance to cetuximab in vitro.
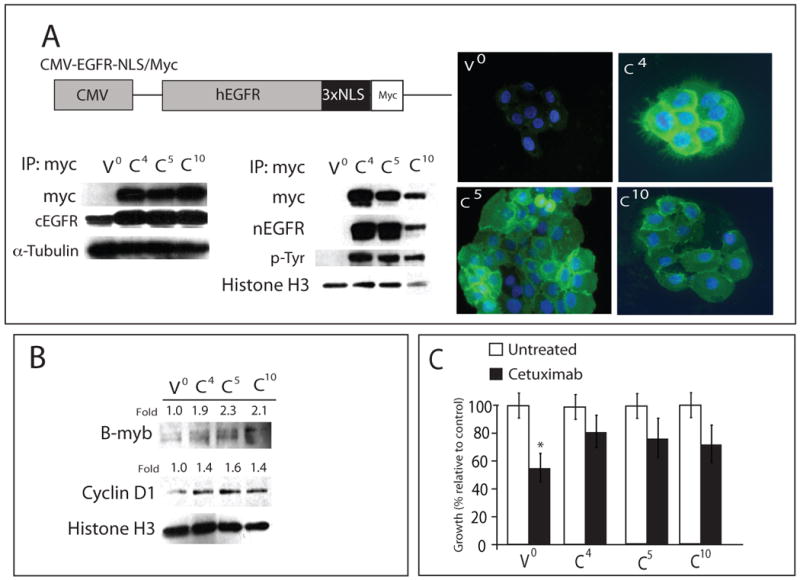
A) A schematic representation of the CMV-EGFR-NLS/Myc construct is shown. EGFR-NLS/Myc was driven by the CMV promoter. The cetuximab-sensitive NSCLC line NCI-H226 was infected with indicated constructs. Represented is three individual clones and vector control (V0; vector only, C4, C5 and C10). Cytoplasmic and nuclear protein from each clone was collected and immunoprecipitated with an anti-myc antibody, fractionated on SDS-PAGE and immunoblotted with the indicated antibodies. α-tubulin and histone H3 were used as loading and purity control for cytosolic and nuclear fractions, respectively. Immunofluorescence of nEGFR staining in CMV-EGFR-NLS/Myc clones. EGFR (green), DNA (blue), stained by PI. V0; vector clone, C4, C5 and C10; CMV-EGFR-NLS/Myc clones. 400× magnification. cEGFR; cytoplasmic EGFR, nEGFR; nuclear EGFR.
B) CMV-EGFR-NLS/Myc expressed in NCI-H226 leads to increased cyclin D1 and B-myb expression. Nuclear protein from EGFR-NLS/myc clones was collected and fractionated by SDS-PAGE followed by immunoblotting for the indicated proteins. histone H3 was used as a loading control. Expression of cyclin D1 and B-myb in CMV-EGFR-NLS/Myc clones (C4, C5 and C10) were quantitated using ImageJ software and normalized against the amounts of those proteins in vector control (V0). V0; vector clone, C4, C5 and C10; CMV-EGFR-NLS/Myc clones.
C) Growth response to cetuximab of three individual clones and vector control (V0; vector only, C4, C5 and C10). CMV-EGFR-NLS/Myc-tag clones (C4, C5 and C10) were treated with 100 nM of cetuximab and growth was measured using the growth proliferation assay and plotted as growth relative to untreated control. Data points are represented as mean +/- SEM. (n=3). *, P<0.05
It has been reported that EGFR activates expression of cyclin D1 and B-myb (15, 31). To determine if the NLS-tagged EGFR had nuclear function, we measured the levels of cyclin D1 and B-myb. Figure 4B indicates that all EGFR-NLS/Myc-expressing clones have a 1.9 to 2.3-fold increase in expression of B-myb and a 1.4 to 1.6-fold increase in cyclin D1 expression. We utilized these clones with increased nEGFR expression and tested the hypothesis that this would result in increased resistance to cetuximab therapy. In Figure 4C we report that, relative to the parental HP controls, each clone exhibited an approximate 50% increase in resistance to cetuximab treatment. Collectively these data suggest that nuclear expression of the EGFR contributes to resistance to cetuximab therapy.
To examine whether these clones with nEGFR expression would be cetuximab-resistant in vivo we inoculated athymic nude mice with HP cetuximab-sensitive parental cells (HP), vector only (V0), and the three EGFR-NLS/Myc-expressing clones (C4, C5, and C10). When tumor volume reached 120-180 mm3 we began treatment with 0.1 mg IgG or 0.1 mg of cetuximab twice weekly. The results of this experiment indicate two major findings. First, tumor cells that express nEGFR grew approximately twice as fast relative to the HP or V0 controls. This is noted by in Figure 5. It took the HP and V0 cells approximately 27 days to reach 100 mm3, whereas all three EGFR-NLS/Myc-expressing clones reached the same tumor volume at approximately 12-13 days. This work was confirmed on a repeat experiment. Secondly, the three EGFR-NLS/Myc-expressing clones showed no statistical difference in growth to the IgG controls in response to cetuximab. Taken together, these results suggest that expression of an NLS-tagged EGFR can contribute to resistance to cetuximab in vivo.
