

Abstract
Objective
Increased activity of Sp family of transcription factors is a frequent and critical event in cancer development and progression. Genes governing tumor growth, invasion and angiogenesis are regulated by Sp factors, like Sp1, Sp3 or Sp4, and are frequently over-expressed in tumors. Targeting Sp factors has been explored as a therapeutic approach. Mithramycin (MTM) is a natural antibiotic that binds DNA and inhibit Sp1-dependent transcription. New analogues, named MTM-SDK and MTM-SK, were recently obtained by genetic engineering of the MTM biosynthetic pathway and have demonstrated improved transcriptional and antiproliferative activity in ovarian cancer cell lines in vitro. In present study we evaluated the activity of the new compounds in human ovarian cancer xenografts.
Methods
Expression of Sp1 and target proteins in ovarian cancer specimens and tumor xenografts was assessed by immunohistochemistry. Drug –induced silencing of Sp1-regulated genes in cells and tumor xenograft samples was assessed by quantitative RT-PCR. Toxicity and antitumor activity of the compounds were investigated in healthy and tumor-bearing immunocompromised mice, respectively.
Results
Expression of Sp1 was frequently increased in human epithelial ovarian cancers. MTM-SDK and MTM-SK acted as potent inhibitors of Sp1-dependent transcription both in vitro and in tumor xenografts. Both compounds were well tolerated even after prolonged administration and delayed growth of ovarian tumor xenografts. MTM-SDK was particularly effective against orthotopic tumors leading to a significant increase of survival and delay of tumor progression.
Conclusions
MTM-SDK and MTM-SK show relevant activity in vivo and represent interesting candidates for treatment of ovarian cancers.
Introduction
Epithelial ovarian cancer is the leading cause of death from gynecological cancers [1-3]. At the advanced stages, surgery is rarely curative and chemotherapy is of limited efficacy [3]. A better understanding of molecular basis of ovarian tumorigenesis may lead to more effective treatment strategies for this disease [4-6]. Sp transcription factors, like Sp1, Sp3 and Sp4, affect multiple cellular processes and have been involved in development various types of cancers, including pancreatic, breast, gastric and thyroid cancer [7]. Diverse strategies, including decoy oligonucleotides, peptide nucleic acids, ribozymes, small-interfering RNAs and low-molecular weight compounds, have been explored to inhibit in particular Sp1 expression or activity for therapeutic applications [8-14]. Analysis of gene regulatory network governing ovarian cancer progression indicates a possible role of the Sp family of transcription factors in this disease and gives reason for development of Sp1 –competing therapeutic approach. [15-25].
The aureolic acid antibiotic, mithramycin (MTM), is a natural polycyclic aromatic polyketide produced by Streptomyces species [26]. MTM binds preferentially to GC-rich sequences in DNA corresponding to Sp binding sites [27, 28], inhibits expression of Sp-regulated genes [29, 30] and has anticancer activity. However, clinical use of MTM was limited by the drug’s side effects [31]. Metabolic engineering of the MTM biosynthetic pathway has been used to produce new derivatives of the natural compound with improved biochemical and pharmacological properties [32-34]. MTM-SDK and MTM-SK (Figure 1) were obtained by targeted inactivation of the ketoreductase MtmW responsible for the last step of MTM biosynthesis [33, 35-37]. In biochemical and cellular assays, the two compounds acted as potent repressors of Sp1-regulated transcription [35]. Moreover, both compounds exhibited potent antiproliferative and pro-apoptotic activity toward cancer cells with minimal effects on growth and viability of normal cells, suggesting that they might be effective agents for treatment of cancer and other diseases with abnormal Sp1 activity [35]. The present work evaluated MTM-SDK and MTM-SK as candidates for treatment of ovarian cancer using human tumor xenograft models in nude mice.
Figure 1.
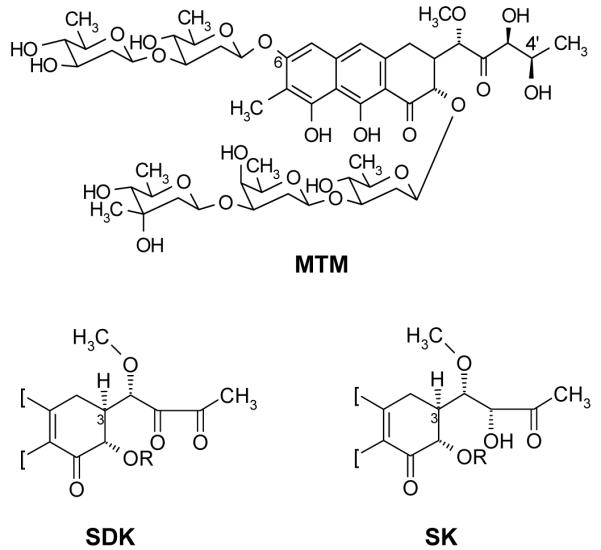
Chemical structure of MTM, MTM-SDK and MTM-SK
Materials and methods
Compounds, Cell Lines and Animals
MTM derivates were isolated from Streptomyces cultures as described [35]. Stock solutions were prepared in sterile DMSO, kept at −20 °C, and diluted in sterile saline solution immediately before use. Human ovarian cancer A2780 cells were maintained under standard conditions [35]. Female CD-1 and athymic female CD-1 nu/nu mice (Charles River Laboratories) were used for toxicity and antitumor activity studies, respectively. Mice were maintained under specific pathogen-free conditions with food and water provided ad libitum. The general health status of the animals was monitored daily. Procedures involving animals and their care were conducted in conformity with the institutional guidelines that are in compliance with national (Legislative Decree 116 of January 27, 1992, Authorization n.169/94-A issued December 19, 1994, by Ministry of Health) and international laws and policies (EEC Council Directive 86/609, OJ L 358. 1, December 12, 1987; Standards for the Care and Use of Laboratory Animals, United States National Research Council, Statement of Compliance A5023-01, November 6, 1998).
