

Abstract
The importance of helicases in nucleic acid metabolism and human disease has raised the bar for understanding how these unique enzymes function to perform their biological roles at the molecular level. Here we will describe experimental procedures and strategies to investigate the functions of helicases. These functional assays have been used to study DNA helicases important for the maintenance of genomic stability and genetically linked to age-related diseases and cancer. We will focus on the description of fluorometric helicase assays, protein displacement assays, and methods to characterize helicase activity on alternate DNA structures (triplex, quadruplex) used by our laboratory. The procedures to study these helicase functions are described in step-by-step detail to enable researchers interested in nucleic acid metabolism and related fields to apply these techniques to their own research questions.
Keywords: helicase, DNA repair, replication, fluorescence, ATPase, quadruplex, triplex, streptavidin, FANCJ, Werner syndrome
INTRODUCTION
Helicases are enzymes that catalytically separate complementary strands of structured nucleic acid molecules in an energy-dependent reaction that is fueled by the hydrolysis of nucleoside triphosphate [1, 2]. The generation of single-strand DNA or RNA is critically important in virtually all aspects of nucleic acid metabolism including DNA replication and repair, recombination, transcription, translation, and chromosome segregation. Therefore, helicases have significant roles in processes of genomic stability and their functions are necessary for preventing age-related diseases and cancer [3–6]. Because of their essential functions in processes that affect cell proliferation, DNA repair, and genome homeostasis, helicases have been proposed to serve as reasonable targets for anti-cancer therapies in which helicase-dependent pathways are targeted for synthetic lethality in cancer cells [7, 8]. However, experimental evidence to support this hypothesis is still lacking.
Many advances have been made in the molecular, cellular and genetic analyses of helicases. However, many details of how helicases function remain to be better characterized. Deciphering the mechanisms of action by helicases at the molecular level will enable researchers to better understand their cellular roles and how their activities are important for the integrity of the genome and prevention of human disease. The work from our laboratory has largely focused on the study of the human RecQ helicases (WRN, BLM, RECQ4, RECQ1, RECQ5) that are implicated in the maintenance of genomic integrity and genetic disorders characterized by premature aging and cancer [3, 9]. We have also investigated the molecular and cellular roles of a DNA helicase (FANCJ) implicated in the hereditary disease Fanconi Anemia [6, 10]. In this Methods article, we will describe some of our experimental approaches and strategies to study molecular aspects of the catalytic activities of human DNA helicases. We will focus on the description of fluorometric helicase assays, protein displacement assays, and methods to characterize helicase activity on alternate DNA structures (triplex, quadruplex) used by our laboratory. The procedures to study these helicase functions are described in step-by-step detail to enable researchers interested in nucleic acid metabolism and related fields to apply these techniques to their own research questions.
1. Fluorescence-based real-time kinetic helicase assays
Fluorometric helicase assays can be used to acquire unique mechanistic information about the helicase under study. We investigated the kinetics of DNA unwinding catalyzed by the Werner syndrome helicase (WRN) using a fluorescence-based real-time kinetic helicase assay [11]. The initial rate of unwinding increased with WRN concentration, indicating that excess enzyme over DNA substrate improved the ability of WRN to unwind the DNA substrate. Under presteady state conditions in which the enzyme is saturated with an excess of DNA substrate, the burst amplitude revealed a 1:1 ratio between WRN and DNA substrate, suggesting an active monomeric form of the helicase [11].
1.1. Design of Fluorescent Molecule Conjugated DNA Substrate
Two complementary DNA oligonucleotides were designed to construct a partial duplex DNA substrate of appropriate duplex and tail length for the particular helicase being studied. In many cases, the helicase does not initiate unwinding from the blunt duplex end of the helicase substrate; consequently, the donor and acceptor fluorophore molecules are positioned at the 3′- and 5′-ends of the blunt end of the substrate attached to paired bases [12]. Upon strand separation by helicase-catalyzed unwinding of the duplex DNA substrate, the fluorophore-quencher pair is separated from one another and a fluorescence increase is observed (Fig. 1A, 1B). This type of helicase unwinding assay measures all-or-none product formation since the oligonucleotide must be completely unwound to observe the fluorescence change. However, fluorescence-based helicase assays have been described in which the fluorescence resonance energy transfer (FRET) probes were placed at the ss-ds-DNA junction to monitor partial DNA unwinding (for example, [13]). In the following section, we will describe a fluorescent DNA substrate with a fluorophore-quencher pair at the blunt end of a forked duplex DNA substrate used to study WRN helicase activity [11].
Fig. 1. Fluorescence-based helicase assays.

Panel A, Schematic of the FRET-based helicase assay to measure helicase activity. Fluorescein (F) is covalently attached to the 3′ blunt end and hexachlorofluorescein (HF) is attached to the 5′ blunt end of the forked duplex DNA substrate. When in close proximity, HF quenches the signal emitted from F upon excitation. Upon unwinding, the emission from F upon excitation is free to be detected because the HF is no longer in close proximity with F. Note that all-or-none unwinding of the DNA substrate is measured using the FRET-based assay. Panel B, Kinetics of fluorescence increase (as a measure of helicase activity on the forked duplex substrate) according to time as measured by the FRET-based helicase assay. Panel C, Schematic of the dye displacement assay to measure helicase activity. The dye molecules are pre-bound to the duplex DNA substrate (gold ovals), causing them to fluoresce. As the dsDNA is unwound, the dye molecules are released which is measured by a decrease in the amount of fluorescence. Note that partial unwinding by a helicase can be measured using the dye displacement assay. Panel D, Kinetics of fluorescence (F) decrease (as a measure of helicase activity on the forked duplex substrate) according to time (T) as measured by the dye-displacement assay.
1.2. Preparation of Fluorescent DNA Substrate
Duplex DNA substrates for the fluorescent helicase studies were prepared in the same way as radiolabeled duplex DNA substrates except that equimolar concentrations of complementary oligonucleotides were used for annealing to avoid effects of excess pre-existing ssDNA on fluorescence measurements taken during the helicase-catalyzed unwinding reaction. To ensure complete annealing of the duplex DNA substrate, annealing reactions using equimolar concentrations of a 32P-labeled oligonucleotide and its complementary strand were performed in parallel and checked on native polyacrylamide gel electrophoresis to determine percent of oligonucleotides that are hybridized to one another.
Equimolar concentrations (500 nM) of two polyacrylamide gel electrophoresis (PAGE) purified oligonucleotides (one with the acceptor molecule (hexachlorofluorescein) and one with the donor molecule (fluorescein)), provided by Midland Certified Reagent Company, were placed in TE (pH 8.0) with 50 mM NaCl.
