

Abstract
Background
Small cell lung cancer (SCLC) is an aggressive malignancy. Current treatments yield dismal survival rates. We have previously demonstrated that histone deacetylase (HDAC) inhibitors can inhibit neuroendocrine tumor growth. Activation of the Notch1 signaling pathway also impairs SCLC cell viability. In this study, we investigated the ability of the HDAC inhibitor valproic acid (VPA) to activate Notch1 signaling and inhibit proliferation in SCLC cells.
Materials and Methods
DMS53 human SCLC cells were treated with VPA (0–10 mM) for two days. Light microscopy was used to examine changes in cell morphology. Western analysis was performed using antibodies against various Notch1 pathway proteins to assess Notch1 activation. Additionally, immunoblotting was performed for two neuroendocrine tumor markers, chromogranin A and achaete-scute complex-like 1 (ASCL-1). Finally, a cell proliferation assay was used to measure the effects of VPA on SCLC growth over eight days.
Results
After treatment with VPA, DMS53 cells underwent dramatic changes in morphology. VPA induced expression of the full-length and active forms of Notch1 protein. Furthermore, VPA suppressed levels of neuroendocrine tumor markers chromogranin A and ASLC-1. Importantly, VPA treatment led to dose-dependent inhibition of SCLC cell proliferation.
Conclusions
The HDAC inhibitor VPA activates Notch1 signaling in SCLC cells. VPA induces changes in cell morphology and suppresses neuroendocrine tumor markers, indicating a change in phenotype. Additionally, VPA profoundly inhibits SCLC cell growth. These results suggest that VPA has potential as a novel therapeutic agent for SCLC.
Keywords: Valproic acid, VPA, histone deacetylase inhibitors, small cell lung cancer, neuroendocrine tumors, Notch1, achaete-scute homolog-1, ASCL-1
Introduction
Lung cancer has the highest mortality of all cancers in the United States (1,2). In 2007, an estimated 213,380 Americans will be diagnosed with lung cancer and 160,390 will die of the disease (1). Small cell lung cancer (SCLC) accounts for approximately 20% of all lung cancer cases (3–6) and is characterized by an aggressive course with early metastasis (3–8). Without treatment, the median survival time with the disease is only 2–3 months (3). There are currently few methods for early detection, and most patients present with symptomatic, late-stage disease (1,3). Upon diagnosis, over 90% of patients with SCLC have metastases to regional lymph nodes or other distant sites, making complete surgical resection possible in less than 10% of cases (7). Treatment of SCLC typically involves an intense regimen of chemotherapy with or without radiotherapy (2,3,7,9). Unfortunately, current treatments yield a dismal 5-year survival rate of only 5–10% (3,9). Clearly, there is a need for novel therapeutic approaches to this disease (8,10).
The Notch1 signaling pathway plays a critical role in the normal embryonic development of the lung and the disseminated neuroendocrine (NE) cell system (5,11,12). Notch1 is a transmembrane receptor which is activated upon ligand binding by a series of proteolytic cleavage events. Once cleaved, the Notch1 intracellular domain (NICD) translocates into the nucleus, where it forms a DNA binding complex and alters transcription of target genes. Notch1 activation then increases expression of hairy-enhancer of split-1 (Hes-1) which in turn down-regulates achaete-scute homolog-1 (ASCL-1) (5,11,12).
Abnormal Notch1 signaling has been implicated in NE tumorigenesis. Notch1 signaling is suppressed in NE tumor (NET) cells, including SCLC cells (5,6,11–14). Expression of exogenous Notch1 resulted in suppression of NET hormone production and inhibition of NET cell growth (11,12), suggesting that Notch1 induction was an attractive strategy for the treatment of these tumors. Until recently, however, there were no known small molecule activators of the Notch1 pathway.
Histone deacetylase (HDAC) inhibitors are a class of molecules that modify chromatin structure and regulate gene transcription and expression (15). HDAC inhibitors have been shown to cause growth inhibition in several malignant cell lines, including SCLC (16,17). Valproic acid (VPA) is an HDAC inhibitor that has been used for decades in the treatment of patients with epilepsy and other neuropsychiatric disorders (18). As the safety profile of VPA is well-established, this HDAC inhibitor is an attractive candidate for development as an anti-cancer agent. We have previously shown that Notch1 signaling is minimal or absent at baseline in several NET cell lines, and that expression of exogenous Notch1 via an inducible construct inhibits NET cell growth (6,14). Additionally, VPA has been reported to activate Notch1 signaling in neuroblastoma, carcinoid, and medullary thyroid cancer cells (13,19,20). We hypothesized, then, that VPA may also activate Notch1 signaling in SCLC cells with subsequent anti-tumor effects. To test this hypothesis we treated human SCLC cells with VPA, and analyzed the effects on Notch1 signaling, cellular morphology, expression of NET markers, and cancer cell proliferation.