Immunohistochemistry
Tissue sections were obtained from paraffin-embedded blocks of epithelial ovarian tumors (n=54), normal ovary samples (n=3) or tumor xenografts. Sections were incubated with primary antibodies (Santa Cruz Biotechnology) against Sp1 (clone sc59; 1:200 dilution), VEGF (clone A20; 1:200 dilution) or c-Myc (clone 9E10; 1:75 dilution). After incubation with secondary antibodies, immunohistochemical staining was performed using ABC-peroxidase or steptavidin-biotin system (ABC kit, Santa Cruz Biotechnology) and detected with diaminobenzidine substrate (DAB). Sections were counterstained with H&E. Immunohistochemical staining was semi-quantitatively assessed as percentage of immunoreactive cells in 10 HPF using a 10% scale. The staining intensity was scored as faint (1+), moderate (2+) and intense (3+). The evaluation was independently performed by two senior pathologists (CC, CR).
Quantitative Real-Time RT-PCR (QRT-PCR)
RNA was isolated from cells treated with 100 nM of compounds for 24 h using Trizol (Invitrogen) and from snap-frozen tumor samples harvested from mice treated with 400 μg/Kg/day × 5 d or 0.4% DMSO using the SV total RNA isolation kit (Promega). RNA was reverse-transcribed with SuperScript Reverse Transcriptase (Applied Biosystems). Real-time PCR primers for c-src, c-myc, VEGF, hTERT, claudin4 (CLDN4), Kruppel-like transcription factor 8 (KLF8), cyclin E1 (CCNE1), X-like apoptosis inhibitor (XIAP) and survivin (BIRC5) are shown in Supplementary Table S1. GAPDH and B2M were used as reference and internal controls. QRT-PCR was performed using SYBRGreen PCR Master Mix (Applied Biosystems) in an ABI PRISM 7000HT machine. Expression levels were calculated as percentage of control gene expression (2(CTcontrol-CTtarget)*100). Mean and standard errors were calculated for each treatment group. Experiments in vitro were done in triplicates. RNA from three mice per group was analyzed separately and experiments were repeated twice. Statistical significance was determined by one-way ANOVA.
Toxicity
Toxicity of MTM derivates was tested in healthy female CD-1 mice. The following doses and schedules of treatment were used: a) 400 μg/Kg administered i.p. daily × 5 (2 mg/Kg total); b) 800 μg/Kg administered i.p. daily × 5 (4 mg/Kg total); c) 400 μg/Kg administered i.p. × 15 (6 mg/Kg total). Control mice received corresponding amounts of DMSO. Body weight, deaths, changes in behaviour, motility, eating and drinking habits, and any other sign of toxicity were recorded.
Antitumor Activity
For subcutaneous xenografts, A2780 cells (8 × 106) were injected into the left flank of athymic CD-1 nu/nu female mice. Treatments started when tumors reached the size of ~100 mm3. Drugs (600 μg/Kg, q2d ×10) or 0.6 % DMSO were administered by i.p. injections in a volume of 10 ml/kg of body weight. Tumor size was measured with a caliper twice a week. For orthotopic xenografts, A2780 cells (8 × 106 in 0.2 ml PBS) were injected in the peritoneal cavity. Treatment started 7 days later with 400 μg/Kg, q2d ×10 or 0.4% DMSO administered i.p. Animals were monitored daily by the veterinary staff, blinded to the experiment, to assess physical and performance status as well as to record the appearance of ascites formation in the peritoneal cavity (abdominal distension), signs of severe sickness and deaths. The endpoints for survival analysis were death or euthanasia. The time of first appearance of clinical signs of ascites was recorded as secondary endpoint. Animals were euthanized when they developed clear signs of severe sickness, including extensive swelling of the abdominal cavity due to massive ascites production leading to severe impairment of motor functions, abnormal breathing, decreased food or water intake, rough hair coat, hunched posture and lethargy. Autopsies were performed to assess the extent of tumor dissemination.