The oligonucleotide annealing reaction mixture was heated at 100 °C for 5 min.
The reaction mixture was moved to a 70 °C heat block which was immediately switched off and allowed to cool to room temperature (approximately 2.5 h).
The DNA substrate was stored at 4 °C until needed for experiments.
1.3. Stopped-Flow Fluorescence Spectrophotometer Setup
A stopped-flow fluorescence spectrophotometer allows for rapid and continuous analysis of a helicase reaction containing a fluorescently-labeled DNA substrate [12]. Reaction components are separated between two syringes to prevent mixing before measurements are taken. The syringe contents are then pushed into a 20 μl flow cell chamber using nitrogen gas under 120 psi. The amount of reaction volume driven through is set using a stop valve. At least 100–150 ul of reaction volume should be driven through the flow cell per run.
A recirculating water bath was turned on and set to the reaction temperature required for helicase activity.
The nitrogen gas supply was opened and set to 120 psi.
The fluorescence spectrophotometer instrument (Applied Photophysics) was turned on and allowed to warm up for 30 min.
The computer and software that monitor the helicase reaction were turned on.
The monochromator was set to a slit width of 1 mm.
Based on the fluorescence properties of the donor and acceptor fluorophores covalently attached to the helicase substrate, the excitation wavelength was set (e.g., 492 nm for fluorescein).
A cut-on filter was used to monitor fluorescence emission changes above a certain wavelength (e.g., 520 nm for fluorescein).
Two syringes containing sterile water were attached to the ports of the instrument and used to wash the tubing and flow cell before conducting helicase experiments.
1.4. Stopped-Flow Fluorescence Spectrophotometer Helicase Reactions
Two 15 ml tubes were placed on ice and 1X helicase reaction salts were added to each one with a volume (typically 150 μl per tube) sufficient for several runs.
In one tube, DNA substrate was added to a concentration of 1.6 nM (since it will be mixed with the contents of the other syringe there will a 2-fold dilution, resulting in a 0.8 nM final DNA substrate concentration).
In the other tube, two-fold concentrations of helicase protein and ATP were added. Note: an ATP stock was prepared from lyophilized powder (GE Healthcare) dissolved in 1X reaction salts. Typically, final 1X ATP concentrations range from 2–5 mM.
The contents of each 15 ml tube were mixed well and drawn up into separate syringes.
The syringes were added to the appropriate ports and washed through the tubing and flow cell to prime the system.
The PM-voltage, number of data points to be collected, and time duration were set based on expected fluorescence values and reaction duration.
Each run was manually started and data was collected for at least four trials so that data could be averaged, generating one curve.
1.5. Helicase Control Reactions for Stopped-Flow Fluorescence Assay
A few controls are needed to normalize fluorescence data to percent substrate unwound [11]. Minimum and maximum fluorescence levels should be determined. These fluorescence levels can be used to normalize the results of the helicase reactions to percent substrate unwound. This allows for direct comparison with a radiometric assay carried out with the same concentrations of enzyme and DNA.
Two 15 ml tubes were set up as above except duplex DNA substrate was omitted from one syringe and replaced with the same concentration of ssDNA oligonucleotide containing the donor molecule.
The 15 ml tube contents were transferred to syringes as above and loaded onto the instrument.
The fluorescence spectrophotometer was set at the same voltage and excitation wavelength as described above.
A flat line was obtained indicating the maximum donor fluorescence possible assuming complete unwinding of the fluorescent DNA substrate (100% substrate unwound).
For the minimum fluorescence value, ATP was omitted from the tube that contains the helicase substrate used for standard fluorometric helicase assays. The flat line obtained in this no ATP control reaction indicated the absence of helicase activity (0% substrate unwound).
The fluorescence change during the course of the helicase reaction was fit to the fluorescence control data to determine percent substrate unwound.
1.6. Fluorescent Dye Displacement Helicase Assay
Fluorescent intercalating dyes can be used to monitor helicase reactions in real-time. Unlike the assay above, the fluorescent molecule is not covalently bound to the DNA molecule but instead intercalates into the dsDNA substrate forming a stably bound complex between the fluorescent dye and the duplex portion of the helicase substrate. Duplex DNA binding by the dye enhances the fluorescence of the dye. Unwinding by the helicase disrupts the dye-duplex DNA interaction as the helicase unwinds the duplex DNA, resulting in a fluorescence decrease (Fig. 1C, 1D). The dye has minimal fluorescence in solution or when bound to ssDNA. One such dye is Hoechst 33258 [14, 15].
An advantage of dye displacement assays compared to strand displacement assays is that partial unwinding of the DNA substrate can be detected. For example, a dye displacement assay can be used to detect partial unwinding of a DNA substrate harboring a DNA lesion (e.g., polyglycol modification to the sugar phosphate backbone) within the duplex region of the DNA substrate. This analysis was performed to address the hypothesis that FANCJ unwound the duplex (16 bp) adjacent to the fork and became trapped at the vicinity of the backbone modification, leaving the remaining duplex (12 bp) intact [15].
The stopped-flow fluorescence spectrophotometer (Applied Photophysics) was set up as previously mentioned with exceptions noted below.
The substrate was made with the same oligonucleotides that were used for the fluorescence molecule conjugated substrate except the oligonucleotides lacked donor and acceptor fluorophores.
For Hoechst 33258, a 344 nm wavelength was used for excitation.
Emissions greater than 400 nm were measured using a 400 nm long pass filter.
One syringe contained 1.6 nM DNA substrate and 200 nM Hoechst 33258 in helicase 1X reaction buffer. The other syringe contained an equal volume of helicase reaction buffer with two-fold concentrations of helicase protein and ATP.
Several curves were generated indicating a decrease in fluorescence intensity upon strand separation by helicase-catalyzed unwinding of the Hoechst 33258-bound duplex DNA substrate.
To normalize the data, similar reactions were performed except the Hoechst 33258 dye was in a syringe with the two unannealed ssDNA molecules to determine the minimum fluorescence that could be achieved with these reactions (100% substrate unwound).
Likewise, reactions were performed with the Hoechst 33258-bound duplex DNA substrate in the absence of ATP to determine maximum fluorescence (0% substrate unwound).
