

Abstract
Eukaryotic DNA replication is a complex process requiring the proper functioning of a multitude of proteins to create error-free daughter DNA strands and maintain genome integrity. Even though synthesis and joining of Okazaki fragments on the lagging strand involves only half the DNA in the nucleus, the complexity associated with processing these fragments is about twice that needed for leading strand synthesis. Flap endonuclease 1 (FEN1) is the central component of the Okazaki fragment maturation pathway. FEN1 cleaves flaps that are displaced by DNA polymerase δ (pol δ), to create a nick that is effectively joined by DNA ligase I. The Pif1 helicase and Dna2 helicase/nuclease contribute to the maturation process by elongating the flap displaced by pol δ. Though the reason for generating long flaps is still a matter of debate, genetic studies have shown that Dna2 and Pif1 are both important components of DNA replication. Our current knowledge of the exact enzymatic steps that govern Okazaki fragment maturation has heavily derived from reconstitution reactions in vitro, which have augmented genetic information, to yield current mechanistic models. In this review we describe both the design of specific DNA substrates that simulate intermediates of fragment maturation and protocols for reconstituting partial and complete lagging strand replication.
INTRODUCTION
Replication of eukaryotic DNA - the “blueprint of life”, is a complex series of events requiring the coordinate effort of a myriad of proteins for successful completion [1]. In order to maintain genome integrity and cell viability the DNA needs to be faithfully replicated. On unwinding the DNA, replication occurs simultaneously on both strands. However, because of the anti-parallel nature of the DNA duplex, replication occurs in a continuous fashion on the leading template and in a discontinuous manner on the lagging template. On the lagging template, short DNA fragments ~ 150 – 200 bp in size, known as Okazaki fragments, are synthesized, which need to be processed further to form a complete, functional duplicated DNA strand. DNA polymerase ε (pol ε ) is the major replicative polymerase that synthesizes the leading strand [2]. On the lagging strand, DNA polymerase α/primase (pol α ) initiates lagging strand synthesis by first synthesizing an RNA primer of 8 – 12 nucleotides (nt) followed by a short stretch of DNA [3]. Okazaki fragment formation is then switched from the priming mode to the extension mode by ATP-dependent replication factor C (RFC) which displaces the pol α, causing the loading and binding of the elongation complex, proliferating cell nuclear antigen (PCNA) and DNA polymerase δ(pol δ) [4]. PCNA, a toroidal homotrimer, acts as a sliding clamp that increases the processivity of pol δ [5]. As a precursor to strand joining, each of the RNA/DNA primers synthesized by the error prone pol must be removed to ensure genomic integrity [1]. The elongation complex, on encountering a downstream Okazaki fragment, performs strand displacement synthesis generating a 5′ flap. Short flaps up to 10 nt in length are generated and cleaved repeatedly at their bases by flap endonuclease 1 (FEN1), creating nicked products [6,7]. After the priming region is removed, the last nick product of FEN1 is sealed by DNA ligase I (Lig I). This pathway is called the “one-nuclease pathway” or the “FEN1- only pathway”.
In some circumstances, flaps escape FEN1 cleavage and become long. The 5′ – 3′ ATP-dependent helicases, Pif1 and Dna2, contribute to the flap elongation, with some flaps achieving lengths that allow binding of the single strand DNA binding protein, replication protein A (RPA). Coating of the flaps with RPA inhibits FEN1 from accessing the flap at its 5′ end and tracking to the flap base and thereby inhibits the cleavage. Dna2, which is also a nuclease, displaces RPA from the long flap and makes multiple endonuclease cuts on the flap. Dna2 is unable to cleave at the base of the flap and leaves behind a flap ~ 5 – 6 nt in length. RPA cannot rebind on this short flap, but FEN1 can effectively cleave at the flap base creating a substrate for Lig I. This is called the “two-nuclease pathway” [7].