Data Analysis
In the orthotopic model survival curves were constructed with the Kaplan-Meyer method and differences in survival time assessed using the log-rank test and the increment of lifespan (ILS) was calculated as: Median survivalTreated – Median survival control/Median survivalcontrol ×100. In the subcutaneous model, tumor volume was calculated as: Tumor volume (mm3)=(length × width2)/2. Differences in tumor growth were evaluated with one-way ANOVA followed by Fisher test. The percentage of tumor growth (T/C %) was calculated as: RTVtreated/RTVcontrol × 100. RTV is the mean relative tumor volume calculated as RTV = Vt/V0, where Vt is the tumor volume at the day of measurement and V0 is the tumor volume at the beginning of the treatment. The percentage of tumor weight inhibition (TWI) was calculated as: 100 - T/C%. The log cell kill (LCK) was calculated as: T-C/3,32 × Td, where T-C is the difference in median time required for the tumors in the treatment (T) and control (C) group to reach a predetermined size (i.e., 1000 mg). Td is the tumor volume doubling time in days, determined in the exponential growth phase of the control group from a best-fit straight line. All data are expressed as the mean values ± standard error (SE). Differences were considered statistically significant at a level of P <0.05.
Results
Sp1 Expression in Ovarian Cancer Tissue Specimens
To confirm the involvement of Sp1 in ovarian tumorigenesis, we assessed expression of this transcription factor in ovarian cancer and normal ovary clinical specimens by immunohistochemistry (Figure 2A). Very few surface epithelial and stromal cells in normal ovary samples (n=3) showed only very weak (faint, 1+) nuclear staining. Epithelial ovarian tumors, including serous (G1-G3), endometriod, mucinous, clear cell, Brenner tumor and undifferentiated carcinomas (n= 52) had an average of 75-80% positive cells with moderate to intense nuclear staining (Figure 2B). G1 and G2 serous carcinomas showed a prevailing moderate (2+) or intense (3+) nuclear staining; in serous G3, endometroid and mucinous neoplasias nuclear staining ranged from faint (1+) to moderate (2+), while clear cells carcinomas and Brenner tumors showed a faint (1+) positivity (Figure 2B). Lower levels of Sp1 positivity (35% of positive cells) were seen in two samples of Mixed Mullerian tumors with a faint (1+) staining. Most epithelial tumors exhibited also focal cytoplasmic staining for VEGF, a known Sp1 target gene (Figure 2B).
Figure 2.

Immunohistochemical analysis of Sp1 and VEGF expression in human ovarian tissue specimens. (A) Representative examples of Sp1 immunostaining of a serous ovarian carcinoma and normal ovary are shown (DAB-H, X 400). Intense nuclear staining is seen in the majority of the tumor cells in the serous carcinoma, while faint and focal staining is present in normal ovary. (B) Summary of the Sp1 and VEGF immunostaining data in epithelial ovarian tumors (n=54). The following scoring system was used to evaluate the intensity of Sp1 and VEGF expression: 1+, faint; 2+, moderate; 3+, intense. ND, not determined.
Inhibition of Sp1-Regulated Genes in Ovarian Cancer Cells In Vitro
We had previously observed that MTM-SDK and MTM-SK were very effective in vitro against multiple human ovarian cancer cell lines [35]. Furthermore, the compounds inhibited Sp1 target genes in cancer cells at relatively low concentrations [35]. Specificity of these compounds toward Sp1 with respect to other transcription factors was demonstrated previously by luciferase reporter assays and microarray-based gene profiling [35]. To further assess the range of activity of these compounds we examined their effects on Sp1 target genes with relevant roles in ovarian cancer. The set included genes involved in cell proliferation (hTERT, CCNE1) [15, 16], apoptosis (BIRC5, XIAP) [17-19], invasion (CLDN4, KLF8) [20-22] and angiogenesis (VEGF) [23]. A2780 cells were treated with 100 nM of compounds for 24 h and gene expression was measured by QRT-PCR (Figure 3A). Expression of c-SRC, VEGF, c-Myc, hTERT, CLDN4 and KLF8 was strongly inhibited by both MTM-SDK and MTM-SK (>80% reduction). CCNE1 and XIAP were reduced by ~60%, while BIRC5 was less affected (~25-30% reduction). All the differences were highly statistically significant (P<0.005). Furthermore, B2M, selected as internal control because is not under the control of Sp1, was not affected by the treatment (97 ± 4% of control). Thus, the MTM derivatives affected a wide range of genes and pathways in ovarian cancer cells. The difference in the degree of down-regulation might reflect the extent of Sp1 involvement in the transcriptional control of the individual genes.
Figure 3.

Inhibition of Sp1 target genes by MTM derivates in vitro and in vivo. Expression of the indicated genes was measured by quantitative real-time RT-PCR and normalized to GAPDH. Similar results were obtained using B2M as reference. Results are presented as percentage of gene expression relative to the control group. (A) Gene expression in A2780 cells treated with the compounds in vitro for 24 h. (B) Gene expression in tumors from mice (n=3 per group) treated with 400 μg/Kg/day × 5 or vehicle and harvested 6 h after the last injection. (C) c-SRC, c-Myc and VEGF expression in tumors from mice (n=3 per group) treated as above and excised 6 and 24 h after the last injection. (D) c-Myc protein level assessed by immunohistochemical staining in tumors from mice (3 per group) treated as above and excised 6 h after last treatment. (DAB-H, X 40). In A, B and C standard errors were less than 10% of mean value. A, P <0.005. B and C, P <0.05.