Materials and Methods
Cell Culture
DMS53 human SCLC cells were obtained from American Type Culture Collection (Manassas, VA) and maintained in Waymouth’s MB752/1 medium (Invitrogen, San Diego, CA), supplemented with 10% fetal bovine serum (Sigma-Aldrich, St. Louis, MO), 100 IU/ml penicillin and 100 μg/ml streptomycin (Invitrogen). The cells were maintained in a humidified atmosphere of 5% CO2 in air at 37° C.
VPA Treatment
DMS53 cells were plated at 50% to 60% confluence in 100-mm cell-culture dishes and incubated overnight. On the following day, cells were treated with VPA (2-propylpentanoic acid; Sigma-Aldrich) for 2 d in doses ranging from 1 to 10 mM. DMSO (0.1%), the solvent used for VPA, was used for control treatments.
Light Microscopy
After treatment with VPA for 2 d, DMS53 cells were examined under light microscopy for changes in cell growth and morphology. Photographs were taken using AxioVision40AC V4.5.0.0 (Carl Zeiss MicroImaging, Oberkochen, Germany) prior to protein isolation for Western blotting.
Western Blotting
Cellular extracts were prepared and quantified with a bicinchoninic (BCA) protein assay kit (Pierce Biotechnology, Rockford, IL) as previously described (14,21). Denatured cellular extracts were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to nitrocellulose membranes (Schleicher and Schuell, Keene, NH). Membranes were blocked 30–60 min in milk solution (1X PBS, 5% nonfat dry milk, 0.05% Tween 20) and incubated at 4° C overnight with primary antibodies. The following primary antibody dilutions were used: 1:1,000 for Notch1 (Santa Cruz Biotechnology, Santa Cruz, CA), Hes-1 (Santa Cruz), mammalian ASCL-1 (BD Biosciences San Diego, CA), and CgA (Zymed Laboratories, San Francisco, CA); and 1:10,000 for G3PDH (Trevigen, Gaithersburg, MD). After primary antibody incubation, membranes were washed either 3 X 5 min or 3 X 10 min in wash buffer (1x PBS, 0.05% Tween-20). The membranes were then incubated with 1:2,000 dilution of horseradish peroxidase conjugated with either goat anti-rabbit or goat anti-mouse secondary antibody (Cell Signaling Technology, Danvers, MA), depending on the source of the primary antibody, at room temperature for 1 h. The membranes were then washed 3 X 5 min or 3 X 10 min in 1X PBS wash buffer and developed by Super Signal West Femto chemiluminescence substrate (Pierce Biotechnology) or Immun-Star (Bio-Rad Laboratories, Hercules, CA) depending on the primary antibody. Protein levels were quantified from the developed films with ImageQuant 5.2 (GE Healthcare Lifesciences, Buckinghamshire, United Kingdom).
MTT Cell Proliferation Assay
Proliferation of DMS53 cells following treatment with control or VPA was measured using the MTT (methylthiazolyldiphenyl-tetrazolium bromide; Sigma-Aldrich) assay. The MTT reagent is a soluble tetrazolium salt that is converted to an insoluble formazan pigment in active, viable cells by the enzymatic activity of a mitochondrial dehydrogenase. The quantity of formazan pigment can then be measured via spectrophotometry (22,23). DMS53 cells were trypsinized and plated in triplicate in 24-well plates and allowed to adhere overnight. Cells were then treated with varying concentrations of VPA or control (0.1% DMSO) for 8 d. Cells were given fresh media containing control or VPA every 2 d. At each time point, media were removed and replaced with 250 μL of medium containing MTT and incubated for 3 h. Then, 750 μL DMSO was added to each well and the plates were shaken at room temperature for 5 min to dissolve the cell membranes. Absorbance was determined using a spectrophotometer (μQuant; Bio-Tek Instruments, Winooski, VT) at a wavelength of 540 nm. Experiments were performed in triplicate at least twice.