In addition to the roles of Dna2 and Pif1 helicase, other helicases are believed to play a significant role in Okazaki fragment processing such as Bloom helicase (BLM) and Werner helicase (WRN). While Dna2 and Pif1 likely participate in creation of the downstream flap generated by pol δ during displacement synthesis, the other helicases may help resolve secondary structures that are formed during the maturation pathway. The RecQ family of helicases, to which the BLM and WRN helicases belong [8], are the most extensively studied helicases anticipated to influence lagging strand processing. It is hypothesized that some of the flaps that escape cleavage by FEN1 will form secondary structures. As FEN1 requires a free 5′ end for cleavage, the secondary structure inhibits FEN1 processing and ultimately ligation. BLM and WRN have been shown to stimulate FEN1 cleavage in vitro, by indirectly removing flap secondary structures [9-11]. BLM [10] and WRN [12] helicases have also been shown to stimulate FEN1 cleavage directly, independent of their helicase activities. This latter mode of stimulation is evident with flap substrates having no secondary structure. In fact, mutant forms of BLM and WRN lacking the helicase domain both co-immunoprecipitate with FEN1 and stimulate FEN1 cleavage activity [10,12]. In addition to their stimulation of FEN1, the RecQ helicases (8-10) also bind numerous other Okazaki fragment processing proteins. For additional information regarding the roles of RecQ helicases in replication and genomic stability, see reviews [8,13,14].
Our initial insights into the replication process came from studies in vitro using cell-free systems to duplicate simian virus 40 (SV40) DNA [15]. Building on these early analyses, many subsequent lagging strand processing protein components were discovered and used to reconstitute a minimal Okazaki fragment processing pathway in vitro. Currently, it is technically impossible to measure the relative use of the two proposed pathways during Okazaki fragment maturation in vivo, and so we need to rely on a combination of genetic information and biochemical reconstitutions to understand why both pathways evolved. These genetic and biochemical approaches have made a strong case for combined contribution of both pathways in the effective resolution of flaps that are created during Okazaki fragment maturation [16]. In this review we provide a brief summary of the various protein components involved in the lagging strand replication pathways, give details of the methods to reconstitute individual events occurring in the pathways, and ultimately describe reconstitution of the entire process of Okazaki fragment maturation. The focus of this review is to provide insights into proper substrate and experimental design in order to derive the most useful information when reconstituting the system in vitro.
DESCRIPTION OF METHOD
The Elongation Complex and the Lagging Strand Polymerase
Pol δ is now considered to be the main replicative polymerase of lagging strand synthesis [16]. In S. cerevisiae, pol δ is a heterotrimeric enzyme, comprised of subunits 125 (Pol3), 55 (Pol31) and 40 kDa (Pol32) [17]. In S. pombe and humans the polymerase is a heterotetramer with a fourth subunit of 18 kDa, which functions to stabilize the complex [18-20]. Experiments in vitro have shown that pol δ can synthesize in the gaps between Okazaki fragments. However, pol δ needs the cooperative actions of PCNA and RFC in order to perform strand displacement synthesis that generates a 5′ flap substrate [7,21].
Design of Substrates Simulating Okazaki Fragments
Pol δ synthesis activity can be measured on either circular or linear DNA. The circular system can be used to assess the coupling of the replication and maturation processes whereas the linear oligonucleotide system helps to resolve the replication intermediates and products [6]. The circular DNA system is designed such that Bluescript + SKII plasmid DNA is cleaved with EcoRI, 32P-labeled on its 3′ end and further digested by ScaI. The labeled 1.14 kb fragment is isolated by preparative agarose gel electrophoresis and hybridized to SKII ssDNA (~2.9 kb) purified from phage generated from the Bluescript +SKII with a helper phage. The labeled circular substrate consisting of the + single strand from the Bluescript and the complementary labeled-strand is preincubated with PCNA and RFC for 1 min and then the reaction is started by adding pol δ. The reaction is terminated at various time points after the addition of pol δ and the products are resolved on a 1% alkaline agarose gel to assess the amount of nick translation [6].
The Burgers group, in a series of elegantly designed experiments, introduced the use of linear DNA substrates to study Okazaki fragment maturation [6]. The substrates they designed were analogous to the previously described T4 system, wherein they created end caps consisting of biotin conjugated with streptavidin to prevent PCNA from sliding off of the double stranded region after it had been loaded by RFC. The Okazaki fragment template is designed such that the template contains biotin at both the 5′ and 3′ ends. Upstream (44 nt) and downstream (60 nt) primers separated by a 6 nt gap are annealed to the template (110 nt), simulating adjacent Okazaki fragments (Figure 1, ii).
Fig 1. Pol dd d Primer Extension.

(A) Model of Pol δ in complex with PCNA and RFC synthesizing in the 5′ – 3′ direction. (B) Model substrates containing biotin-conjugated streptavidin on the 5′ and 3′ ends of the template strand. Primer extension substrate model (i) and Okazaki fragment substrate or strand displacement synthesis substrate model (ii). (C) Assessment of primer extension. The asterisk indicates the position of the radiolabel.