Inhibition of Sp1-Regulated Genes in Ovarian Tumor Xenografts
Next, we determined the ability of MTM-SDK and MTM-SK to interfere with Sp1-dependent transcription in vivo. Mice with subcutaneous tumors were treated with the compounds (400 μg/Kg/day ×5). Tumors were excised 6 and 24 h after the last injection to measure gene expression by QRT-PCR. As shown in Figure 3B, the level of all the genes tested was greatly reduced in drug treated mice (> 90 % reduction, P<0.05). For most genes (hTERT, CLDN4, KLF8, CCNE1, XIAP, BIRC5, c-SRC, c-Myc) the inhibition was equal or even greater than that observed in vitro, probably due to the cumulative effect of the prolonged in vivo treatment. The effect on VEGF expression was less pronounced in vivo than in vitro (Figure 3C), probably reflecting the complexity and redundancy of the mechanisms governing expression of this gene in the in vivo environment. Interestingly, similar levels of gene down-regulation were observed for c-SRC, c-Myc and VEGF at 6 and 24 h after the last treatment indicating that the effect was not readily reversed (Figure 3C). Treatment with MTM-SDK and MTM-SK resulted also in reduction of protein level as indicated by semi-quantitative assessment of c-Myc immunohistochemical staining in tumor tissues from mice treated with 100, 200 and 400 μg/Kg/day or 0,4% DMSO for 5 days and taken 6 h after the last treatment. Tumors from control mice showed clear positive staining for c-Myc, while marked dose-dependent reduction of c-Myc immunostaining was seen in drug treated mice with maximal effect at the highest dose (Figure 3D).
Toxicity in Non-Tumor Bearing Mice
Prior to evaluate antitumor activity, we assessed the tolerability of the compounds given by multiple i.p. injections in healthy female CD-1 mice. Three different treatment schedules were tested: 1) 400 μg/Kg/day ×5 (total dose 2 mg/Kg), 2) 800 μg/Kg/day ×5 (total dose 4 mg/Kg) and 3) 400 μg/Kg/day ×15 (total dose 6 mg/Kg). There were no signs of toxicity or deaths with any of the schedules of treatment. Body weight was not different between control and drug treated mice even at the highest cumulative dose (Figure 4). Thus, the compounds could be given safely within this range of doses and with prolonged treatment schedules.
Figure 4.

Body weights of mice treated with MTM, MTM-SDK and MTM-SK. Healthy female CD-1 mice (4 per group) were treated with i.p. injections of 400 μg/Kg/day ×15 (6 mg/Kg total). The control group received an equivalent amount of DMSO. Each point represents the mean ± SD of 4 mice.
Tumor Growth Inhibition in Subcutaneous Xenografts
To evaluate in vivo antitumor activity, tumor-bearing mice were treated with i.p. injections of vehicle or MTM-SDK and MTM-SK at 600 μg/Kg, q2d ×10 over a 20-day period. Dosage and regime of treatment were based on the results of the toxicity assays and tumor growth dynamics. Treatment with MTM-SDK and MTM-SK induced a statistically significant delay of tumor growth compared to the control group (P <0.05, Figure 5A). TWI% of 45 and 48 were achieved for MTM-SDK and MTM-SK with LCK of 0.3 and 0.2, respectively. Both compounds were well-tolerated with this treatment schedule without signs of toxicity or body weight loss (Figure 5B).
Figure 5.

Antitumor activity of MTM-SDK and MTM-SK in subcutaneous ovarian tumor xenograft. Mice (10 per group) were inoculated with A2780 cells and treated with 600 μg/Kg, q2d ×10, or vehicle (DMSO) given by intraperitoneal injections over a 20-day period. (A) Tumor size was measured twice weekly. Results are expressed as mean ± SD of 10 mice. Difference in tumor size is statistically significant after 10 days of treatment (** P <0.05). (B) Body weight was measured twice weekly. Data are presented as relative body weight, i.e., the difference between mean body weight at start of the treatment and at the indicated time points.
Tumor Growth Inhibition in Orthotopic Xenografts
Antitumor activity of MTM-SDK and MTM-SK was evaluated also in orthotopic ovarian tumor xenografts. Since intraperitoneal administration of drugs to tumors disseminated in the peritoneal cavity was expected to be more effective, a lower dose was used in these experiments (400 μg/Kg/day). Survival and abdominal distension, which reflects both ascites accumulation and increasing tumor burden, were monitored daily for >100 days. Vehicle treated mice became sick and died within a short time interval with a median survival of 55 days (Figure 6A). Treatment with MTM-SDK increased the median survival to 80.5 days (P <0.005; ILS, 46 %). The median survival of MTM-SDK treated mice was significantly higher than that of MTM treated mice (P <0.05). Interestingly, 4 out of 10 mice in the MTM-SDK treatment group were alive at 100 days after tumor cell inoculation and apparently tumor-free, as indicated by the absence of clinical signs of disease like ascites and intraperitoneal tumor masses at the authopsy. MTM-SDK delayed also the appearance of ascites and abdominal distension. The median time to detect abdominal distension was 70 days in mice treated with MTM-SDK compared to 51 days in control mice (P <0.005). At the autopsy, control mice had massive bloody ascites and numerous tumor implants disseminated on the surface of the peritoneum, intraperitoneal organs and diaphragm (Figure 6B). In comparison, MTM-SDK treated mice had greatly reduced ascites and no or minimal signs of intraperitoneal tumor dissemination. Consistent with the data in non tumor-bearing mice, there were no signs of toxicity in MTM-SDK treated mice. MTM-SK was less effective than MTM-SDK in the orthotopic model, similar to its reduced potency in cell culture studies. Treatment with MTM SK had little effect on survival and the survival gain was not statistically significant compared to the control group (66 vs. 55 days of control mice; P = 0.135). MTM SK also had no effect on abdominal distension (P = 0.342). At the dose tested here, MTM did not have any effect on median survival (58 days vs. 55 days of control group; P = 0.336) and ascites accumulation (52 versus 51 days of control group, P = 0.404).