2. Biotinylated DNA-streptavidin displacement assays
Helicases can be characterized for their ability to translocate along single- or double-stranded DNA substrates [16] and remove protein obstacles in their path [17]. One experimental approach to examine helicase-catalyzed protein displacement from a DNA substrate is to take advantage of the very stable interaction of streptavidin bound to a biotinylated DNA molecule (for review, see [18]). In this assay, a single-stranded DNA oligonucleotide is used that contains a biotin covalently attached to a defined nucleotide of the polynucleotide chain. The position of the biotin is determined based on the directionality of translocation by the helicase. Traditionally, helicase directionality is inferred from results of helicase assays with DNA directionality substrates; however, newer assays are being developed to assay directionality of helicase translocation [19]. In its simplest version, a 5′ to 3′ helicase would displace streptavidin bound to a biotin moiety covalently attached to the 3′ end of the oligonucleotide, whereas a 3′ to 5′ helicase would displace streptavidin bound to the biotin attached to the 5′ end (Fig. 2A). The helicase may be able to disrupt the biotinylated DNA-streptavidin complex or be blocked by the streptavidin bound tightly to the DNA molecule [16–18]. This information can be used to better understand how motor ATPases and helicases disrupt biologically relevant protein-DNA interactions. For example, the human Bloom’s syndrome helicase (BLM) [20], FANCJ [17], and RECQ5 [21] helicases effectively strip RAD51 from DNA, an activity that is important for motor ATPase-driven regulation of homologous recombinational repair.
Fig. 2. Streptavidin-bound biotinylated DNA substrates as a tool to study helicase-related functions.




Panel A, Schematic of the biotinylated single-stranded DNA streptavidin displacement assay to determine directionality of single-stranded DNA translocation by a helicase. A 5- to 3′ helicase displaces streptavidin bound to a biotin attached to the 3′ end of the single-stranded oligonucleotide. A 3′ to 5′ helicase displaces streptavidin bound to a biotin attached to the 5′ end of the single-stranded oligonucleotide. Note that this assay only can be applied to determine ssDNA translocation of a helicase that has the ability to disrupt the high affinity interaction of streptavidin bound to the biotinylated oligonucleotide. Panel B, A 3′ to 5′ helicase that unwinds a 3′-single-stranded DNA-tailed duplex substrate with streptavidin bound to the end of the 3′-ssDNA tail does not require a free DNA end to unwind the duplex. Panel C, A 3′ to 5′ helicase may be completely blocked by streptavidin bound to the 3′-ssDNA tail just upstream of the single-stranded/double-stranded DNA junction of a simple 3′-tailed DNA substrate. Panel D, The ability of a helicase to efficiently unwind the forked duplex with streptavidin bound just upstream of the junction suggests that the helicase recognizes elements of the fork structure to initiate unwinding. Panel E, The ability of a helicase to bypass the internal streptavidin block on the 5′-flap substrate and unwind the downstream duplex suggests that the helicase interacts differently with the 5′-flap structure compared with the 3′-ssDNA tailed duplex to initiate unwinding. Panel F, The ability of a helicase to effectively stimulate FEN-1 cleavage on a flap DNA substrate with streptavidin bound to the terminal 3′ nucleotide at the end of the upstream duplex suggests that the helicase does not require a free upstream end to stimulate FEN-1 cleavage of the 5′ flap substrate.
The streptavidin displacement assay can also be used to learn mechanistic information about the DNA substrate preference and recognition for initiation of helicase unwinding. WRN unwound a 3′-single-stranded DNA-tailed duplex substrate with streptavidin bound to the end of the 3′-ssDNA tail, suggesting that WRN does not require a free DNA end to unwind the duplex (Fig. 2B); however, WRN was completely blocked by streptavidin bound to the 3′-ssDNA tail 6 nucleotides upstream of the single-stranded/double-stranded DNA junction (Fig. 2C) [16]. WRN efficiently unwound the forked duplex with streptavidin bound just upstream of the junction (Fig. 2D), suggesting that WRN recognizes elements of the fork structure to initiate unwinding. WRN was also able to bypass the internal streptavidin block on the 5′-flap substrate (Fig. 2E), suggesting that WRN interacts differently with the 5′-flap structure compared with the 3′-ssDNA tailed duplex to initiate unwinding [16]. The ability of WRN to target DNA replication/repair intermediates may be relevant to its role in genome stability maintenance.
To characterize the mechanism for WRN stimulation of structure-specific cleavage by Flap Endonuclease I (FEN-1), we also employed streptavidin-bound DNA substrates [22]. WRN effectively stimulated FEN-1 cleavage on a flap DNA substrate with streptavidin bound to the terminal 3′ nucleotide at the end of the upstream duplex (Fig. 2F), indicating that WRN does not require a free upstream end to stimulate FEN-1 cleavage of the 5′ flap substrate. These results indicate that the mechanism whereby WRN stimulates FEN-1 cleavage is distinct from that proposed for the functional interaction between proliferating cell nuclear antigen (PCNA) and FEN-1. The ability of WRN to efficiently target the 5′ flap substrate for unwinding or facilitate cleavage by FEN-1 suggests a specialized role of WRN in Okazaki fragment processing during cellular DNA replication.
2.1. Design of Biotinylated DNA Substrate
Oligonucleotides can be designed with a biotin covalently attached to a nucleotide within the polynucleotide chain at the 5′ end, 3′ end, or a defined internally positioned nucleotide. We have purchased biotinylated oligonucleotides from Lofstrand Labs or Midland Certified Reagent Company. The position of the biotin should also account for the minimum number of bases needed for the helicase to bind or recognize the substrate; otherwise, lack of streptavidin displacement activity may be a consequence of the helicase inefficiently binding to the ssDNA region adjacent to the streptavidin-bound biotin and with the appropriate polarity.
2.2. Preparation of Radiolabeled Biotinylated DNA Substrate
For ssDNA and other substrates, 10 pmol of biotinylated oligonucleotide was 5′-32P-end labeled in a 20 μl reaction with 30 μCi [γ-32P]ATP and T4 Polynucleotide Kinase (New England Biolabs) in 1X T4 PNK reaction salts at 37 °C for 1 h.
The kinase was inactivated by incubating the reaction at 65 °C for 20 min.
After heat inactivation, unincorporated ATP was removed by centrifuging the sample at 700 × g for 2 min in a G-25 spin column (GE Healthcare).
The collected sample was either stored at 4 °C (ssDNA) or annealed to a complementary oligonucleotide and then stored at 4 °C.