Primer Extension and Strand Displacement Synthesis by Pol δ
To assess primer extension by pol δ, the downstream primer is eliminated so that the substrate only contains a 5′ labeled upstream primer (44 nt) annealed to the template (110 nt) (Figure 1B, i). Prior to starting the reaction, the substrate is incubated with a 5 fold excess of streptavidin over substrate for 20 min on ice. Increasing concentrations of pol δ are incubated along with the substrate in the reconstitution buffer (Table 1) either in the presence or absence of PCNA and RFC for 10 min at 37 °C. The reaction is terminated by adding 20 μl of 2X termination dye (Table 2) and boiled for 5 mins. The boiled samples are fractionated by electrophoresis in a 22.5%/7 M urea denaturing polyacrylamide gel for 1 hour and 30 mins at 80 W. The gel is then dried and exposed to a phosphor screen overnight, scanned using a PhosphorImager and analyzed using ImageQuant software. The amount of synthesis that occurred is analyzed as described (Table 3). An example of a synthesis reaction is shown in Figure 1C.
TABLE 1.
Reaction Buffer Compositions
| Nuclease Buffer | 50 mM | Tris-HCl [pH 8.0] |
| 2 mM | DTT | |
| 30 mM | NaCl | |
| 0.1mg/ml | BSA | |
| 5% | Glycerol | |
| 2 mM | MgCl2* | |
| 4 mM | ATP * | |
| Ligation Buffer | 30 mM | HEPES [pH 7.6] |
| 0.01% | Nonidet P-40 | |
| 40 mM | KCl | |
| 0.1mg/ml | BSA | |
| 8 mM | MgCl2 | |
| 0.1 mM | ATP | |
| ATPase Buffer | 40 mM | Tris-HCl [pH 7.5] |
| 2.5 mM | DTT | |
| 25 mM | NaCl | |
| 0.1mg/ml | BSA | |
| 5% | Glycerol | |
| 5 mM | MgCl2** | |
| Helicase Buffer | 50 mM | Tris-HCl [pH 8.0] |
| 2 mM | DTT | |
| 30 mM | NaCl | |
| 0.5mg/ml | BSA | |
| 5% | Glycerol | |
| 2 mM | MgCl2** | |
| 0.5 mM | ATP ** | |
| EMSA Buffer | 50 mM | Tris-HCl [pH 8.0] |
| 2 mM | DTT | |
| 30 mM | NaCl | |
| 0.1 mg/ml | BSA | |
| 5% | Glycerol | |
| 2 mM | MgCl2*** | |
| 10 μM | ATP | |
| Okazaki Reconstitution Buffer | 50 mM | Tris-HCl [pH 7.5] |
| 2 mM | DTT | |
| 75 mM | NaCl | |
| 0.25 | mg/ml | BSA |
| 5% | Glycerol | |
| 4 mM | MgCl2 | |
| 1 mM | ATP | |
| 50 μM | dNTPs |
For Dna2 nuclease assays, increase MgCl2 to 8mM and decrease ATP to 2mM
For Dna2 helicase assays, increase ATP to 8mM and decrease MgCl2 to 2mM
If FEN1 and Dna2 are being used in the EMSA, either MgCl2 can be eliminated from the buffer or can be replaced by 2mM CaCl2 or 0.2 mM EDTA
TABLE 2.
Loading Dyes
| 2X Termination Dye | ||
| 90% | Formamide [v/v] | |
| 10mM | EDTA | |
| 0.01% | Bromophenol Blue | |
| 0.01% | Xylene Cyanole | |
| 6X Helicase Dye | ||
| 50mM | EDTA | |
| 0.9% | SDS | |
| 0.125% | Bromophenol Blue | |
| 0.125% | Xylene Cyanole | |
| 30% | Glycerol | |
| EMSA Loading Dye | ||
| 18mM | EDTA | |
| 0.04% | Bromophenol Blue | |
| 0.04% | Xylene Cyanole | |
| 25% | Glycero | |
TABLE 3.