Figure 6.

Antitumor activity of MTM SDK, MTM SK and MTM in orthotopic ovarian tumor xenografts. Mice (10 per group) were inoculated intraperitoneally with A2780 cells and treatment started 7 days later. Vehicle or compounds (400 μg/Kg/day) were given by intraperitoneal injections for 10 days. (A) Kaplan-Meier survival curves. (B) Abdominal distension and intraperitoneal tumor masses (arrows) in representative mice of the control (left) and MTM-SDK (right) group sacrificed at day 50 from cell implant.
Discussion
In this study, we found increased expression of the Sp1 transcription factor in epithelial ovarian cancer. This finding justified testing of novel Sp1-targeting compounds for ovarian cancer treatment. Targeting over-active transcription factors, like Sp1, which can act as a nodal point of multiple signalling pathways involved in cancer initiation and progression, could be a very effective strategy for this rapidly progressive and lethal cancer. Here we tested for the first time the new MTM analogues MTM SDK and MTM SK in vivo in human ovarian tumor xenografts. These new analogues of the aromatic polyketide MTM were generated by genetic engineering of the biosynthetic pathway of this natural antibiotic in the producing microorganism. We had previously shown that MTM-SDK and MTM-SK exhibited improved pharmacological properties, inhibited transcription of multiple genes regulated by the Sp family of transcription factors, and had potent antiproliferative and pro-apoptotic activity in human cancer cell lines in vitro. The present work shows that these compounds are very effective inhibitors of Sp1-dependent transcription also in vivo and have relevant effects on tumor growth and survival at the relatively low and non-toxic doses.
We had shown previously the ability of MTM-SDK and MTM-SK to inhibit Sp1 dependent transcription in cells [35]. A gene profiling study showed that these compounds had the potential to inhibit transcription of multiple genes and affect multiple downstream pathways critical for ovarian tumorigenesis [35]. To confirm this hypothesis we selected a set of Sp1-regulated genes known to be over-expressed in ovarian tumors. The selected genes contributed to various aspects of ovarian tumorigenesis, including proliferation, replicative potential, survival, tumor angiogenesis, invasion and metastasis [2, 5, 15-23, 38-40]. Moreover, each gene individually has been proposed as a relevant therapeutic target. We measured gene expression in cells and tumor samples following treatment with MTM-SDK and MTM-SK and found that the level of expression of these genes was reduced, albeit to different extents, upon treatment with the compounds both in vitro and in vivo.
Treatment with MTM-SDK and MTM-SK resulted in significant delay of growth of subcutaneous tumor xenografts and increased survival in orthotopic xenografts. Administration of the compounds was well tolerated and prolonged treatment was not associated with toxicity. Interestingly, MTM-SDK was particularly effective in the orthotopic model leading to a substantial improvement in survival and delay in tumor progression. The orthotopic model mimics closely the advanced stages of the human disease for which intraperitoneal spread is the major mechanism of progression [41, 42]. Following intraperitoneal inoculation of ovarian cancer cells mice develop generally within 4-6 weeks disseminated intraperitoneal carcinomatosis followed by massive ascites production and pronounced abdominal swelling [41]. MTM-SDK treatment delayed the clinical onset of abdominal distension compared to the control group, reflecting reduced carcinomatosis and ascites. At the same dose, MTM did not have any effect compared to control mice and MTM-SK induced a marginal, not statistically significant improvement in survival, confirming the greater activity of MTM-SDK in this setting. Further study will be required to determine the reason of the higher activity of MTM-SDK in this context. MTM-SDK could be very effective due to its rapid cellular uptake [35] and greater ability to interfere with genes essential for survival, spreading and peritoneal implantation of ovarian cancer cells [2, 42]. Intraperitoneal chemotherapy has been proposed as an effective treatment option for advanced ovarian cancer based on randomized clinical studies showing similar or greater benefits compared to standard intravenous therapy [43, 44]. Thus, intraperitoneal administration of a highly effective drug like MTM-SDK might be a valid approach to control disease progression by increasing exposure of cancer cells to the compound.
To our knowledge, MTM-SDK and MTM-SK are the first MTM derivatives to be tested in vivo and to show improved pharmacological properties compared to the parent compound. In this study we show that the MTM compounds are well tolerated and exhibit significant transcriptional and antitumor effects in ovarian tumor xenografts. Taken together, these findings highlight the importance of Sp transcription factors in ovarian tumorigenesis and indicate that new MTM based analogues are valid drug candidates for this disease.