2.3. Biotinylated DNA-Streptavidin Displacement Assay
Streptavidin displacement assays were performed in the same way as helicase assays except that the biotinylated oligonucleotide was preincubated with streptavidin. Recombinant streptavidin was purchased as a lyophilized powder (Sigma) and dissolved in 25 mM HEPES (pH 7.4), 20% glycerol, and 10 mM NaCl. Dissolved streptavidin stock aliquots (100 μM streptavidin) were frozen in liquid nitrogen and stored at −80 °C.
20 μl reaction mixtures were made containing 1X helicase reaction salts, nucleoside triphosphate (typically 2–5 mM), and 0.5 nM DNA substrate.
The substrate in the reaction mixture was preincubated with 100 nM streptavidin for 10 min at 37 °C in the helicase reaction salts.
A control reaction mixture was set up that did not include streptavidin, as well as a control with streptavidin but without helicase.
After 10 min, helicase was added to each reaction mixture immediately followed by the addition of 1 μM biotin to capture any displaced or free streptavidin in the reaction.
The reactions were carried out for an additional 15 min at the standard helicase reaction temperature.
Reactions were quenched with 10 μl of stop buffer (50 mM EDTA, 40% glycerol, 0.9% SDS, 0.05% bromophenol blue, and 0.05% xylene cyanol).
Reaction products were resolved on nondenaturing 12% polyacrylamide gels (18 × 16 cm) run for 2 hr at 180 volts.
Phosphorimager screens were used to detect radioactive DNA on gels and were scanned with a Typhoon scanner (GE Healthcare).
ImageQuant software was used to analyze the bands on the gel, the slower migrating band representing streptavidin-bound DNA and the faster migrating band representing streptavidin-free DNA.
3. Triplex DNA substrate preparations and unwinding assays
Triplexes are formed when a third strand lies in the major groove of duplex DNA and can occur most readily on polypurine–polypyrimidine sequences [23]. The third strand may be composed of either pyrimidines or purines, and the stability of the resulting triplex structure is dictated by the specific sequence. Triplexes form when an appropriate sequence partially melts with one of the single strands folding back to complex with an adjacent duplex (intramolecular triplex) or from a different molecule to form an intermolecular triplex structure. Triplexes have been shown to exist in chromosomes and nuclei [24, 25], and are proposed to be a source of genomic instability [26]. Triplex formation by the Friedreich’s ataxia (GAA)n repeat inhibits DNA replication based on observations that the repeated sequence inhibits DNA polymerization in vitro [27, 28] and replication forks stall at (GAA)n repeats in vivo [29]. The ability to form triplexes using oligonucleotides directed against specific target sequences has prompted their consideration as gene-targeting reagents [30]. However, triplex formation in vivo may be at least partly inhibited by triplex-destabilizing proteins or helicases that exist in cells [31–34].
In the following section, we will describe simple protocols to prepare triplex DNA substrates that can be tested for unwinding by DNA helicases in vitro. Examples of triplex DNA substrates are shown in Table 1.
Table 1.
Triplex DNA Substrates Used for In Vitro Helicase Studiesa
| Triplex DNA | Structure | Details | Nucleotide Sequence (5′–3′) |
|---|---|---|---|
| 3′ Tail Blunt Triplex | 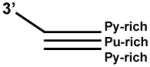 |
For helicases that require 3′ tail to load, (e.g. BLM, WRN) | TC30W: TTTCTTTTTTCTTCTTTTCTTTCTTTTTCT TC30C: AGAAAAAGAAAGAAAAGAAGAAAAAAGAAA TC30 3′ tailb: TCTTTTTCTTTCTTTTCTTCTTTTTTCTTTTCACGCTCCGTACGA |
| 5′ Tail Blunt Triplex | 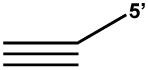 |
For helicases that require 3′ tail to load, (e.g. FANCJ) | TC30 5′ tailc: TGACGCTCCGTACGATCTTTTTCTTTCTTTTCTTCTTTTTTCTTT TC30W and TC30C used to make duplex |
| 5MeC 3′ Tail Blunt Triplex | 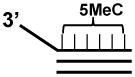 |
More stable at physiological pH | 5MeC TC30 3′ tailb,d: TCTTTTTCTTTCTTTTCTTCTTTTTTCTTTTCACGCTCCGTACGA TC30W and TC30C oligonucleotides used to make duplex |
| Plasmid-based Triplex |  |
This substrate has a long (4 kb) underlying duplex | Plasmid pSupF5 is linearized by digestion with Nde I, and the third strand is annealed to triplex target site in duplex. TC30 3′ tail oligonucleotide used as third strand. |
| Purine-rich Third Strand Triplex | 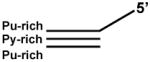 |
Triplex can be formed at neutral pH | 5′-15nt-TC30PURc: TGACGCTCCGTACGAAGGAAGGGGGGGGAGGAGGGGGAGGGGGAG PUR30W: GGGGGAGGGGGAGGAGGGGGGGGAAGGA PYR30C: TCCTTCCCCCCCCTCCTCCCCCTCCCCCTC |
| Intramolecular H-DNA Triplex | Forms intramolecular triplex, protected from Ear I digestion | TC100-foldbacke: TCTTTTTCTTTCTTTTCTTCTCTTTTCTTTTTTTTTTTCTTTTCTCTTCT↓TT TCTTTCTTTTTCTTTTTTAGAAAAAGAAAGAAAAGAAGAGAA AAGAAA |
See text for details.
3′ overhang ssDNA tail sequence is underlined.
5′ overhang ssDNA tail sequence is underlined.
Cytosines in the 5MeC-TC30 3′ tail oligonucleotide are 5-methylcytosines (5MeC), indicated in bold.
Digested site of Ear I is marked by an arrow.
3.1. Preparation of Oligonucleotide-based Triplex DNA Substrate
The flush triplex substrates contain triplex DNA regions without underlying duplex extensions on either side of the triplex site. The triplex forming third strand is either a 30-mer or 60-mer annealed to a 30 bp duplex target, respectively (Table 1) [31]. A triplex substrate that is more stable at physiological pH can be made using a pyrimidine motif third strand containing 5-MethylCytosine (5MeC) throughout the sequence. In the buffer used for the triplex destabilization assays (see below), the Tm of the triple helix with 5MeC-TC30 was 57.9 °C compared to 54.0 °C for the nonmethylated TC30 triplex substrate [34].
The 30 bp blunt DNA duplex is prepared by incubating equimolar amounts of oligonucleotides TC30W and TC30C (6 pmol each) in an annealing buffer (TE (pH 8.0), 50 mM NaCl) for 5 min at 100 °C, followed by slow cooling over a period of several hours to room temperature.