Figure 5. Overexpression of a NLS tagged EGFR in cetuximab-resistant cells confers resistance to cetuximab in vivo.

Male athymic nude mice were injected subcutaneously with 1×106 cetuximab-sensitive parental cells (HP) or CMV-EGFR-NLS/Myc clone cells (Vector only, Clone 4, Clone 5 and Clone 10) into the dorsal flank. Once tumors reached a volume 120-180mm3 mice were treated with 0.1 mg IgG or cetuximab twice weekly. Tumor diameters were measured serially with calipers and tumor volumes were calculated. Points, mean tumor volume of eight mice per group; bars, SD. T-test was used to compare tumor volumes between cetuximab treated and control IgG mice. *, P<0.05
Cells that have nuclear EGFR are intrinsically resistant to cetuximab
H226 lines represent a NSCLC model of acquired resistance to cetuximab. However, intrinsic resistance to EGFR-targeted therapy is a significant clinical problem. We were interested to determine if other tumor cell lines with known responses to cetuximab in vitro could be correlated with nEGFR expression. To test this hypothesis, we utilized nine established HNSCC lines: SCC1, SCC4Y, SCC6, SCC11A, SCC14A, SCC17B, SCC22b, SCC37A and SCC1483. Cytoplasmic and nuclear protein from these cell lines were fractionated and blotted for EGFR (Figure 6A). Cells were treated with vehicle or 5nM of cetuximab for 72 hours and MTT assays performed (Figure 6B). The results of this experiment indicated that cells that have intrinsic resistance to cetuximab therapy, in vitro, have increased nEGFR (SCC4Y, SCC6, SCC11A, and SCC14A) and those with low nEGFR have a higher sensitivity to cetuximab treatment (SCC1, SCC17B, SCC37A and SCC1483). SCC22B was used as an EGFR negative expressing line, and thus was not responsive to cetuximab therapy.
Figure 6. Nuclear EGFR expression in head and neck squamous cell carcinoma lines with intrinsic resistance to cetuximab.
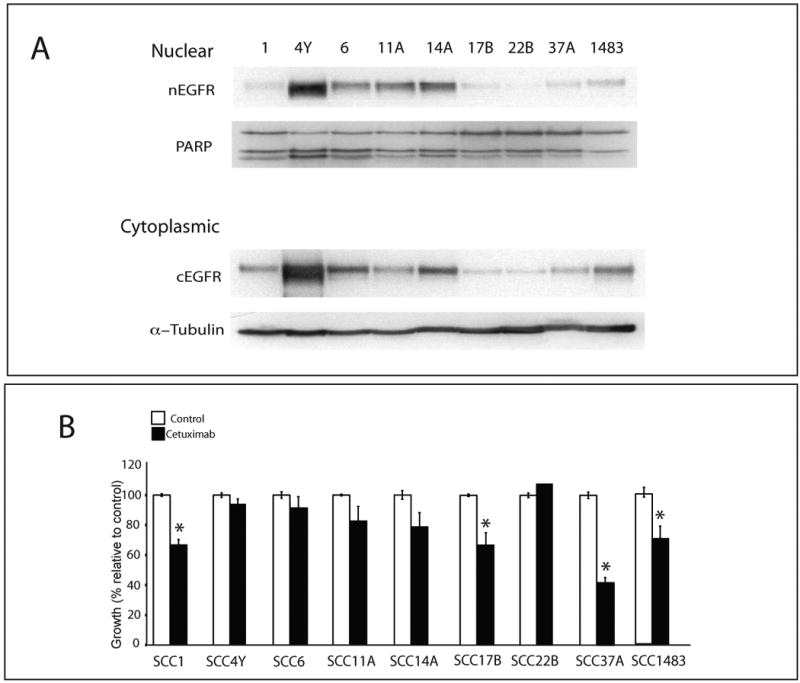
A) Immunoblot analysis of cytoplasmic (cEGFR) and nuclear (nEGFR) expression in head and neck squamous cancer cell (HNSCC) lines. Cells were harvested and cytoplasmic and nuclear protein collected and fractionated by SDS-PAGE followed by immunoblotting for EGFR. α-tubulin and PARP were used as loading and purity control for cytosolic and nuclear fractions, respectively.
B) Cetuximab growth response using HNSCC lines. HNSCC lines were treated with 5 nM cetuximab for 72 hours. Growth was measured using the MTT assays and plotted as a percentage of growth relative to the untreated control cells. Data points are represented as mean +/- SEM. (n=3).