Statistical Analysis
Statistical analyses were preformed using the SPSS statistical package (SPSS 10.0, SPSS Inc., Chicago, IL). Analysis of variance (ANOVA) was used for the MTT growth assay and quantified Western blot data. P-values less than 0.05 were considered statistically significant.
Results
VPA Induces Notch1 Signaling in SCLC Cells
Given that VPA has been reported to activate Notch1 signaling in several NET cell lines (13,20), we were interested in determining the ability of VPA to activate Notch1 signaling in SCLC cells. To assess this, we measured protein levels of full-length Notch1 and NICD after treatment with various concentrations of VPA. Notch1 proteins were not detectable in untreated cells. However, VPA treatment resulted in a dose-dependent induction of both full-length Notch1 and NICD (Figure 1). Hes-1 is a well-established target of Notch1 signaling and is positively regulated by Notch1 (5,11,12). Treatment of SCLC cells with VPA in concentrations as low as 1mM led to increased cellular levels of Hes-1 (Figure 1). These results indicate that VPA induces Notch1 signaling in SCLC cells.
FIG 1.
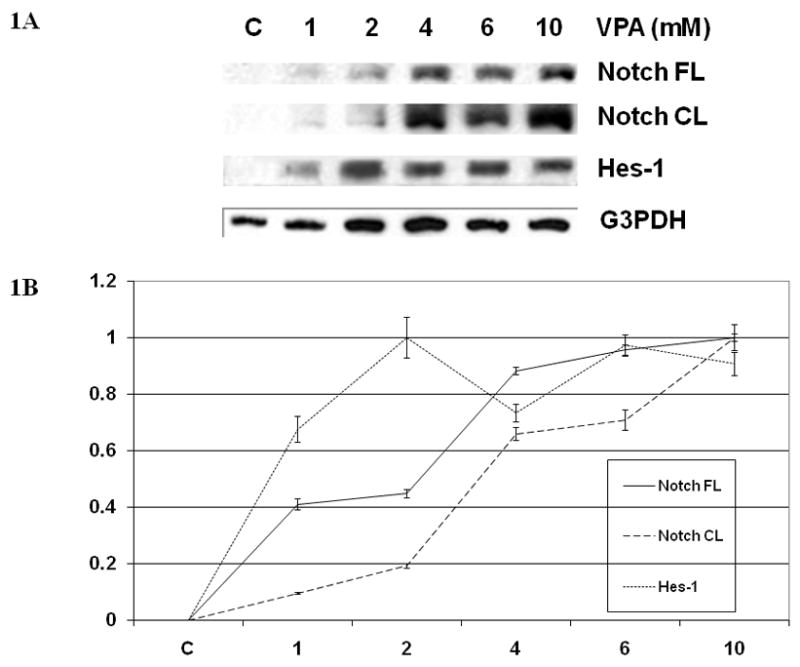
(A) VPA Induces Notch1 Signaling in SCLC Cells. Western blots of cell extracts from DMS53 cells treated with control (DMSO 0.1%) or VPA (1 to 10 mM) for 2 d showed increased expression of full-length Notch1 and the cleaved, active Notch intracellular domain (NICD). Levels of Hes-1, a downstream target of Notch1, were also elevated from baseline with VPA treatments. Equal protein loading was confirmed with antibodies against G3PDH. (B) After quantification with image analysis software, there were statistically significant differences between control and treatment groups with full-length Notch, NICD, and Hes-1 expression (p-value <0.001).
VPA Induces Morphologic Differentiation in SCLC Cells
We next examined SCLC cells under light microscopy to evaluate for changes in cellular appearance after treatment with VPA. Cells cultured in control media grew in clumps and sheets with poorly defined cellular borders (Figure 2A). There were indefinite points of cell-to-cell contact between tumor cells in the control plates, representing typical uninhibited tumor cell growth. Cells treated with increasing concentrations of VPA showed marked morphologic changes and reduced cell density. At VPA concentrations as low as 4 mM (Figure 2B), cells had clearly defined cellular margins and a characteristic spindle shaped morphology with neuronal-like projections suggestive of differentiation. Distinctly noticeable in the photomicrograph is the lack of cell-to-cell contact between the tumor cells, as was previously seen in the control plates. When VPA concentrations were increased to10 mM (Figure 2C), there was a further decrease in cell viability and further decreases in points of cell-to-cell contact. With increasing concentrations of VPA, the cells became more differentiated in appearance, assuming a spindle-shaped morphology with increases in the size of gaps between the once confluent tumor cells.