3 pmol 5′-32P-labeled TC30 oligonucleotide was incubated overnight at room temperature with the 30-bp annealed duplex (TC30W and TC30C) in a buffer containing 33 mM Tris–Acetate (pH 5.5), 66 mM KOAc, 100 mM NaCl, 10 mM MgCl2, and 0.1 mM spermine. The 2:1 ratio of annealed duplex: radiolableled third strand insures that all the labeled third strand is annealed to the duplex target site.
The 5′-tailed blunt triplex substrate with a pyrimidine third stand, which contained a triplex region without underlying duplex extensions on either side of the triplex site, consisted of a 30-bp duplex (TC30W and TC30C; Table 1) annealed to TC30 5′ tail under the annealing conditions described above.
The 3′-tailed blunt triplex was prepared in a similar manner to the 5′-tailed triplex substrate except a TC30 3′ tail was used as the third strand for the triplex annealing reaction (Table 1).
3.2. Preparation of Plasmid-based Triplex DNA Substrate
For plasmid-based triplex DNA substrates with a pyrimidine third strand, the plasmid pSupF5, which contains a duplex sequence that serves as a target for a third strand designated TC30, was used (Table 1) [31]. The plasmid-based triplex DNA substrate characterized by a 30-mer third strand annealed to a 4 kb duplex fragment provides an advantage that it has a long underlying duplex extension on each side of the triplex site, enabling the investigator to study triplex unwinding by a helicase on a DNA substrate without nearby blunt duplex ends. Therefore, the plasmid-based triplex may represent a more physiological DNA substrate for the helicase.
pSupF5 double-stranded plasmid was cleaved with NdeI, yielding fragments of 4 and 0.6 kilobase pairs as determined by agarose gel electrophoresis. The triplex site lies 1800 bp from one end of the large fragment.
Triplexes were prepared by incubation of 3 pmol of the indicated 5′-32P-labeled TC30 oligonucleotide or TC30 with a 15-nt 5′-ssDNA tail, designated 5′-15nt-TC30 (Table 1) overnight at room temperature with 6 pmol of NdeI-cleaved plasmid in triplex annealing buffer containing 33 mM Tris Acetate (pH 5.5), 66 mM KOAc, 100 mM NaCl, 10 mM MgCl2, and 0.1 mM spermine.
DNA complexes were separated from unbound oligonucleotide by gel filtration chromatography using Bio-Gel A-5 M resin (Bio-Rad) equilibrated in triplex annealing buffer described above. Triplex DNA substrates were stored at 4 °C for two weeks and found to be stable for functional assays.
3.3. Verification of pSupF5 Plasmid-based Triplex
The pSupF5 plasmid-based triplex DNA substrate was verified for its integrity by XbaI restriction digestion. Restriction protection of the XbaI site, which resides in the triplex target site, demonstrated that the third strand had annealed to its target site [31].
Triplex complexes isolated by gel chromatography were ethanol precipitated in the presence of 2.5 M ammonium acetate, washed with 70% ethanol, and allowed to dry.
Precipitated DNA was resuspended in 50 mM NaCl, 10 mM Tris-HCl (pH 7.0), 10 mM MgCl2, 1 mM dithiothreitol, 0.1 mM spermine, and 100 μg/ml bovine serum albumin.
DNA was digested with PstI and XbaI at 37 °C for 1 hour, and products analyzed by electrophoresis on 1% agarose gels. Restriction protection of the XbaI site which resides in the triplex target site was performed and demonstrated for the restriction fragment based triplex substrate as previously described [31].
3.4 Preparation of Oligonucleotide-based Triplex with Purine-rich Third Strand
Triplexes characterized by a pyrimidine-rich third strand annealed to the Watson-Crick duplex target site, as described above, cannot be formed at neutral pH unless cytosines of the third strand are modified (e.g. methylated). In contrast, triplexes can be formed at neutral pH in the presence of bivalent metals from an incoming purine-rich third strand that is annealed to a duplex that consists of a pyrimidine-rich strand and a purine-rich strand (Table 1). This type of triplex may be more relevant to physiological conditions.
To prepare a 5′-tailed blunt triplex DNA substrate, a 30-bp duplex (25 pmol, PUR30W and PUR30C; Table 1) was annealed to 10 pmol of radiolabeled 5′-15nt-TC30PUR. Annealing was performed in the presence of 30 mM PIPES (pH 7.0), 10 mM MgCl2, 100 mM LiCl, 0.1 mM spermine at room temperature overnight.
Products of annealing reactions were resolved on native 8% polyarcylamide gels to resolve the triplex DNA substrate with a purine third strand from G-quadruplex, which migrated much more slowly on the native gel. The following Running buffer was used for electrophoresis: 30 mM PIPES (pH 7.0), 10 mM MgCl2, 100 mM LiCl. The triplex DNA substrate was gel-purified and electroeluted in Running buffer described above using Midi D-Tube Dialyzer (6–8 kD MWCO, Novagen). The concentration of triplex DNA substrate was determined using the specific activity of the radiolabeled oligonucleotide (5′-15nt-TC30PUR) as determined using a liquid scintillation counter (MicroBeta TriLux, Perkin Elmer).
3.5. Triplex DNA Unwinding Assay
Helicase-catalyzed triplex DNA unwinding assays are based on the same principal as helicase assays with more conventional duplex DNA substrates. Differential mobility of intact substrate and released strand on native polyacrylamide gels enables one to visualize the all-or-none products of helicase activity on a triplex DNA substrate.
The specified radiolabeled triplex DNA substrate was incubated with helicase and ATP under reaction conditions typically suitable for unwinding assays with conventional duplex DNA substrates. Reactions were initiated by the addition of protein and typically incubated at 30 °C for 15 min.
At the end of the incubation period, a 10 μl aliquot of loading buffer (40% glycerol, 0.9% SDS, 0.1% bromophenol blue, 0.1% xylene cyanol) was added to the reaction mixture, and the sample was immediately loaded on to 18 cm × 16 cm nondenaturing polyacrylamide gels (10% or 12% acrylamide, 40 mM Tris–acetate (pH 5.5), 5 mM MgCl2, 25% glycerol) and electrophoresed at 4 °C for 4 hr.
Radiolabeled DNA species on polyacrylamide gels were visualized with a PhosphorImager and quantitated using ImageQuant software (Amersham Biosciences).