Discussion
Cetuximab has shown great promise in clinical oncology, but is limited by intrinsic as well as acquired resistance. These resistance issues present a clinical obstacle for the optimal advancement of this promising molecular targeting agent. In this study we investigated mechanisms of acquired resistance to cetuximab using previously established cetuximab-resistant tumor clones following long-term exposure to cetuximab (Wheeler et al., 2008). Sub-cellular analysis of EGFR localization in cetuximab-resistant clones indicated a marked increase in nuclear expression of the EGFR (Figure 1B-D). Emerging evidence over the last decade has indicated that nEGFR has been detected in highly proliferative tissues and linked with poor clinical outcome in breast, oropharyngeal SCC, and ovarian cancer (Lo and Hung, 2006; Psyrri et al., 2005; Xia et al., 2008). Furthermore, nEGFR functions as a transcription factor for cyclin D1, iNOS, B-myb and Aurora Kinase A (Cao et al., 1995; Hanada et al., 2006; Hung et al., 2008; Lin et al., 2001; Lo et al., 2005a; Lo et al., 2006b; Lo and Hung, 2006; Marti et al., 1991) as well as phosphorylating and stabilizing PCNA (Wang et al., 2006). These data suggest that nEGFR provides a “second compartment” of proliferation mediated by the EGFR, the first compartment being the classical membrane-bound EGFR signaling through the RAS/RAF/MEK/MAPK pathway. Analysis of cetuximab-resistant clones indicated a marked upregulation of cyclin D1, B-myb and PCNA demonstrating that nEGFR was functional in cetuximab-resistant clones. Together, these observations suggest a potential model of resistance to cetuximab therapy (Figure 7).
Figure 7. Potential mechanism for resistance to cetuximab.

A) Cetuximab-sensitive cells depend on classical EGFR membrane signaling. B) Tumor cells that acquire resistance to cetuximab gain nEGFR as a second compartment of proliferation. C) Cetuximab can abrogate signals from plasma membrane EGFR but not nEGFR; nEGFR continues to send proliferative signals by modulation of Cyclin D1, B-myb, Aurora kinase K and regulation of PCNA. D) The SFK inhibitor dasatinib inhibits nuclear translocation of the EGFR from the plasma membrane leading to increased EGFR on the plasma membrane and restoring sensitivity to cetuximab.
It has been reported that EGF stimulation can induce the accumulation of EGFR in the nucleus (Hsu and Hung, 2007). This finding led us to investigate the levels of ligand expression in cetuximab-resistant cells (Figure 2A). In addition to EGF, we found several EGFR ligands to be increased. Furthermore, we found that all ligands that were upregulated had the potential to induce translocation of the EGFR to the nucleus with varying degrees (Figure 2A). These data suggest that deregulation of EGFR ligands have the potential to drive EGFR to the nucleus. Furthermore, blockade of TACE, the enzyme necessary for release of EGFR ligands from the membrane (Borrell-Pagès et al., 2003; Sunnarborg, 2002; Sunnarborg et al., 2002), dramatically decreased nEGFR in all cetuximab-resistant clones, further strengthening a role for ligands in this process (Figure 2B). If EGFR ligands can drive nEGFR, then ligand alone should induce moderate levels of resistance to cetuximab. In Figure 2D we tested this hypothesis by adding individual ligands or ligand plus cetuximab and measured the resistance. Each ligand resulted in a varying degree of resistance but appeared that HB-EGF and β-cellulin, which induced the more accumulation of EGFR in the nucleus (Figure 2A), correlated with the most robust resistance (Figure 2D). Taken together these findings indicate that EGFR ligands may be a critical determinant of inducing resistance to cetuximab and may be a viable screening tool for therapeutic response to cetuximab. Similar findings have been reported for additional HER family ligands in the resistance to HER family inhibitor therapy (Rajput et al., 2007; Ritter et al., 2007). Collectively our results and these reports suggest that surveillance of HER family ligands levels may provide an indicator of therapeutic response to cetuximab and other HER family inhibitors.
Recent experiments have shown that EGF can induce nuclear translocation of the EGFR. In Figure 3A, we expanded on this finding and showed that EGF, HB-EGF, AR, and β-cellulin drive EGFR to the nucleus. Furthermore, we showed that dasatinib could block this translocation. These findings suggest that EGFR movement to the nucleus is dependent, in part, on SFK, and this may be an early signal in translocation. Dasatinib treatment of cetuximab-resistant clones led to a dramatic decrease in the nEGFR (Figure 3B). Taken together these findings suggest that overexpression of EGFR ligands initiate movement of the EGFR to the nucleus, mediated by SFK. Blockade of SFK may represent a unique therapeutic target to block nEGFR in patients.
Our results also imply that blockade of SFK led to increased EGFR on the plasma membrane and not the nucleus (Figure 3C). This is most likely due to the fact that SFK's are an initial signal for movement of the EGFR from the membrane to the nucleus. We postulated that increased membrane EGFR and loss of the “second compartment” of EGFR signaling in the nucleus after SFK blockade may restore sensitivity to cetuximab. This was seen in Figure 3D where the combinatorial therapy of dasatinib and cetuximab had greater impact than either agent alone. The overall findings from this series of experimentations indicate that targeting both the EGFR and SFK may provide a valuable clinical strategy to explore. Investigations into which Src family member may regulate EGFR translocation to the nucleus remain to be elucidated.