Supplementary Material
Supplementary Table S1. Genes and primer sets for gene expression analysis by real-time RT-PCR.
Acknowledgements
This work was supported by grants from the Ticino Foundation for Cancer Research (to C.V.C.), the National Institutes of Health (R01 CA091901-06A1 to J.R.) and the Fondazione Nerina e Mario Mattioli (to S.P. and M.B.).
Footnotes
Conflict of Interest Statement There are no conflicts of interest to disclose
References
- [1].Foster T, Brown TM, Chang J, Menssen HD, Blieden MB, Herzog TJ. A review of the current evidence for maintenance therapy in ovarian cancer. Gynecol Oncol. 2009 doi: 10.1016/j.ygyno.2009.07.026. [DOI] [PubMed] [Google Scholar]
- [2].Ozols RF, Bookman MA, Connolly DC, Daly MB, Godwin AK, Schilder RJ, Xu X, Hamilton TC. Focus on epithelial ovarian cancer. Cancer Cell. 2004;5:19–24. doi: 10.1016/s1535-6108(04)00002-9. [DOI] [PubMed] [Google Scholar]
- [3].Cannistra SA. Cancer of the ovary. N Engl J Med. 2004;351:2519–29. doi: 10.1056/NEJMra041842. [DOI] [PubMed] [Google Scholar]
- [4].Bast RC, Jr., Hennessy B, Mills GB. The biology of ovarian cancer: new opportunities for translation. Nat Rev Cancer. 2009;9:415–28. doi: 10.1038/nrc2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Landen CN, Jr., Birrer MJ, Sood AK. Early events in the pathogenesis of epithelial ovarian cancer. J Clin Oncol. 2008;26:995–1005. doi: 10.1200/JCO.2006.07.9970. [DOI] [PubMed] [Google Scholar]
- [6].Yap TA, Carden CP, Kaye SB. Beyond chemotherapy: targeted therapies in ovarian cancer. Nat Rev Cancer. 2009;9:167–81. doi: 10.1038/nrc2583. [DOI] [PubMed] [Google Scholar]
- [7].Safe S, Abdelrahim M. Sp transcription factor family and its role in cancer. Eur J Cancer. 2005 doi: 10.1016/j.ejca.2005.08.006. [DOI] [PubMed] [Google Scholar]
- [8].Abdelrahim M, Samudio I, Smith R, 3rd, Burghardt R, Safe S. Small inhibitory RNA duplexes for Sp1 mRNA block basal and estrogen-induced gene expression and cell cycle progression in MCF-7 breast cancer cells. J Biol Chem. 2002;277:28815–22. doi: 10.1074/jbc.M203828200. [DOI] [PubMed] [Google Scholar]
- [9].Ishibashi H, Nakagawa K, Onimaru M, Castellanous EJ, Kaneda Y, Nakashima Y, Shirasuna K, Sueishi K. Sp1 decoy transfected to carcinoma cells suppresses the expression of vascular endothelial growth factor, transforming growth factor beta1, and tissue factor and also cell growth and invasion activities. Cancer Res. 2000;60:6531–6. [PubMed] [Google Scholar]
- [10].Lou Z, O’Reilly S, Liang H, Maher VM, Sleight SD, McCormick JJ. Down-regulation of overexpressed sp1 protein in human fibrosarcoma cell lines inhibits tumor formation. Cancer Res. 2005;65:1007–17. [PubMed] [Google Scholar]
- [11].Abdelrahim M, Baker CH, Abbruzzese JL, Safe S. Tolfenamic acid and pancreatic cancer growth, angiogenesis, and Sp protein degradation. J Natl Cancer Inst. 2006;98:855–68. doi: 10.1093/jnci/djj232. [DOI] [PubMed] [Google Scholar]
- [12].Chadalapaka G, Jutooru I, Chintharlapalli S, Papineni S, Smith R, 3rd, Li X, Safe S. Curcumin decreases specificity protein expression in bladder cancer cells. Cancer Res. 2008;68:5345–54. doi: 10.1158/0008-5472.CAN-07-6805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Chintharlapalli S, Papineni S, Ramaiah SK, Safe S. Betulinic acid inhibits prostate cancer growth through inhibition of specificity protein transcription factors. Cancer Res. 2007;67:2816–23. doi: 10.1158/0008-5472.CAN-06-3735. [DOI] [PubMed] [Google Scholar]
- [14].Papineni S, Chintharlapalli S, Abdelrahim M, Lee SO, Burghardt R, Abudayyeh A, Baker C, Herrera L, Safe S. Tolfenamic acid inhibits esophageal cancer through repression of specificity proteins and c-Met. Carcinogenesis. 2009;30:1193–201. doi: 10.1093/carcin/bgp092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Bermudez Y, Yang H, Saunders BO, Cheng JQ, Nicosia SV, Kruk PA. VEGF- and LPA-induced telomerase in human ovarian cancer cells is Sp1-dependent. Gynecol Oncol. 2007;106:526–37. doi: 10.1016/j.ygyno.2007.05.005. [DOI] [PubMed] [Google Scholar]