Calculations
| % Fully Synthesized Product : {(b)/(b+c+a)} * 100 |
| Where, “b” is the fully synthesized product, “c” are the intermediate synthesis products and “a” is the remaining unsynthesized substrate. |
| % Cleaved Product : {(b)/(b+a)} * 100 |
| Where, “b” is the cleaved product and “a” is the remaining uncleaved substrate. |
| % Ligated Product: {(b)/(b+a)} * 100 |
| Where, “b” is the ligated product and “a” is the remaining unligated substrate. |
| % Bound Product: {(b)/(b+a)} * 100 |
| Where, “b” is the bound product and “a” is the remaining unbound substrate. |
| % Unwound Product: {(b)/(b+a)} * 100 |
| Where, “b” is the unwound product and “a” is the remaining unwound substrate Strand Melting is measured in a similar manner. |
| % Annealed Product: {(b)/(b+a)} * 100 |
| Where, “b” is the annealed product and “a” is the remaining unannealed substrate |
|
Calculation of Dissociation Constants: After EMSA, the curves are fit using nonlinear least square regression of the hyperbolic equation: |
| y = Bmax*[Protein]/(Kd + [Protein]) |
| where, y is the percent of the oligonucleotide bound, [Protein] is the concentration of protein in nM, Bmax is the maximum binding and Kd is the equilibrium dissociation constant. |
For measuring the efficiency of pol δ strand displacement synthesis, a substrate (described above) containing a 5′ labeled upstream primer and a downstream primer annealed to the template is employed (Figure 1B, ii). Streptavidin conjugated substrate is incubated with the elongation complex components and the reaction is fractionated by electrophoresis on a denaturing gel and analyzed as described above. As the upstream primer extends and meets the downstream primer, a strong pause site will be observed on the gel and any synthesis beyond that into the downstream primer is strand displacement synthesis. The profile of synthesis will represent the strand displacement capability of pol δ on an Okazaki fragment substrate.
The central role of FEN1
Okazaki fragment maturation requires the complete removal of the RNA primer that is synthesized by pol α/primase. Flaps created by pol δ strand displacement synthesis are resolved by FEN1. FEN1 is a Mg2+ dependent, structure-specific nuclease that is central to both DNA replication and repair. In S. cerevisiae, FEN1 is a single polypeptide about 42 kDa in size. FEN1 contains 5′ – 3′ endonuclease with minor 5′ – 3′ exonuclease activity. Most of the minimum protein components required for Okazaki fragment processing have been shown to enhance FEN1 cleavage activity in vitro, including PCNA [22], RFC [23], and Dna2 [6,24]. While RPA stimulates FEN1 cleavage activity on short flaps (~ 2 – 6 nt) [25], when RPA can stably bind to longer flaps (> 22nt), it inhibits the FEN1 cleavage [26]. FEN1 is both acetylated and phosphorylated during the cell cycle, with both modifications inhibiting its nuclease functions [27,28]. While inactivation or haploinsufficiency of FEN1 is detrimental in human cells and often leads to tumor progression and malignancies [29,30], null mutants of FEN1 in yeast are viable, though not robust, suggesting inefficient backup by another endonuclease for resolution of flap structures [31].
Design of Flap Substrates
FEN 1 is a tracking enzyme, which must access the flap 5′ end and traverse the flap in order to exhibit cleavage activity at the flap base. For optimal enzymatic binding [32] and cleavage [33] activity, FEN1 prefers a substrate containing a 5′ downstream flap and a one nt 3′ upstream overhang. Biochemical analysis has demonstrated that FEN1 preferentially cleaves one nt into the double-stranded annealed region of the downstream 5′ flap when presented with the preferred substrate [33]. FEN1 can cleave both RNA and DNA single strand flap substrates [34,35]. As FEN1 is structure-specific for binding and catalysis, features of the substrate can modulate the activity of FEN1 in vitro. When overlapping flaps are complementary to the template, the flaps will equilibrate until the optimal one nt 3′ flap is formed, at which point FEN1 cleaves one nt into the duplex DNA of the downstream annealed region [33]. Cleavage is progressively diminished if the upstream primer is intentionally shortened in a 3′ to 5′ direction, and the rate of cleavage is minimal when no upstream primer is present. Blocking the 5′ end of the flap with a primer or protein inhibits FEN1 since it interferes with 5′ entry and tracking. While a 5′ block inhibits cleavage, FEN1 can still bind the substrate [36]. Inhibition of FEN1 cleavage occurs when secondary structures form on the 5′ flap, including hairpins, recombinant intermediates, bubbles, or annealed, complementary segments of DNA [9,11,35,37].