It is difficult to determine if nEGFR itself can drive resistance since the presence of ligand appears to be necessary. We took steps to distinguish these two processes by overexpressing EGFR tagged to a nuclear localization sequence (EGFR-NLS) in the cetuximab-sensitive HP parental lines in vitro and in vivo (Figure 4 and 5). Three different EGFR-NLS/Myc-expressing clones, was localized to the nucleus, was tyrosine phosphorylated and resulted in the upregulation of Cyclin D1 and B-myb (Figure 4A and B). Each EGFR-NLS/Myc-expressing clone had increases in resistance to cetuximab in vitro (Figure 4C). In addition, when these tumor cells were utilized in mouse xenograft models and challenged repeatedly with cetuximab, all three clones showed no significant difference in growth relative to the IgG control, whereas the HP and the vector only controls had statistically significant tumor growth control with cetuximab (Figure 5). Collectively these data support the hypothesis that nEGFR, in part, can play a role in resistance to cetuximab therapy.
Nuclear EGFR has been strongly correlated with poor overall survival in several tumor types (Lo et al., 2005b; Psyrri et al., 2008; Xia et al., 2008). In summary, this is the first report indicating that nEGFR may play a functional role in the response to molecular therapeutics agents. The findings reported herein have clinical implications for the design of optimal therapeutic strategies. In this model, we demonstrate that HER family ligands are upregulated in the cells with acquired-resistance to cetuximab. This in turn enhances the translocation of the EGFR to the nucleus. This process appears to be dependent on SFK. Once in the nucleus, EGFR performs several functions that mediate proliferation of tumor cells. In patients where this mechanism may be prominent, SFK inhibitor, dasatinib represents an approach inhibit nuclear translocation, leading to removal of nEGFR and its functions with re-sensitization to cetuximab therapy. Furthermore, these findings suggest that nEGFR may prove a viable molecular target. This may be undertaken by targeting nEGFR-expressing tumors with EGFR TKI (erlotinib and gefitinib) as they can diffuse across the membrane and abrogate nEGFR kinase function such as phosphorylation of PCNA. Strategies along these lines have already been suggested (Wheeler et al., 2008). In addition, targeting with dasatinib and cetuximab either concomitantly or sequentially may be a viable clinical approach for individuals with nuclear expression of EGFR.
Materials and Methods
Cell Culture
The human NSCLC line NCI-H226 was purchased from ATCC (Manassas, VA) and maintained in 10% fetal bovine serum in RPMI1640 (Mediatech Inc., Manassas, VA) with 1% penicillin and streptomycin. The development of cells with acquired resistance has been previously described (Wheeler et al., 2008). UM-SCC1, UM-SCC4Y, UM-SCC6, UM-SCC11A, UM-SCC14A, UM-SCC17B, UM-SCC-22B, and UM-SCC1483 cells were obtained from University of Michigan and University of Pittsburgh School of Medicine provided PCI-37a (SCC37A) cells. The cells were maintained in 10% fetal bovine serum (FBS, Invitrogen, Carlsbad, CA) in Dulbecco's modified Eagle's medium (DMEM, Mediatech Inc.) supplemented with 1% hydrocortisone.
Compounds
Cetuximab (C225, Erbitux™) was provided by ImClone Systems Inc., (New York, NY). Dasatinib (BMS-354825, Sprycel™) was provided by Bristol-Meyers Squibb (New York, NY). EGF, HB-EGF, human amphiregulin (AR) and β-cellulin were purchased from R&D Systems (Minneapolis, MN). TAPI2 was purchased from Peptide International (Louisville, KY). All other chemicals were purchased from Sigma (St. Louis, MO).
Plasmid constructs and transfection
Wild-type human EGFR cDNA was subcloned into the NcoI/NotI restriction sites of the pCMV/myc/nuc vector to create the EGFR-NLS/myc construct (Invitrogen). This was then amplified via the polymerase chain reaction and subcloned into the pQCXIP retroviral vector (Clontech, Mountain View, CA). Virus was generated according to the manufacturer's recommendations. Infected cells were grown in RPMI1640 medium containing puromycin (2 μg/mL). Stable clones were selected and maintained in RPMI1640 with 0.5 μg/mL of puromycin.
Cell proliferation assay
Exponentially grown cells were seeded in 6-well plates. Following treatment, monolayers were washed with PBS and fixed/stained with 0.5% crystal violet. Plates were air dried overnight and dye was eluted with 0.1 M sodium citrate (pH 4.2) in ethanol (1:1). Elution was transferred to 96-well plates, and the absorbance was read at 540 nm to determine cell growth. The percentage cell growth was calculated by comparison of the A540 reading from treated versus control cells. The anti-proliferative effect of cetuximab (Figure 6B) was evaluated using MTT assay (12, 34(Wheeler et al., 2009)
Cellular Fractionation and Immunoblotting analysis
Whole cell protein lysate was obtained with lysis buffer (Wheeler et al., 2008). Cellular fractionation was performed as described previously (Hsu and Hung, 2007). Protein was quantitated using the Bradford method (Bio-Rad Laboratories, Hercules, CA). Western blotting was performed as described previously (Wheeler et al., 2008). The NIH ImageJ program was used to measure densitometry of the Western bands. The antibodies used in this study were as follows: EGFR, p-EGFR (Tyr1173), CyclinD1, B-Myb, HRP-conjugated goat-anti-rabbit IgG and goat-anti-mouse IgG were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA). PCNA, PARP and histone H3 were obtained from Cell Signaling Technology (Beverly, MA). Phospho-tyrosine (4G10) was purchased from Millipore (Billerica, MA), alpha-tubulin from Calbiochem (San Diego, CA), and c-Myc from Invitrogen.