3.6. Preparation of Intramolecular Triplex DNA Substrate and Helicase Activity Analysis
Intramolecular H-DNA triplex substrate may represent a more physiological structure that would form naturally. Therefore, it may be desired to prepare an intramolecular DNA substrate using a designed oligonucleotide (TC100-foldback) that can fold back on itself [34] (Table 1).
The TC100-foldback oligonucleotide was 5′ 32P end-labeled and allowed to anneal to form an intramolecular triplex under the conditions described above.
The TC100-based triplex DNA substrate was verified for its integrity by EarI restriction digestion and analysis on 10% polyacrylamide gels. Restriction protection of the EarI site, which resides in the triplex target site, was used to determine the extent that the third strand had annealed to its target site.
The intramolecular triplex DNA substrate was incubated with helicase and ATP under reaction conditions typically suitable for unwinding assays with conventional duplex DNA substrates. Reactions were initiated by the addition of helicase and typically incubated at 30 °C for 15 min.
At the end of the incubation period, the DNA was digested with EarI and the products were resolved on 10% polyacrylamide gels.
4. G-Quadruplex DNA substrate preparation and unwinding assays
G-quadruplex (G4) DNA structures form by the planar arrays of four hydrogen-bonded guanines [35]. Genomic DNA sequences which can form G-quadruplexes in vitro under physiological conditions can be found in the G-rich 3′ overhangs of telomeres such as those of Tetrahymena, ribosomal DNA, immunoglobulin heavy chain switch regions, and promoters [36]. The unique metabolism of G- quadruplexes may influence a number of biological processes including immunoglobulin gene rearrangements, promoter activation, and telomere maintenance [37]. Several helicases which can unwind G-quadruplex DNA structures have been reported [38–41].
The structures and stabilities of G-quadruplexes are known to be dependent on the presence and concentrations of monovalent cations K+ or Na+, because the cations can coordinate the carbonyl oxygen of the guanine and promote stacking interactions of the G-quartets [42]. Below are procedures used by our group for G4 DNA substrate preparation and G4 unwinding assays [41]. Polyacrylamide gel electrophoresis-purified oligonucleotides were used for the preparation of DNA substrates. Some commonly used G4 DNA substrates are listed in Table 2. The TP sequence is a consensus repeat from the murine immunoglobulin Sγ2b switch region; CGG represents the Fragile X syndrome triplet repeat; human telomeric DNA and ribosome DNA sequence are G-rich sequences which can form G4 structures. Participation of specific guanines in the G4 DNA structure can be confirmed by methylation protection analysis at the N-7 of guanine [42].
Table 2.
G-Quadruplex DNA Substrates Used for In Vitro Helicase Studiesa
| G-Quadruplex DNA | Structure | Details | Nucleotide Sequence (5′–3′)b |
|---|---|---|---|
| 3′-Tail d(CGG)7-G4 | 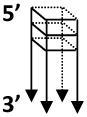 |
- Fragile X syndrome CGG repeats - For helicases that require 3′ tail to load, (e.g. BLM, WRN) |
3′-Tail d(CGG)7: CGGCGGCGGCGGCGGCGGCGGCGTGGA CTG |
| 5′-Tail d(CGG)7-G4 | 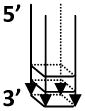 |
- Fragile X syndrome CGG repeats - For helicases that require 5′ tail to load, (e.g. FANCJ) |
5′-Tail d(CGG)7: GTCAGGTGCGGCGGCGGCGGCGGCGGCGGC |
| TP-G4 | 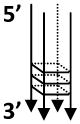 |
- A consensus repeat from murine immunoglobulin heavy chain switch region - Both ends contains tails, 5′-21 nt tail; 3′-7 nt tail |
TP: TGGACCAGACCTAGCAGCTATGGGGGAGCTGGGGAAGGTGGGAATGTGA |
| rD4-G4 | 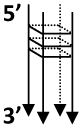 |
- A sequence from human 28 S ribosomal RNA gene (acc. No. M11167) - Both ends contains tails, 5′-10 nt tail; 3′-25 nt tail |
rD4: TTGAAAATCCGGGGGAGAGGGTGTAAATCTCGCGCCGCGCCGTACC |
| Z33-G4 |  |
- A complementary sequence to bases 159–191 of the supF gene in plasmid pZ189 - Both ends contain 14 nt tails |
Z33: AAAGTGATGGTGGTGGGGGAAGGATTTCGAACC |
| 5′-15ntTeR2-G4 |  |
- Human telomere sequence - 5′ end contain 15 nt tail |
5′-15ntTeR2: TCATGACTAGACATGTTAGGGTTAGGGTTA |
See text for details.
The guanine residues that compose the G-quartet structures in oligonucleotides are underlined.
DNA helicases such as WRN and BLM with 3′ to 5′ directionality of unwinding require a 3′ ssDNA tail to unwind the adjacent G-quadruplex structure [40] (Fig. 3A). In contrast, the 5′ to 3′ FANCJ helicase requires a 5′ ssDNA tail to unwind the adjacent G-quadruplex structure [41] (Fig. 3A). Here we describe the 5′ tailed TP-G4 DNA substrate preparation and unwinding assays by FANCJ helicase. One can also adapt this protocol for other G4 DNA substrates and helicases or G4-destabilizing proteins.
Fig. 3. G-quadruplex DNA unwinding by FANCJ helicase.

Panel A, Schematic of G-quadruplex DNA substrates with either a 3′ single-stranded DNA tail or 5′ single-stranded DNA tail that can be unwound by specialized 3′ to 5′ helicases (e.g., WRN, BLM)[40] or 5′ to 3′ helicases (e.g., FANCJ)[41], respectively. Panel B, Helicase-catalyzed G-quadruplex DNA unwinding assays are based on the same principal as helicase assays with more conventional duplex DNA substrates. Differential mobility of intact substrate and released strand on native polyacrylamide gels enables one to visualize the all-or-none products of helicase activity on a G-quadruplex DNA substrate. Shown is a gel image from the products of FANCJ helicase reactions using a human telomere sequence G-quadruplex DNA substrate (see Table 2)[41]. Products were resolved on a native 8% polyacrylamide gel. M, radiolabeled 5′-15ntTeR2 single-stranded oligonucleotide.
4.1 Preparation of G-Quadruplex DNA Substrate
Single-stranded oligonucleotides TP (Table 2) (10 pmol) was 5′-end-labeled with T4 polynucleotide kinase (New England Biolabs) and [γ-32P]ATP (PerkinElmer) in 30-μl reactions containing 10 mM Tris-HCl, pH 7.4, 10 mM MgCl2, 30 μCi [γ-32P]ATP, and 10 units of kinase at 37 °C for 30 min. Unincorporated [γ 32P]ATP was removed using a Sephadex G-25 spin column (GE Healthcare), and labeled DNAs were collected as the flow-through fractions. A small volume (5 μl) of labeled oligonucleotide was saved to use as a marker.