Binding and Cleavage Properties of FEN1
Analysis of FEN1 cleavage typically involves measurements over time or concentration. FEN1 is incubated with 5 fmol of 32P-labeled DNA flap substrate in a total reaction volume of 20 μl of nuclease buffer (Table 1). Concentration-dependent reactions are generally incubated at 37 °C for 10 min. The reaction is terminated and fractionated by electrophoresis on a denaturing gel as described earlier (pol δ section). Stimulation of FEN1 cleavage by components of the Okazaki fragment maturation complex can be assessed by comparing cleavage by FEN1 alone to cleavage in presence of other Okazaki fragment maturation protein components. Similarly, inhibition of FEN1 activity by a post-translational modification can be measured by comparing the cleavage of unmodified and modified (either acetylated or phosphorylated) FEN1. Altering the structure of the substrate, as described in the design of flap substrate section, can also greatly influence the activity of FEN1. Optimal FEN1 activity is visualized at pH 8.0 [32]. In addition to activating FEN1 by the addition of Mg2+, assays in vitro can utilize Mn2+ to enable FEN1 nuclease activity. Conversely, FEN1 cleavage is inhibited by Ca2+, Zn2+, and divalent metal chelating agents such as EDTA [34]. Monovalent cations, such as Na+ and K+, are inhibitory to FEN1 activity [32]. The cleavage pattern of FEN1 is analyzed using the calculations described in Table 3. An example of the FEN1 cleavage assay is shown in Figure 2.
Fig 2. FEN1 is structure specific nuclease.
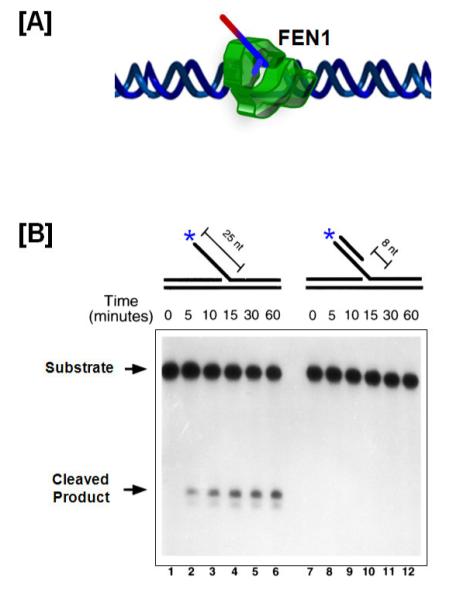
(A) FEN1 after it completes tracking to the base of a flap, and just before cleavage. (B) FEN1 flap cleavage is measured on a denaturing gel using an unblocked flap (left panel) and 5′ blocked flap (right panel) substrate. The asterisk indicates the position of the radiolabel.
Fig 2B was originally published in Journal of Biological Chemistry by Murante R.S., et al., “Calf 5′ to 3′ Exo/Endonuclease Must Slide from a 5′ End of the Substrate to Perform Structure-specific Cleavage”, J Biol. Chem., (1995); 270:30377-30383 © the American Society for Biochemistry and Molecular Biology
The dissociation constant (Kd) for FEN1 binding to a nick flap substrate (0 nt 3′ overhang on the upstream primer) in the absence of Mg2+ is 7.5 nM [38]. To assess FEN1-DNA binding, refer to the electromobility gel shift assay (EMSA) procedure utilized for RPA binding using the EMSA reaction buffer (Table 1), and to determine the dissociation constant refer to the calculation section in Table 3. For FEN1 binding assays excluding MgCl2, replacing the MgCl2 with CaCl2, or adding 0.2 mM EDTA will effectively prevent nuclease activity. The fraction of the protein bound to DNA is assessed by autoradiography measurements as described in Table 3.
Using an Okazaki fragment substrate labeled at the 3′ end of the downstream primer, the stimulatory effect of FEN1 on strand displacement synthesis can also be measured using the strand displacement assay as described above. To effectively make the measurement, a constant amount of pol δ and increasing concentrations of FEN1 need to be used in the assay. FEN1 presumably promotes displacement by repeatedly removing the displaced nucleotides, preventing them from rebinding the template in a way that would interfere with the polymerase. The greater rate of shortening of the downstream primer with increasing FEN1 is an indication of the effectiveness with which FEN1 cleavage promotes displacement.