Immunoprecipitation
Nuclear lysates containing 0.2 mg of protein were incubated overnight with 2 μg of anti-Myc antibody. After adding 30 μl of protein A/G agarose beads, nuclear lysates were incubated for another 2 hours at 4°C. The immunoprecipitates were pelleted by centrifugation and washed three times with lysis buffer and three times with PBS. The captured immunocomplexes were then eluted by boiling the beads in 2× SDS sample buffer for 5 minutes and subjected to immunoblot analysis.
Flow cytometric analysis
Cells were washed in PBS and harvested with trypsin-EDTA (0.05% solution, Gibco, Carlsbad, CA). A cell suspension containing 1 × 106 cells in 100 μL staining buffer (SB-PBS containing 1% BSA) was incubated with 2 μg of either control IgG or anti-EGFR FITC-conjugated antibodies (Santa Cruz Biotechnology Inc.) for 1h at 4°C. Cells were washed and resuspended in 300μl of SB. Propidium iodide (PI) at final concentration of 5 μg/ml was added to each sample just prior to analysis on the cytometer. Samples were analyzed on a FACSCalibur flow cytometer (BD biosciences, San Jose, CA) and a minimum of 20,000 events per sample was acquired. Histogram analysis was performed with FlowJo software (Tree Star, Inc., Ashland, OR). Data was restricted to live events based on PI exclusion.
Immunofluorescence assay
Approximately 2 × 103 cells were seeded on a four-well glass chamber slide (Nalgene Nunc, Naperville, IL) overnight. Forty-eight hours later, cells were washed 3 times with PBS and fixed with 2% formaldehyde for 15 minutes at room temperature. Cells were incubated in ice-cold 100% methanol for 10 minutes at -20°C and blocked with 5% normal serum in PBS with 0.3% Triton X100 solution for 1 hour at room temperature and incubated with anti-EGFR antibody overnight at 4°C. Next, cells were incubated with FITC-conjugated appropriate secondary antibody in PBS containing 0.3% Triton X100 and 1% BSA for 2 hours. Slides were mounted using ProLong gold with DAPI (Invitrogen). Fluorescence microscopy, photography and measurement intensity were carried out using BD Pathway 855 (BD Biosciences).
Real-time quantitative PCR analysis
Total RNA was isolated using the TRIzol reagent (Life Technologies, Gaithersburg, MD). cDNAs from total RNA of HP, HC1, HC4 and HC8 were synthesized using Superscript III First-Strand Synthesis System (Invitrogen Corp, Carlsbad, CA). Real-time PCR analysis was performed using a Bio-Rad iQ5 Real-Time PCR Detection System (Bio-Rad Laboratories, Hercules, CA) using the iQ Supermix as recommended by manufacturer. All reactions were performed in triplicate. The sequences of the primer sets used for this analysis are as follows: EGF (Fw) 5′-AGCTAACCCATTATGGCAACA-3′, (Rv) 5′-AGTTTTCACTGAGTCAGCTCCAT-3′, and (Probe) 5′-FAM-AGGGCCCTGGACCCACCAC-TAMRA-p-3′; amphiregulin (AR) (Fw) 5′-ATATCACATTGGAGTCACTGCCCA-3′, (Rv) 5′-GGGTCCATTGTCTTATGATCCAC-3′, and (Probe) 5′-FAM-AGCCATAAATGATGAGTCGGTCCTCTTTCC-TAMIRA-p-3′; heparin-binding EGF (HB-EGF) (Fw) 5′-GAAAGACTTCCATCTAGTCACAAAGA-3′, (Rv) 5′-GGGAGGCCCAATCCTAGA-3′, and (Probe) 5′-FAM-TCCTTCGTCCCCAGTTGCCG-TAMRA-p-3′; beta-cellulin (BTC) (Fw) 5′-TGCCCCAAGCAATACAAGC-3′, (Rv) 5′-CGTCTGCTCGGCCACC-3′, and (Probe) 5′-FAM-AAGCGGCATCTCCCTTTGATGCAGTAA-TAMRA-p-3′. Fold increases or decreases in gene expression were determined by quantitation of cDNA from target samples (HC1, HC4 and HC8) relative to a calibrator sample (HP). Human beta-actin gene was used as the endogenous control for normalization of initial RNA levels. To determine this normalized value, 2-ΔΔCT values were compared between target and calibrator samples, where the change in crossing threshold (ΔCt)= Ct target gene − Ct GAPDH and ΔΔCt = ΔCt HP − ΔCt HC1, HC4 or HC8.