FIG 2.
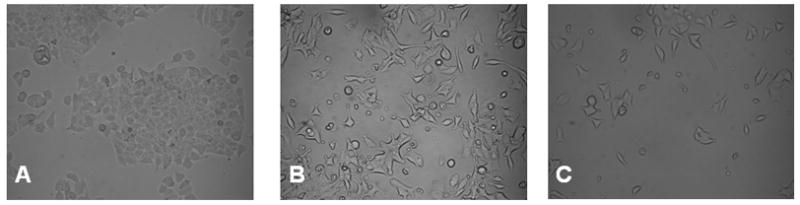
VPA Decreases Growth and Induces Morphologic Differentiation in SCLC Cells. DMS53 cells were treated with VPA or control (DMSO 0.1%) for 2 d and examined with light microscopy under 20x magnification. Control plates (A) grew in confluent sheets with ill-defined cellular borders. Cells treated with VPA in 4 mM (B) or 10 mM (C) concentrations progressively assumed a flatter, rounder, and spindle shaped conformation with neuronal-like projections, decreased points of cell-to-cell contact, and more distinct cellular borders.
VPA suppresses production of NE tumor markers in SCLC cells
The observed changes in SCLC cellular morphology after VPA treatment suggested an alteration in phenotype. Previous studies utilizing an inducible Notch1 construct in NET cells, including SCLC, have demonstrated that Notch1 induction leads to a decrease in NET markers such as ASCL-1 and chromogranin (CgA) (5,11–13). We performed Western blot analysis to assess whether VPA treatment would reproduce these effects. As shown in Figure 3, high levels of the NET marker ASCL-1 were present in SCLC cells at baseline. Treatment with increasing concentrations of VPA suppressed ASCL-1 levels in a dose-dependant manner, with complete suppression of ASCL-1 at the 10 mM concentration.
FIG 3.
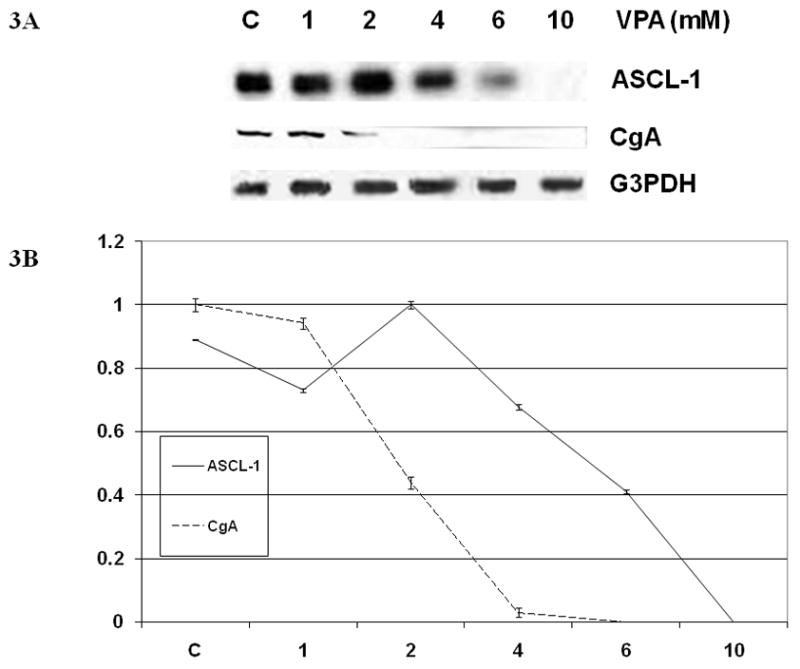
(A) Induction of Notch1 Signaling by VPA Regulates Levels of the NET Markers ASCL-1 and CgA. Western analysis of cell extracts from DMS53 cells treated with control (DMSO 0.1%) or VPA (1 to 10 mM) for 2 d show decreases in NET markers ASCL-1 and CgA. Equal protein loading was confirmed with G3PDH. (B) After quantification with image analysis software, there were statistically significant differences between control and treatment groups with ASCL-1 and CgA expression (p-value <0.001).
Similar results were seen with CgA, a NET marker that is clinically used in the diagnosis and monitoring of NETs. Significant quantities of CgA were observed in control cells. However, VPA treatment in concentrations of 2 and 4 mM led to a significant decrease in CgA levels, and the tumor marker was undetectable after the cells were exposed to 6 and 10 mM concentrations of VPA (Figure 3).