NaCl was added to a final concentration of 1M according to pooled volume of labeled oligonucleotide.
Samples were heated for 5 min at 100 °C, and immediately placed in a 70 °C heat block for overnight incubation.
The heat block was turned off in the morning of the second day and the samples were allowed to slowly cool to room temperature over a period of 2–3 hours.
A preparative non-denaturing 8% polyacrylamide gel that contained 0.5X TBE and 10 mM KCl was prepared.
Equal volume of 2X G4 DNA loading buffer (74% glycerol, 0.01% xylene cyanol, 0.01% bromophenol blue, 10 mM KCl, 20 mM EDTA) was added to samples, and the DNA substrate molecules were resolved on a non-denaturing 8% polyacrylamide gel using 0.5X TBE/10 mM KCl as Running buffer. A lane with the radiolabeled ssDNA was also loaded on the gel to serve as a marker.
Upon completion of electrophoresis, the gel was exposed to X-ray film for 15–30 min, and the film was developed. The position of G4 DNA was compared to the radiolabeled ssDNA marker. Approximately 80% of the radiolabeled G-rich oligonucleotide migrated as a single species corresponding to the G4 DNA substrate.
The gel slice corresponding to the more slowly migrating G4 DNA molecules was excised and the slice was chopped up with a razor blade. The minced polyacrylamide was placed in 0.4 ml TE with10 mM KCl and allowed to gently rotate overnight at 4 °C.
The soaked polyacrylamide gel sample was centrifuged at 12, 000 × g for 5 min at 4 °C. The supernatant containing the G4 DNA was transferred to a fresh tube, and stored at 4 °C.
The concentration of TP-G4 DNA substrate was determined using the specific activity of the radiolabeled oligonucleotide (TP) as determined using a liquid scintillation counter (MicroBeta TriLux, Perkin Elmer). Since G4 DNA was formed by four strand oligonucleotides, four oligonucleotide molecules constitute one G4 DNA molecule.
4.2. G-Quadruplex DNA Unwinding Assay
Helicase-catalyzed quadruplex DNA unwinding assays are based on the same principal as helicase assays with more conventional duplex DNA substrates. Differential mobility of intact substrate and unwound single-stranded DNA on native polyacrylamide gels enables one to visualize the all-or-none products of helicase activity on a quadruplex DNA substrate. Fig. 3B shows a gel image from the products of a FANCJ G-quadruplex DNA substrate helicase assay resolved on a native polyacrylamide gel.
Reaction mixtures (20 μl) contained 40 mM Tris-HCl (pH 7.4), 25 mM KCl, 5 mM MgCl2, 2 mM dithiothreitol, 2% glycerol, 100 ng/μl bovine serum albumin, 10 fmol of the specified G-quadruplex (0.5 nM DNA substrate concentration), 2–5 mM ATP, and increasing helicase concentrations.
Reactions were initiated by the addition of helicase and then incubated at 30 °C for 15 min.
A 20 μl aliquot of loading buffer (74% glycerol, 0.01% xylene cyanol, 0.01% bromophenol blue, 10 mM KCl, 20 mM EDTA) was added to the reaction mixture at the end of incubation, and the products were incubated with proteinase K (0.5 mg/ml, Invitrogen) for an additional 15 min at 30 °C.
Helicase reaction products were subsequently resolved on nondenaturing 8% polyacrylamide gels. Radiolabeled DNA species on polyacrylamide gels were visualized with a PhosphorImager and quantitated using ImageQuant software (GE Healthcare). For example, see Fig. 3B.
Concluding remarks
In this Methods article, we have described experimental approaches and strategies to better understand the molecular functions and mechanisms of DNA helicases. In the fluorometric helicase assay section, we have focused the discussion on an all-or-none real-time assay based on the separation of a fluorophore-quencher pair and also a dye displacement assay that enables one to study partial unwinding of the DNA substrate. In the protein displacement assay section, we have described a convenient biotinylated DNA-streptavidin displacement assay to study this novel motor ATPase function and that can also be applied to investigate mechanistic aspects of DNA substrate recognition and initiation of unwinding by a helicase. Finally, we have described helicase assays used in the laboratory to examine the ability of specialized DNA helicases to unwind triplex and quadruplex alternate DNA structures that can be sources of genomic instability and perturb cellular processes of nucleic acid metabolism. We have provided detailed procedures for the assays so that helicase specialists and non-specialists may be able to employ these protocols in a practical manner. Lastly, we recognize the great advances made by helicase researchers in the field that have enabled us to apply the knowledge acquired by their work to address new scientific questions. We apologize to those whose work we have not cited due to space limitations.