Mouse xenograft model
Athymic nude mice (4 to 6-week-old males) were obtained from the Harlan laboratories (Indianapolis, IN). All animal procedures and maintenance were conducted in accordance with the institutional guidelines of the University of Wisconsin. Cetuximab-sensitive parental cells (HP) vector only control, or CMV-EGFR-NLS/Myc clones (C4, C5 and C10) were injected bilaterally in the dorsal flanks of the mice at day 0 (2 × 106 cells). Once tumors reached volumes 120-180 mm3 eight mice per group were randomized to IgG or cetuximab. Mice received 0.1 mg intraperitoneal cetuximab or IgG injections twice per week. Tumor volume measurements were evaluated by digital calipers and calculated by the formula (π)/6 × (large diameter) × (small diameter)2.
Statistical analysis
Student t-test was used to evaluate the significance of change in percent growth and EGFR cell surface expression. Statistical analysis to compare tumor sizes in xenograft-bearing mice was also done with a t-test. Differences between clones were considered statistically significant if P≤0.05.
Acknowledgments
We would like to thank Shyhmin Huang, Eric A. Armstrong, and Sergio Benavente for their initial work in the establishment of cetuximab-resistant H226 cell lines. Cetuximab and dasatinib were kindly provided by ImClone and Bristol Myers Squibb, respectively.
The abbreviations used are
- AR
amphiregulin
- cEGFR
cytoplasmic epidermal growth factor receptor
- DMSO
dimethyl sulfoxide
- EGF
epidermal growth factor
- EGFR
epidermal growth factor receptor
- HB-EGF
heparin-binding epidermal growth factor
- HNSCC
head and neck squamous cell carcinoma
- IF
immunofluorescence
- mCRC
metastatic colorectal cancer
- mAbs
monoclonal antibodies
- nEGFR
nuclear epidermal growth factor receptor
- NLS
nuclear localization sequence
- NSCLC
non-small cell lung cancer
- PCNA
proliferating cell nuclear antigen p-Tyr, phospho-tyrosine
- RTK
receptor tyrosine kinase
- SCC
squamous cell carcinoma
- SFK
Src-family kinases
- TACE
tumor necrosis factor alpha converting enzyme
- TGFα
transforming growth factor alpha
- TKD
tyrosine kinase domain
- TKI
tyrosine kinase inhibitor
Footnotes
Conflict of interest: The authors declare no conflict of interest.
References
- Arteaga CL. EGF receptor as a therapeutic target: patient selection and mechanisms of resistance to receptor-targeted drugs. J Clin Oncol. 2003;21:289s–291s. doi: 10.1200/JCO.2003.10.523. [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay D, Mandal M, Adam L, Mendelsohn J, Kumar R. Physical interaction between epidermal growth factor receptor and DNA-dependent protein kinase in mammalian cells. J Biol Chem. 1998;273:1568–73. doi: 10.1074/jbc.273.3.1568. [DOI] [PubMed] [Google Scholar]
- Bianco R, Troiani T, Tortora G, Ciardiello F. Intrinsic and acquired resistance to EGFR inhibitors in human cancer therapy. Endocr Relat Cancer. 2005;12:S159–171. doi: 10.1677/erc.1.00999. [DOI] [PubMed] [Google Scholar]
- Borrell-Pagès M, Rojo F, Albanell J, Baselga J, Arribas J. TACE is required for the activation of the EGFR by TGF-alpha in tumors. EMBO J. 2003;22:1114–24. doi: 10.1093/emboj/cdg111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camp ER, Summy J, Bauer TW, Liu W, Gallick GE, Ellis LM. Molecular Mechanisms of Resistance to Therapies Targeting the Epidermal Growth Factor Receptor. Clin Cancer Res. 2005;11:397–405. [PubMed] [Google Scholar]
- Cao H, Lei ZM, Bian L, Rao CV. Functional nuclear epidermal growth factor receptors in human choriocarcinoma JEG-3 cells and normal human placenta. Endocrinology. 1995;136:3163–72. doi: 10.1210/endo.136.7.7540549. [DOI] [PubMed] [Google Scholar]
- Dittmann K, Mayer C, Fehrenbacher B, Schaller M, Raju U, Milas L, et al. Radiation-induced epidermal growth factor receptor nuclear import is linked to activation of DNA-dependent protein kinase. J Biol Chem. 2005;280:31182–9. doi: 10.1074/jbc.M506591200. [DOI] [PubMed] [Google Scholar]
- Friedmann BJ, Caplin M, Savic B, Shah T, Lord CJ, Ashworth A, et al. Interaction of the epidermal growth factor receptor and the DNA-dependent protein kinase pathway following gefitinib treatment. Mol Cancer Ther. 2006;5:209–18. doi: 10.1158/1535-7163.MCT-05-0239. [DOI] [PubMed] [Google Scholar]