VPA Inhibits Proliferation of SCLC Cells
After demonstrating that VPA activates Notch1 signaling in SCLC cells leading to changes in cellular morphology and suppression of NET markers, we were interested in objectively measuring the effect of VPA treatment on SCLC cell growth. VPA has been shown to inhibit growth in several tumor cell lines (13,19,20,24). To date, the effect of VPA treatment on SCLC cell growth has not been characterized. In order to assess the impact of VPA on SCLC cells, we utilized the MTT cell proliferation assay, a measure cell mitochondrial activity. Treatment of SCLC cells with VPA concentrations as low as 1 mM significantly impaired cell mitochondrial activity (Figure 4). Higher concentrations of VPA actually resulted in a decrease in activity compared to baseline, suggesting arrest of cell proliferation and possible cell death.
FIG 4.
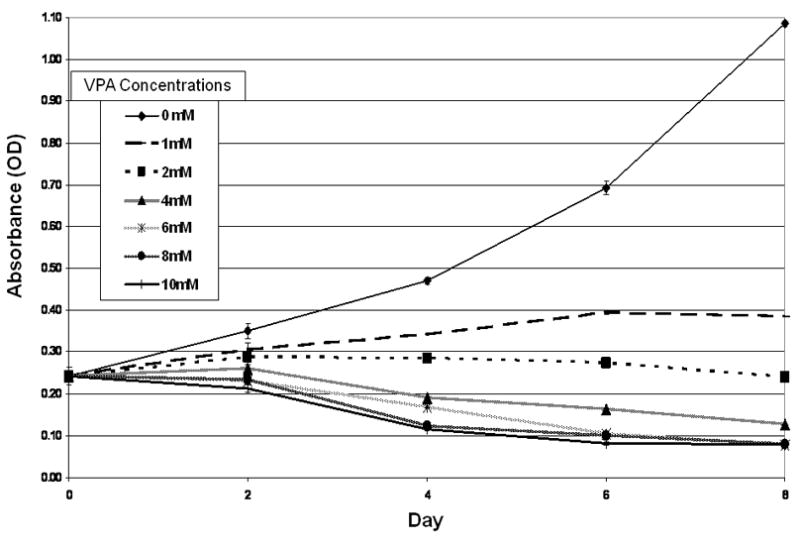
VPA Inhibits Proliferation of SCLC Cells. DMS53 cells were treated with control (DMSO) or varying doses of VPA (1 to 10 mM) for up to 8 d and cell proliferation was measured by the MTT assay. VPA inhibited cell proliferation in a dose-dependent and statistically-significant manner.
Discussion
Lung cancer is the leading cause of cancer deaths worldwide, with nearly one million deaths occurring each year (2,3,25). Lung cancer by itself accounts for more deaths than colorectal, breast, and prostate cancers combined (4). SCLC is the most aggressive type of lung cancer, and is characterized by rapid spread to regional lymph nodes as well as other distant sites of metastasis (3–8). Current treatment regimens yield unsatisfactory results, and new treatment options are sorely needed (8,10).
Notch1 signaling has been identified as a potential therapeutic target for SCLC. The Notch1 signaling pathway is silenced in many NE malignancies, including SCLC (5,6,11–14). Induction of Notch1 with an inducible construct led to inhibition of tumor cell growth and NET marker levels. This suggested that activation of Notch1 signaling could have anti-tumor effects in patients with SCLC. Until recently, however, no small molecule activators of Notch1 signaling were available.
In 2005, Stockhausen and colleagues reported the ability of VPA, a branched-chain fatty acid that has long been used in the treatment of patients with epilepsy, to activate Notch1 signaling in neuroblastoma cells (19). We subsequently treated human carcinoid tumor and medullary thyroid cancer cells with VPA, and found that Notch1 was also activated in these cell lines, with a resultant decrease in cell proliferation and NET marker expression (13,20). VPA is an attractive potential anticancer drug for neuroendocrine tumors because it is approved by the U.S. Food and Drug Administration (FDA) and has a favorable toxicity profile compared to most cytotoxic chemotherapeutic agents.