DNA Ligase I – Sealing the Nicks
DNA ligases play an essential role in maintaining the physical structure of DNA by sealing nicked DNA created during DNA replication and repair. DNA Ligase I (Lig I), associated with DNA replication, functions by catalyzing the formation of phosphodiester bonds between adjacent 5′ phosphoryl and 3′ hydroxyl termini at single strand nicked sites. Human Lig I is ATP-dependent, about 102 kDa in size, and indispensable for cell viability [39,40]. RFC interacts and binds to Lig I, reducing its ligase activity [41]. On being post-translationally modified by phosphorylation, human Lig I loses its ability to interact with RFC, thereby relieving the inhibitory effect of RFC on Lig I [42]. Mammalian Lig I has the unique ability to encircle the DNA, stabilize distorted structure, and position its active site near the nick to seal the DNA [43]. Flaps displaced by the elongation complex are cleaved by FEN1 creating a nick, which needs a functional Lig I to complete the maturation of the Okazaki fragment. In the absence of a functional FEN1, pol δ maintains a ligatable nick by a process known as “idling”, wherein the 3′ – 5′ exonuclease activity of pol δ can back up and stabilize the polymerase at a position that is efficient for ligation. PCNA plays a critical role in interacting and recruiting Lig I to the sites of Okazaki fragment maturation and DNA repair. However, there are contradictory reports of both stimulation [44] and inhibition [45,46] of the activity of Lig I in the presence of PCNA. Ligation is the final step in both the “one-nuclease” and “two-nuclease” pathways.
Design of Substrates for Analyzing the Ligation Step
The main requirement for ligation is the presence of a 3′ hydroxyl and a 5′ phosphoryl group on the nicked site. Therefore, when designing nicked substrates it is essential that the downstream primer contains a 5′ phosphate. An upstream primer (40 nt) and a 5′ phosphorylated downstream primer (70 nt) are annealed to a template (110 nt) to create a nicked substrate for ligation. The substrate can either contain a 5′ labeled upstream primer or a 3′ labeled downstream primer for effective quantitation. If nicked DNA is created by the Okazaki protein components in a dynamic reconstitution (i.e. with pol δ, PCNA, RFC and FEN1) using an Okazaki fragment substrate (as described in the pol δ section), then the 5′ phosphate on the downstream primer will be generated in the course of the reaction.
Ligation of Nicked Substrates
Ligation efficiency can be assessed on either a pre-created substrate or a product of FEN1. Lig I is incubated with 5 fmol of 32P-labeled DNA nicked substrate in a total reaction volume of 20 μl of ligation buffer (Table 1). Concentration-dependent reactions are generally incubated at 37 °C for 10 mins. The reaction is terminated and fractionated by electrophoresis on a denaturing gel as described earlier (pol δ section). The percentage of ligated substrate can be determined using calculations described in Table 3. An example of a ligation assay is shown in Figure 3.
Fig 3. Lig I seals DNA nicks.
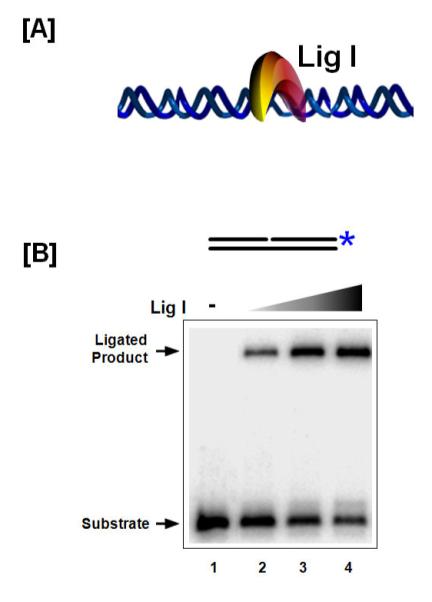
(A) Model of Lig I binding and sealing nicked DNA. (B) Denaturing gel assessing DNA Lig I ligation efficiency on a nicked DNA substrate. The asterisk indicates the position of the radiolabel.