- [16].Farley J, Smith LM, Darcy KM, Sobel E, O’Connor D, Henderson B, Morrison LE, Birrer MJ. Cyclin E expression is a significant predictor of survival in advanced, suboptimally debulked ovarian epithelial cancers: a Gynecologic Oncology Group study. Cancer Res. 2003;63:1235–41. [PubMed] [Google Scholar]
- [17].Liguang Z, Peishu L, Hongluan M, Hong J, Rong W, Wachtel MS, Frezza EE. Survivin expression in ovarian cancer. Exp Oncol. 2007;29:121–5. [PubMed] [Google Scholar]
- [18].Lee TJ, Jung EM, Lee JT, Kim S, Park JW, Choi KS, Kwon TK. Mithramycin A sensitizes cancer cells to TRAIL-mediated apoptosis by down-regulation of XIAP gene promoter through Sp1 sites. Mol Cancer Ther. 2006;5:2737–46. doi: 10.1158/1535-7163.MCT-06-0426. [DOI] [PubMed] [Google Scholar]
- [19].Shaw TJ, Lacasse EC, Durkin JP, Vanderhyden BC. Downregulation of XIAP expression in ovarian cancer cells induces cell death in vitro and in vivo. Int J Cancer. 2008;122:1430–4. doi: 10.1002/ijc.23278. [DOI] [PubMed] [Google Scholar]
- [20].Honda H, Pazin MJ, Ji H, Wernyj RP, Morin PJ. Crucial roles of Sp1 and epigenetic modifications in the regulation of the CLDN4 promoter in ovarian cancer cells. J Biol Chem. 2006;281:21433–44. doi: 10.1074/jbc.M603767200. [DOI] [PubMed] [Google Scholar]
- [21].Agarwal R, D’Souza T, Morin PJ. Claudin-3 and claudin-4 expression in ovarian epithelial cells enhances invasion and is associated with increased matrix metalloproteinase-2 activity. Cancer Res. 2005;65:7378–85. doi: 10.1158/0008-5472.CAN-05-1036. [DOI] [PubMed] [Google Scholar]
- [22].Wang X, Urvalek AM, Liu J, Zhao J. Activation of KLF8 transcription by focal adhesion kinase in human ovarian epithelial and cancer cells. J Biol Chem. 2008;283:13934–42. doi: 10.1074/jbc.M709300200. [DOI] [PubMed] [Google Scholar]
- [23].Duncan TJ, Al-Attar A, Rolland P, Scott IV, Deen S, Liu DT, Spendlove I, Durrant LG. Vascular endothelial growth factor expression in ovarian cancer: a model for targeted use of novel therapies? Clin Cancer Res. 2008;14:3030–5. doi: 10.1158/1078-0432.CCR-07-1888. [DOI] [PubMed] [Google Scholar]
- [24].Abdelrahim M, Baker CH, Abbruzzese JL, Sheikh-Hamad D, Liu S, Cho SD, Yoon K, Safe S. Regulation of vascular endothelial growth factor receptor-1 expression by specificity proteins 1, 3, and 4 in pancreatic cancer cells. Cancer Res. 2007;67:3286–94. doi: 10.1158/0008-5472.CAN-06-3831. [DOI] [PubMed] [Google Scholar]
- [25].Higgins KJ, Abdelrahim M, Liu S, Yoon K, Safe S. Regulation of vascular endothelial growth factor receptor-2 expression in pancreatic cancer cells by Sp proteins. Biochem Biophys Res Commun. 2006;345:292–301. doi: 10.1016/j.bbrc.2006.04.111. [DOI] [PubMed] [Google Scholar]
- [26].Lombo F, Menendez N, Salas JA, Mendez C. The aureolic acid family of antitumor compounds: structure, mode of action, biosynthesis, and novel derivatives. Appl Microbiol Biotechnol. 2006;73:1–14. doi: 10.1007/s00253-006-0511-6. [DOI] [PubMed] [Google Scholar]
- [27].Barcelo F, Scotta C, Ortiz-Lombardia M, Mendez C, Salas JA, Portugal J. Entropically-driven binding of mithramycin in the minor groove of C/G-rich DNA sequences. Nucleic Acids Res. 2007;35:2215–26. doi: 10.1093/nar/gkm037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Sastry M, Patel DJ. Solution structure of the mithramycin dimer-DNA complex. Biochemistry. 1993;32:6588–604. doi: 10.1021/bi00077a012. [DOI] [PubMed] [Google Scholar]
- [29].Blume SW, Snyder RC, Ray R, Thomas S, Koller CA, Miller DM. Mithramycin inhibits SP1 binding and selectively inhibits transcriptional activity of the dihydrofolate reductase gene in vitro and in vivo. J Clin Invest. 1991;88:1613–21. doi: 10.1172/JCI115474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Snyder RC, Ray R, Blume S, Miller DM. Mithramycin blocks transcriptional initiation of the c-myc P1 and P2 promoters. Biochemistry. 1991;30:4290–7. doi: 10.1021/bi00231a027. [DOI] [PubMed] [Google Scholar]
- [31].Calabresi P. The pharmacological basis of therapeutics. Pergamon Press; Oxford, U.K.: 1991. Antineoplastic agents. [Google Scholar]