VPA is a member of the HDAC inhibitor drug class. HDAC inhibitors antagonize histone deacetylase (HDAC) enzymes, which remove acetyl groups from DNA histone proteins. Inhibition of HDAC enzymes results in increased histone acetylation due to the unopposed activity of histone acetyl transferase, which leads to changes in chromatin structure and modification of transcriptional activity (26,27). In an earlier study we treated SCLC cells with trichostatin A (TSA), an HDAC inhibitor (16). TSA treatment resulted in inhibition of SCLC cell proliferation through both apoptosis and cell cycle arrest.
In the current study we assessed the effects of VPA treatment on SCLC cells. Previous research has demonstrated that Notch1 signaling is minimal or absent in SCLC (5,6,11,12). Consistent with these earlier observations, we found no detectable full-length or cleaved Notch1 protein by Western blot in untreated SCLC cells. As Notch1 is suppressed in SCLC, we hypothesized that activation of this signaling pathway may have antitumor effects.
VPA treatment of SCLC cells resulted in induction of Notch1. The exact mechanism by which VPA is able to affect Notch1 protein levels has yet to be described. Previous studies have demonstrated that although there is minimal Notch1 protein in NET cells at baseline, Notch1 mRNA is present (14,28). It is possible that VPA interferes with Notch1 protein degradation, which is mediated by proteins such as Sel-10, Itch, c-CBL, and Deltex (29). The mechanism by which VPA activates Notch1 signaling is a subject of ongoing investigation.
Previous experiments showed that expression of exogenous Notch1 in NET cells resulted in increased levels of Hes-1. Increased Hes-1 gene expression resulted in transcriptional repression of the ASCL-1 gene, thus decreasing cellular levels of ASCL-1 protein (5,6,10–12). Additionally, the presence of Notch1 protein itself has been correlated with increases in ASCL-1 protein ubiquitination and degradation (11). Thus, control of ASCL-1 protein levels in SCLC cells is regulated by both Hes-1 dependent and Hes-1 independent mechanisms. In the present study, Notch1 induction with VPA was associated with increased expression of Hes-1 and down-regulation of ASCL-1. Similar to ASCL-1, the NET marker CgA was also suppressed by VPA treatment. These changes in NET marker expression, combined with the observed alterations in cellular morphology, suggest that VPA exerts a profound effect on the NE phenotype of SCLC cells.
An important finding of the current study is that VPA causes inhibition of proliferation in SCLC cells. Histologic changes diagramed in the photomicrographs indicate decreased cell density and increased differentiation of the VPA treated cells when compared to control (DMSO). NETs such as SCLC have been shown to be less aggressive when increased histologic differentiation is noted (30,31). Growth inhibition was further supported by an MTT proliferation assay, where doses as low as 1 mM of VPA, after only 4 days of treatment, significantly inhibited cell growth. Higher concentrations of VPA and extended duration of treatments led to an even more dramatic suppression of cancer cell growth. VPA treatment led to a dose-dependent inhibition of mitochondrial activity as measured by the MTT assay, suggesting a decrease in cell viability or cell proliferation. It should be noted, however, that the MTT assay measures mitochondrial activity of the cell, and not the number of living cells (32). It is possible that VPA treatment induces cell cycle arrest or other metabolic changes specific to the mitochondria rather than causing overt cell death in SCLC. We hypothesize that a combination of the former mechanisms may be at work when SCLC is treated with VPA. Additional research is needed to further elucidate the mechanism by which VPA inhibits SCLC cell proliferation.
Furthermore, we have previously demonstrated in a mouse xenograft model of NET disease that non-toxic concentrations of VPA can activate Notch1 signaling and inhibit tumor growth (13). The mean blood level of VPA in treated mice was within the therapeutic range for human patients treated for epilepsy. The ability of VPA to activate Notch1 and inhibit NET proliferation both in vitro and in vivo is encouraging, and suggests that clinical trials are warranted.
In summary, the HDAC inhibitor VPA activates Notch1 signaling, suppresses NET markers, and inhibits growth in SCLC cells. As the safety profile of VPA in humans is well established, we conclude that VPA is a potential new therapeutic agent for SCLC, worthy of further investigation.
Acknowledgments
This study was supported by T35 training grant DK062709 from the National Institutes of Health. The authors thank Abram Vaccaro for his technical assistance.
Financial Support: American Cancer Society Research Scholars Grant 05-08301TBE; National Institutes of Health Grants T35-DK062709, R21-DK066169, and R01-CA109053; American College of Surgeons George H.A. Clowes Jr. Memorial Research Career Development Award; Vilas Foundation Research Grant; Carcinoid Cancer Foundation Research Grant; Association for Academic Surgery Karl Storz Endoscopy Research Grant; Doctors Cancer Foundation Award and the Society of Surgical Oncology Clinical Investigator Award.