- Giri DK, Ali-Seyed M, Li LY, Lee DF, Ling P, Bartholomeusz G, et al. Endosomal transport of ErbB-2: mechanism for nuclear entry of the cell surface receptor. Mol Cell Biol. 2005;25:11005–18. doi: 10.1128/MCB.25.24.11005-11018.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta AK, McKenna WG, Weber CN, Feldman MD, Goldsmith JD, Mick R, et al. Local recurrence in head and neck cancer: Relationship to radiation resistance and signal transduction. Clinical Cancer Research. 2002;8:885–892. [PubMed] [Google Scholar]
- Hanada N, Lo HW, Day CP, Pan Y, Nakajima Y, Hung MC. Co-regulation of B-Myb expression by E2F1 and EGF receptor. Mol Carcinog. 2006;45:10–7. doi: 10.1002/mc.20147. [DOI] [PubMed] [Google Scholar]
- Hsu SC, Hung MC. Characterization of a novel tripartite nuclear localization sequence in the EGFR family. J Biol Chem. 2007;282:10432–40. doi: 10.1074/jbc.M610014200. [DOI] [PubMed] [Google Scholar]
- Hung LY, Tseng JT, Lee YC, Xia W, Wang YN, Wu ML, et al. Nuclear epidermal growth factor receptor (EGFR) interacts with signal transducer and activator of transcription 5 (STAT5) in activating Aurora-A gene expression. Nucleic Acids Res. 2008;36:4337–51. doi: 10.1093/nar/gkn417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi S, Boggon TJ, Dayaram T, Janne PA, Kocher O, Meyerson M, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352:786–92. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- Liao HJ, Carpenter G. Role of the Sec61 translocon in EGF receptor trafficking to the nucleus and gene expression. Mol Biol Cell. 2007;18:1064–72. doi: 10.1091/mbc.E06-09-0802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SY, Makino K, Xia W, Matin A, Wen Y, Kwong KY, et al. Nuclear localization of EGF receptor and its potential new role as a transcription factor. Nat Cell Biol. 2001;3:802–8. doi: 10.1038/ncb0901-802. [DOI] [PubMed] [Google Scholar]
- Lo HW, Ali-Seyed M, Wu Y, Bartholomeusz G, Hsu SC, Hung MC. Nuclear-cytoplasmic transport of EGFR involves receptor endocytosis, importin beta1 and CRM1. J Cell Biochem. 2006a;98:1570–83. doi: 10.1002/jcb.20876. [DOI] [PubMed] [Google Scholar]
- Lo HW, Hsu SC, Ali-Seyed M, Gunduz M, Xia W, Wei Y, et al. Nuclear interaction of EGFR and STAT3 in the activation of the iNOS/NO pathway. Cancer Cell. 2005a;7:575–89. doi: 10.1016/j.ccr.2005.05.007. [DOI] [PubMed] [Google Scholar]
- Lo HW, Hsu SC, Hung MC. EGFR signaling pathway in breast cancers: from traditional signal transduction to direct nuclear translocalization. Breast Cancer Res Treat. 2006b;95:211–8. doi: 10.1007/s10549-005-9011-0. [DOI] [PubMed] [Google Scholar]
- Lo HW, Hung MC. Nuclear EGFR signalling network in cancers: linking EGFR pathway to cell cycle progression, nitric oxide pathway and patient survival. Br J Cancer. 2006;94:184–8. doi: 10.1038/sj.bjc.6602941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo HW, Xia W, Wei Y, Ali-Seyed M, Huang SF, Hung MC. Novel prognostic value of nuclear epidermal growth factor receptor in breast cancer. Cancer Res. 2005b;65:338–48. [PubMed] [Google Scholar]
- Lu Y, Li X, Liang K, Luwor R, Siddik ZH, Mills GB, et al. Epidermal growth factor receptor (EGFR) ubiquitination as a mechanism of acquired resistance escaping treatment by the anti-EGFR monoclonal antibody cetuximab. Cancer Res. 2007;67:8240–7. doi: 10.1158/0008-5472.CAN-07-0589. [DOI] [PubMed] [Google Scholar]
- Marti U, Burwen SJ, Wells A, Barker ME, Huling S, Feren AM, et al. Localization of epidermal growth factor receptor in hepatocyte nuclei. Hepatology. 1991;13:15–20. [PubMed] [Google Scholar]
- Massie C, Mills IG. The developing role of receptors and adaptors. Nat Rev Cancer. 2006;6:403–9. doi: 10.1038/nrc1882. [DOI] [PubMed] [Google Scholar]
- Mukohara T, Engelman JA, Hanna NH, Yeap BY, Kobayashi S, Lindeman N, et al. Differential effects of gefitinib and cetuximab on non-small-cell lung cancers bearing epidermal growth factor receptor mutations. J Natl Cancer Inst. 2005;97:1185–94. doi: 10.1093/jnci/dji238. [DOI] [PubMed] [Google Scholar]
- Ni CY, Murphy MP, Golde TE, Carpenter G. gamma -Secretase cleavage and nuclear localization of ErbB-4 receptor tyrosine kinase. Science. 2001;294:2179–81. doi: 10.1126/science.1065412. [DOI] [PubMed] [Google Scholar]