Acknowledgments
This work was supported by the Intramural Research program of the NIH, National Institute on Aging and the Fanconi Anemia Research Fund (RMB).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lohman TM, Tomko EJ, Wu CG. Nat Rev Mol Cell Biol. 2008;9:391–401. doi: 10.1038/nrm2394. [DOI] [PubMed] [Google Scholar]
- 2.Patel SS, Donmez I. J Biol Chem. 2006;281:18265–18268. doi: 10.1074/jbc.R600008200. [DOI] [PubMed] [Google Scholar]
- 3.Brosh RM, Jr, Bohr VA. Nucleic Acids Res. 2007;35:7527–7544. doi: 10.1093/nar/gkm1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chu WK, Hickson ID. Nat Rev Cancer. 2009;9:644–654. doi: 10.1038/nrc2682. [DOI] [PubMed] [Google Scholar]
- 5.Lehmann AR. Biochimie. 2003;85:1101–1111. doi: 10.1016/j.biochi.2003.09.010. [DOI] [PubMed] [Google Scholar]
- 6.Wu Y, Suhasini AN, Brosh RM., Jr Cell Mol Life Sci. 2009;66:1209–1222. doi: 10.1007/s00018-008-8580-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aggarwal M, Brosh RM., Jr J Cell Biochem. 2009;106:758–763. doi: 10.1002/jcb.22048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gupta R, Brosh RM., Jr Anticancer Agents Med Chem. 2008;8:390–401. doi: 10.2174/187152008784220339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sharma S, Doherty KM, Brosh RM., Jr Biochem J. 2006;398:319–337. doi: 10.1042/BJ20060450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu Y, Brosh RM., Jr Curr Mol Med. 2009;9:470–482. doi: 10.2174/156652409788167159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choudhary S, Sommers JA, Brosh RM., Jr J Biol Chem. 2004;279:34603–34613. doi: 10.1074/jbc.M401901200. [DOI] [PubMed] [Google Scholar]
- 12.Bjornson KP, Amaratunga M, Moore KJ, Lohman TM. Biochemistry. 1994;33:14306–14316. doi: 10.1021/bi00251a044. [DOI] [PubMed] [Google Scholar]
- 13.Cheng W, Hsieh J, Brendza KM, Lohman TM. J Mol Biol. 2001;310:327–350. doi: 10.1006/jmbi.2001.4758. [DOI] [PubMed] [Google Scholar]
- 14.Eggleston AK, Rahim NA, Kowalczykowski SC. Nucleic Acids Res. 1996;24:1179–1186. doi: 10.1093/nar/24.7.1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gupta R, Sharma S, Doherty KM, Sommers JA, Cantor SB, Brosh RM., Jr Nucleic Acids Res. 2006;34:6673–6683. doi: 10.1093/nar/gkl964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brosh RM, Jr, Waheed J, Sommers JA. J Biol Chem. 2002;277:23236–23245. doi: 10.1074/jbc.M111446200. [DOI] [PubMed] [Google Scholar]
- 17.Sommers JA, Rawtani N, Gupta R, Bugreev DV, Mazin AV, Cantor SB, Brosh RM., Jr J Biol Chem. 2009;284:7505–7517. doi: 10.1074/jbc.M809019200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morris PD, Tackett AJ, Raney KD. Methods. 2001;23:149–159. doi: 10.1006/meth.2000.1116. [DOI] [PubMed] [Google Scholar]
- 19.Shin JH, Kelman Z. BMC Mol Biol. 2006;7:43. doi: 10.1186/1471-2199-7-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bugreev DV, Yu X, Egelman EH, Mazin AV. Genes Dev. 2007;21:3085–3094. doi: 10.1101/gad.1609007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hu Y, Raynard S, Sehorn MG, Lu X, Bussen W, Zheng L, Stark JM, Barnes EL, Chi P, Janscak P, Jasin M, Vogel H, Sung P, Luo G. Genes Dev. 2007;21:3073–3084. doi: 10.1101/gad.1609107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brosh RM, Jr, Driscoll HC, Dianov GL, Sommers JA. Biochemistry. 2002;41:12204–12216. doi: 10.1021/bi026031j. [DOI] [PubMed] [Google Scholar]
- 23.Manor H, Rao BS, Martin RG. J Mol Evol. 1988;27:96–101. doi: 10.1007/BF02138367. [DOI] [PubMed] [Google Scholar]
- 24.Agazie YM, Burkholder GD, Lee JS. Biochem J. 1996;316:461–466. doi: 10.1042/bj3160461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee JS, Burkholder GD, Latimer LJ, Haug BL, Braun RP. Nucleic Acids Res. 1987;15:1047–1061. doi: 10.1093/nar/15.3.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang G, Vasquez KM. Proc Natl Acad Sci U S A. 2004;101:13448–13453. doi: 10.1073/pnas.0405116101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gacy AM, Goellner GM, Spiro C, Chen X, Gupta G, Bradbury EM, Dyer RB, Mikesell MJ, Yao JZ, Johnson AJ, Richter A, Melancon SB, McMurray CT. Mol Cell. 1998;1:583–593. doi: 10.1016/s1097-2765(00)80058-1. [DOI] [PubMed] [Google Scholar]
- 28.Ohshima K, Montermini L, Wells RD, Pandolfo M. J Biol Chem. 1998;273:14588–14595. doi: 10.1074/jbc.273.23.14588. [DOI] [PubMed] [Google Scholar]
- 29.Krasilnikova MM, Mirkin SM. Mol Cell Biol. 2004;24:2286–2295. doi: 10.1128/MCB.24.6.2286-2295.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Seidman MM, Puri N, Majumdar A, Cuenoud B, Miller PS, Alam R. Ann N Y Acad Sci. 2005;1058:119–127. doi: 10.1196/annals.1359.020. [DOI] [PubMed] [Google Scholar]
- 31.Brosh RM, Jr, Majumdar A, Desai S, Hickson ID, Bohr VA, Seidman MM. J Biol Chem. 2001;276:3024–3030. doi: 10.1074/jbc.M006784200. [DOI] [PubMed] [Google Scholar]
- 32.Kopel V, Pozner A, Baran N, Manor H. Nucleic Acids Res. 1996;24:330–335. doi: 10.1093/nar/24.2.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maine IP, Kodadek T. Biochem Biophys Res Commun. 1994;204:1119–1124. doi: 10.1006/bbrc.1994.2578. [DOI] [PubMed] [Google Scholar]
- 34.Wu Y, Rawtani N, Thazhathveetil AK, Kenny MK, Seidman MM, Brosh RM., Jr Biochemistry. 2008;47:5068–5077. doi: 10.1021/bi702102d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huppert JL. Chem Soc Rev. 2008;37:1375–1384. doi: 10.1039/b702491f. [DOI] [PubMed] [Google Scholar]
- 36.Huppert JL. Biochimie. 2008;90:1140–1148. doi: 10.1016/j.biochi.2008.01.014. [DOI] [PubMed] [Google Scholar]
- 37.Maizels N. Nat Struct Mol Biol. 2006;13:1055–1059. doi: 10.1038/nsmb1171. [DOI] [PubMed] [Google Scholar]
- 38.Fry M, Loeb LA. J Biol Chem. 1999;274:12797–12802. doi: 10.1074/jbc.274.18.12797. [DOI] [PubMed] [Google Scholar]
- 39.London TB, Barber LJ, Mosedale G, Kelly GP, Balasubramanian S, Hickson ID, Boulton SJ, Hiom K. J Biol Chem. 2008;283:36132–36139. doi: 10.1074/jbc.M808152200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mohaghegh P, Karow JK, Brosh RM, Jr, Bohr VA, Hickson ID. Nucleic Acids Res. 2001;29:2843–2849. doi: 10.1093/nar/29.13.2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu Y, Shin-Ya K, Brosh RM., Jr Mol Cell Biol. 2008;28:4116–4128. doi: 10.1128/MCB.02210-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sen D, Gilbert W. Nature. 1990;344:410–414. doi: 10.1038/344410a0. [DOI] [PubMed] [Google Scholar]