References
- 1.American Cancer Society. Cancer Facts & Figures 2007. Atlanta: American Cancer Society; 2007. [Google Scholar]
- 2.Spiro SG, Silvestri GA. One hundred years of lung cancer. Am J Respir Crit Care Med. 2005;172:523–529. doi: 10.1164/rccm.200504-531OE. [DOI] [PubMed] [Google Scholar]
- 3.Spiro SG, Porter JC. Lung Cancer – Where are we today? Current advances in staging and nonsurgical treatment. Am J Respir Crit Care Med. 2002;166:1166–1196. doi: 10.1164/rccm.200202-070SO. [DOI] [PubMed] [Google Scholar]
- 4.Kunnimalaiyaan M, Chen H. The Raf-1 pathway: a molecular target for treatment of select neuroendocrine tumors? Anticancer Drugs. 2006;17:139–142. doi: 10.1097/00001813-200602000-00004. [DOI] [PubMed] [Google Scholar]
- 5.Ball DW. Achaete-scute homolog-1 and Notch in lung neuroendocrine development and cancer. Cancer Lett. 2004;204:159–169. doi: 10.1016/S0304-3835(03)00452-X. [DOI] [PubMed] [Google Scholar]
- 6.Kunnimalaiyaan M, Chen H. Tumor suppressor role of Notch1 signaling in neuroendocrine tumors. The Oncologist. 2007;12:535–542. doi: 10.1634/theoncologist.12-5-535. [DOI] [PubMed] [Google Scholar]
- 7.Lewiński T, Żuławki M, Czesław T, Pietraszek A. Small cell lung cancer I-III A: cytoreductive chemotherapy followed by resection with continuation of chemotherapy. Eur J Cardiothorac Surg. 2001;20:391–398. doi: 10.1016/s1010-7940(01)00787-4. [DOI] [PubMed] [Google Scholar]
- 8.Sattler M, Salgia R. Molecular and cellular biology of small cell lung cancer. Semin Oncol. 2003;30:57–71. doi: 10.1053/sonc.2003.50019. [DOI] [PubMed] [Google Scholar]
- 9.Greco FA, Saijo N. Current perspectives in the treatment of small cell lung cancer – Introduction. Semin Oncol. 2001;28:1–2. [Google Scholar]
- 10.Chen H, Thiagalingam A, Chopra H, Borges MW, Feder JN, Nelkin BD, Baylin SB, Ball DW. Convervation of the Drosophila lateral inhibition pathway in human lung cancer: a hairy-related protein (HES-1) directly represses achaete-scute homolog-1 expression. Proc Natl Acad Sci USA. 1997;94:5355–5360. doi: 10.1073/pnas.94.10.5355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sriuranpong V, Borges MW, Ravi RK, Arnold DR, Nelkin BD, Baylin SB, Ball DW. Notch signaling induces cell cycle arrest in small cell lung cancer cells. Cancer Res. 2001;61:3200–3205. [PubMed] [Google Scholar]
- 12.Sriuranpong V, Borges MW, Strock CL, Nakakura EK, Watkins DN, Blaumueller CM, Nelkin BD, Ball DW. Notch signaling induces rapid degradation of achaete-scute homolog 1. Mol Cell Biol. 2002;22:3129–3139. doi: 10.1128/MCB.22.9.3129-3139.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Greenblatt DY, Vaccaro AM, Jaskula-Sztul R, Ning L, Haymart M, Kunnimalaiyaan, Chen H. Valproic acid activates Notch-1 signaling and regulates the neuroendocrine phenotype in carcinoid cancer cells. Oncologist. 2007;8:942–51. doi: 10.1634/theoncologist.12-8-942. [DOI] [PubMed] [Google Scholar]
- 14.Kunnimalaiyaan M, Traeger K, Chen H. Conservation of the Notch1 signaling pathway in gastrointestinal carcinoid cells. Am J Gastrointest Liver Physiol. 2005;289:G636. doi: 10.1152/ajpgi.00146.2005. [DOI] [PubMed] [Google Scholar]
- 15.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 16.Platta CS, Greenblatt DY, Kunnimalaiyaan M, Chen H. The HDAC inhibitor Trichostatin A inhibits growth of small cell lung cancer cells. J Surg Res. 2007;142:219–226. doi: 10.1016/j.jss.2006.12.555. [DOI] [PubMed] [Google Scholar]