- [32].Remsing LL, Garcia-Bernardo J, Gonzalez A, Kunzel E, Rix U, Brana AF, Bearden DW, Mendez C, Salas JA, Rohr J. Ketopremithramycins and ketomithramycins, four new aureolic acid-type compounds obtained upon inactivation of two genes involved in the biosynthesis of the deoxysugar moieties of the antitumor drug mithramycin by Streptomyces argillaceus, reveal novel insights into post-PKS tailoring steps of the mithramycin biosynthetic pathway. J Am Chem Soc. 2002;124:1606–14. doi: 10.1021/ja0105156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Remsing LL, Gonzalez AM, Nur-e-Alam M, Fernandez-Lozano MJ, Brana AF, Rix U, Oliveira MA, Mendez C, Salas JA, Rohr J. Mithramycin SK, a novel antitumor drug with improved therapeutic index, mithramycin SA, and demycarosyl-mithramycin SK: three new products generated in the mithramycin producer Streptomyces argillaceus through combinatorial biosynthesis. J Am Chem Soc. 2003;125:5745–53. doi: 10.1021/ja034162h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Trefzer A, Blanco G, Remsing L, Kunzel E, Rix U, Lipata F, Brana AF, Mendez C, Rohr J, Bechthold A, Salas JA. Rationally designed glycosylated premithramycins: hybrid aromatic polyketides using genes from three different biosynthetic pathways. J Am Chem Soc. 2002;124:6056–62. doi: 10.1021/ja017385l. [DOI] [PubMed] [Google Scholar]
- [35].Albertini V, Jain A, Vignati S, Napoli S, Rinaldi A, Kwee I, Nur-e-Alam M, Bergant J, Bertoni F, Carbone GM, Rohr J, Catapano CV. Novel GC-rich DNA-binding compound produced by a genetically engineered mutant of the mithramycin producer Streptomyces argillaceus exhibits improved transcriptional repressor activity: implications for cancer therapy. Nucleic Acids Res. 2006;34:1721–34. doi: 10.1093/nar/gkl063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Remsing LL, Bahadori HR, Carbone GM, McGuffie EM, Catapano CV, Rohr J. Inhibition of c-src transcription by mithramycin: structure-activity relationships of biosynthetically produced mithramycin analogues using the c-src promoter as target. Biochemistry. 2003;42:8313–24. doi: 10.1021/bi034091z. [DOI] [PubMed] [Google Scholar]
- [37].Gibson M, Nur-e-alam M, Lipata F, Oliveira MA, Rohr J. Characterization of kinetics and products of the Baeyer-Villiger oxygenase MtmOIV, the key enzyme of the biosynthetic pathway toward the natural product anticancer drug mithramycin from Streptomyces argillaceus. J Am Chem Soc. 2005;127:17594–5. doi: 10.1021/ja055750t. [DOI] [PubMed] [Google Scholar]
- [38].Orsulic S, Li Y, Soslow RA, Vitale-Cross LA, Gutkind JS, Varmus HE. Induction of ovarian cancer by defined multiple genetic changes in a mouse model system. Cancer Cell. 2002;1:53–62. doi: 10.1016/s1535-6108(01)00002-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Honda H, Pazin MJ, D’Souza T, Ji H, Morin PJ. Regulation of the CLDN3 gene in ovarian cancer cells. Cancer Biol Ther. 2007;6:1733–42. doi: 10.4161/cbt.6.11.4832. [DOI] [PubMed] [Google Scholar]
- [40].Chen CH, Shen J, Lee WJ, Chow SN. Overexpression of cyclin D1 and c-Myc gene products in human primary epithelial ovarian cancer. Int J Gynecol Cancer. 2005;15:878–83. doi: 10.1111/j.1525-1438.2005.00150.x. [DOI] [PubMed] [Google Scholar]
- [41].Shaw TJ, Senterman MK, Dawson K, Crane CA, Vanderhyden BC. Characterization of intraperitoneal, orthotopic, and metastatic xenograft models of human ovarian cancer. Mol Ther. 2004;10:1032–42. doi: 10.1016/j.ymthe.2004.08.013. [DOI] [PubMed] [Google Scholar]
- [42].Naora H, Montell DJ. Ovarian cancer metastasis: integrating insights from disparate model organisms. Nat Rev Cancer. 2005;5:355–66. doi: 10.1038/nrc1611. [DOI] [PubMed] [Google Scholar]
- [43].Armstrong DK, Bundy B, Wenzel L, Huang HQ, Baergen R, Lele S, Copeland LJ, Walker JL, Burger RA. Intraperitoneal cisplatin and paclitaxel in ovarian cancer. N Engl J Med. 2006;354:34–43. doi: 10.1056/NEJMoa052985. [DOI] [PubMed] [Google Scholar]
- [44].Markman M, Walker JL. Intraperitoneal chemotherapy of ovarian cancer: a review, with a focus on practical aspects of treatment. J Clin Oncol. 2006;24:988–94. doi: 10.1200/JCO.2005.05.2456. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table S1. Genes and primer sets for gene expression analysis by real-time RT-PCR.