- Offterdinger M, Schofer C, Weipoltshammer K, Grunt TW. c-erbB-3: a nuclear protein in mammary epithelial cells. J Cell Biol. 2002;157:929–39. doi: 10.1083/jcb.200109033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, et al. Acquired Resistance of Lung Adenocarcinomas to Gefitinib or Erlotinib Is Associated with a Second Mutation in the EGFR Kinase Domain. PLoS Med. 2005;2:1–11. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Psyrri A, Egleston B, Weinberger P, Yu Z, Kowalski D, Sasaki C, et al. Correlates and determinants of nuclear epidermal growth factor receptor content in an oropharyngeal cancer tissue microarray. Cancer Epidemiol Biomarkers Prev. 2008;17:1486–92. doi: 10.1158/1055-9965.EPI-07-2684. [DOI] [PubMed] [Google Scholar]
- Psyrri A, Yu Z, Weinberger PM, Sasaki C, Haffty B, Camp R, et al. Quantitative determination of nuclear and cytoplasmic epidermal growth factor receptor expression in oropharyngeal squamous cell cancer by using automated quantitative analysis. Clin Cancer Res. 2005;11:5856–62. doi: 10.1158/1078-0432.CCR-05-0420. [DOI] [PubMed] [Google Scholar]
- Rajput A, Koterba AP, Kreisberg JI, Foster JM, Willson JK, Brattain MG. A novel mechanism of resistance to epidermal growth factor receptor antagonism in vivo. Cancer Res. 2007;67:665–73. doi: 10.1158/0008-5472.CAN-06-2773. [DOI] [PubMed] [Google Scholar]
- Ritter CA, Perez-Torres M, Rinehart C, Guix M, Dugger T, Engelman JA, et al. Human Breast Cancer Cells Selected for Resistance to Trastuzumab In vivo Overexpress Epidermal Growth Factor Receptor and ErbB Ligands and Remain Dependent on the ErbB Receptor Network. Clin Cancer Res. 2007;13:4909–4919. doi: 10.1158/1078-0432.CCR-07-0701. [DOI] [PubMed] [Google Scholar]
- Sunnarborg S. Tumor Necrosis Factor-alpha Converting Enzyme (TACE) Regulates Epidermal Growth Factor Receptor Ligand Availability. Journal of Biological Chemistry. 2002;277:12838–12845. doi: 10.1074/jbc.M112050200. [DOI] [PubMed] [Google Scholar]
- Sunnarborg SW, Hinkle CL, Stevenson M, Russell WE, Raska CS, Peschon JJ, et al. Tumor necrosis factor-alpha converting enzyme (TACE) regulates epidermal growth factor receptor ligand availability. J Biol Chem. 2002;277:12838–45. doi: 10.1074/jbc.M112050200. [DOI] [PubMed] [Google Scholar]
- Viloria-Petit A, Crombet T, Jothy S, Hicklin D, Bohlen P, Schlaeppi JM, et al. Acquired resistance to the antitumor effect of epidermal growth factor receptor-blocking antibodies in vivo: a role for altered tumor angiogenesis. Cancer Res. 2001;61:5090–101. [PubMed] [Google Scholar]
- Viloria-Petit AM, Kerbel RS. Acquired resistance to EGFR inhibitors: mechanisms and prevention strategies. International Journal of Radiation Oncology, Biology, Physics. 2004;58:914–26. doi: 10.1016/j.ijrobp.2003.09.091. [DOI] [PubMed] [Google Scholar]
- Wang SC, Lien HC, Xia W, Chen IF, Lo HW, Wang Z, et al. Binding at and transactivation of the COX-2 promoter by nuclear tyrosine kinase receptor ErbB-2. Cancer Cell. 2004;6:251–61. doi: 10.1016/j.ccr.2004.07.012. [DOI] [PubMed] [Google Scholar]
- Wang SC, Nakajima Y, Yu YL, Xia W, Chen CT, Yang CC, et al. Tyrosine phosphorylation controls PCNA function through protein stability. Nat Cell Biol. 2006;8:1359–68. doi: 10.1038/ncb1501. [DOI] [PubMed] [Google Scholar]
- Wheeler DL, Huang S, Kruser TJ, Nechrebecki MM, Armstrong EA, Benavente S, et al. Mechanisms of acquired resistance to cetuximab: role of HER (ErbB) family members. Oncogene. 2008;27:3944–56. doi: 10.1038/onc.2008.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler DL, Iida M, Kruser TJ, Nechrebecki MM, Dunn EF, Armstrong EA, et al. Epidermal growth factor receptor cooperates with Src family kinases in acquired resistance to cetuximab. Cancer Biol Ther. 2009;8:696–703. doi: 10.4161/cbt.8.8.7903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia W, Wei Y, Du Y, Liu J, Chang B, Yu YL, et al. Nuclear expression of epidermal growth factor receptor is a novel prognostic value in patients with ovarian cancer. Mol Carcinog. 2008 doi: 10.1002/mc.20504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, Hung MC. Nuclear localization of p185neu tyrosine kinase and its association with transcriptional transactivation. Biochem Biophys Res Commun. 1994;203:1589–98. doi: 10.1006/bbrc.1994.2368. [DOI] [PubMed] [Google Scholar]
- Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2:127–37. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]