- 17.Vigushin DM, Coombes RC. Histone deacetylase inhibitors in cancer treatment. Anticancer drugs. 2002;12:1–13. doi: 10.1097/00001813-200201000-00001. [DOI] [PubMed] [Google Scholar]
- 18.Henry TR. The history of valproate in clinical neuroscience. Psychopharmacol Bull. 2003;37(suppl 2):5–16. [PubMed] [Google Scholar]
- 19.Stockhausen MT, Sjolund J, Manetopoulos C, Axelson H. Effects of the histone deacetylase inhibitor valproic acid on Notch signaling in human neuroblastoma cells. Br J Cancer. 2005;92:751–759. doi: 10.1038/sj.bjc.6602309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Greenblatt DY, Cayo MA, Adler JT, Ning L, Haymart M, Kunnimalaiyaan M, Chen H. Valproic acid activates Notch1 signaling and induces apoptosis in medullary thyroid cancer cells. Ann Surg. doi: 10.1097/SLA.0b013e3181758d0e. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sippel RS, Carpenter JE, Kunnimalaiyaan M, et al. Raf-1 activation suppresses neuroendocrine marker and hormone levels in human gastrointestinal carcinoid cells. Am J Physiol Gastrointest Liver Physiol. 2003;285:G245. doi: 10.1152/ajpgi.00420.2002. [DOI] [PubMed] [Google Scholar]
- 22.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 23.Gerlier D, Thomasset N. Use of MTT colorimetric assay to measure cell activation. J Immunol Methods. 1986;94:57–63. doi: 10.1016/0022-1759(86)90215-2. [DOI] [PubMed] [Google Scholar]
- 24.Catalano MG, Fortunati N, Pugliese M, Constantino L, Poli R, Bosco O, Boccuzzi G. Valproic acid induces apoptosis and cell cycle arrest in poorly differentiated thyroid cancer cells. J Clin Endocrinol Metab. 2005;90:1383–1389. doi: 10.1210/jc.2004-1355. [DOI] [PubMed] [Google Scholar]
- 25.Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55:74–108. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- 26.Liu T, Kuljaca S, Tee A, Marshall GM. Histone deacetylase inhibitors: Multifunctional anticancer agents. Cancer Treat Rev. 2006;32:157–165. doi: 10.1016/j.ctrv.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 27.Shao Y, Gao Z, Marks PA, Jiang X. Apoptotic and autophagic cell death induced by histone deacetylase inhibitors. Proc Natl Acad Sci USA. 2004;101:18030–18035. doi: 10.1073/pnas.0408345102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nakakura EK, Sriuranpong VR, Kunnimalaiyaan M, Hsiao EC, Schuebel KE, Borges MW, Jin N, Collins BJ, Nelkin BD, Chen H, Ball DW. Regulation of neuroendocrine differentiation in gastrointestinal carcinoid tumor cells by notch signaling. J Clin Endocrinol Metab. 2005;90:4350–4356. doi: 10.1210/jc.2005-0540. [DOI] [PubMed] [Google Scholar]
- 29.Miele L. Notch signaling. Clin Cancer Res. 2006;12:1074–1079. doi: 10.1158/1078-0432.CCR-05-2570. [DOI] [PubMed] [Google Scholar]
- 30.Chen H, Carson-Walter EB, Baylin SB, Nelkin BD, Ball DW. Differentiation of medullary thyroid cancer by C-Raf-1 silences expression of the neural transcription factor human achaete-scute homolog-1. Surgery. 1996;120:168–172. doi: 10.1016/s0039-6060(96)80284-4. [DOI] [PubMed] [Google Scholar]
- 31.Sippel RS, Carpenter JE, Kunimalaiyaan M, Chen H. The role of human achaete-scute homolog-1 in medullary thyroid cancer cells. Surgery. 2003;134:886–871. doi: 10.1016/s0039-6060(03)00418-5. [DOI] [PubMed] [Google Scholar]
- 32.Givens KT, Kitada S, Chen AK, Rothschiller J, Lee DA. Proliferation of human ocular fibroblasts. An assessment of in vitro colorimetric assays. Invest Ophthalmol Vis Sci. 1990;31:1856–1862. [PubMed] [Google Scholar]