Pathway Switching by RPA
The ssDNA binding protein, RPA, coats ssDNA and protects it from nuclease damage and also prevents the formation of secondary structures, thereby maintaining the integrity of the DNA during replication. RPA functions as the central protein in switching the Okazaki fragment maturation process from the “one-nuclease” pathway to the “two-nuclease” pathway. When long flaps are generated either in vivo or in vitro, they have the potential to be coated by RPA. Coated flaps require the coordinated action of components of the two-nuclease pathway for effective resolution. In addition to regulating protein utilization of flaps, RPA is considered to act as the “fidelity-clamp” for pol α/primase by working as an auxiliary factor stabilizing the polymerase and improving accurate nucleotide addition [47] and also influences strand displacement synthesis by pol δ [48]. RPA is composed of three subunits, 70, 32, and 14 kDa in S. cerevisiae and 70, 34, and 11 kDa in humans. RPA32 is phosphorylated during UV damage, resulting in reduced affinity for pol /primase and delocalization from replication centers [49], representing a regulatory shift from DNA replication to repair.
Design of substrates to assess RPA binding
In addition to binding and stabilizing ssDNA, RPA also possesses strand melting and low-efficiency strand annealing properties [50]. Therefore, in considering the design of substrates to assess RPA binding that simulate the flap-intermediates created during Okazaki fragment processing, it is imperative that RPA does not melt the upstream or downstream primer annealed to the template. This melting would create a fully single-stranded substrate capable of binding RPA, which would produce unanticipated products, and also deplete the intended substrate (as shown in Figure 4).
Fig 4. RPA binds single stranded DNA and melts short double stranded DNA segments.

(A) Model of RPA binding the single stranded DNA region of a flap (top arrow) or melting and binding the substrate (bottom arrow). (B) RPA binding is visualized using an EMSA. Lane 1 shows the labeled flap substrate as a control. Lanes 1 – 4 indicate RPA bound to intact substrate forming shifted complexes. Lanes 5 – 7 indicate RPA bound to melted substrate forming alternate shifted complexes. The asterisk indicates the position of the radiolabel.
Binding characteristics of RPA
Depending on the length of the ssDNA, RPA exhibits various modes of binding, the weakest occurring on ssDNA about 8 to 10 nt long and the strongest on ssDNA greater than 23 nt in length [49]. RPA binding to a flap substrate can be measured using EMSA. For this assay the labeled substrate is mixed with protein, and then the mixture is fractionated on a native gel. The binding of protein retards the substrate compared to unbound substrate. A lane containing the substrate alone serves as a control to identify the substrate band in the experimental lanes (lane 1; Figure 4B). While EMSA is a non-equilibrium binding technique, it is known to yield generally accurate results because the protein does not significantly equilibrate off of the substrate during migration in the gel [51]. As increasing concentrations of RPA are incubated with the substrate, more RPA binds, causing a progressive darkening of the shifted bands. Atlow concentration of RPA, a single molecule of RPA binds to the substrate (lane 2) and at higher concentrations multiple RPA molecules might bind, as evident from a super-shift in the gel (lane 4). As discussed above, if RPA unwinds the substrate, various RPA-bound ssDNA species can be created, producing a confusing array of bands (lanes 5, 6, 7). Typically, increasing concentrations of RPA are incubated with 5 fmol of substrate (either 3′ or 5′ 32P-labeled) in a reaction volume of 20 μl for 10 mins at 37 °C in the EMSA reaction buffer (Table 1). The reactions are mixed with 4 μl EMSA loading dye (Table 2) and loaded onto a pre-run 6% polyacrylamide (native) gel in 0.5X TBE buffer. Gels are subjected to electrophoresis at 150 V for 30 – 45 mins after which they are vacuum dried. The gels are then exposed to a phosphor screen overnight. The image is analyzed on a PhosphorImager and the amount of binding of RPA to the substrate determined as described in Table 3. A hypothetical binding assay is depicted in Figure 4.
Determining Kd values
The dissociation constant of RPA on 53 nt flap substrate is 0.35 ± 0.10 nM [52]. For measurement of dissociation constants the amount of labeled substrate used in the reaction is lowered to 1 fmol and a wider range of RPA titration is carried out. The wider concentration range allows for more accurate measurement of the binding constant and also provides for more data points that can be fit to a curve. The dissociation constant is determined using the calculations described in Table 3.
RPA strand annealing and strand melting properties can be measured using the annealing and helicase assay described below in the Pif1 section.
Role of Dna2 in the Two Nuclease Pathway
Results in vitro show that long flap intermediates coated by RPA are resistant to cleavage by FEN1 because RPA blocks the ability of FEN1 to enter the flap from the 5′ end and track to the flap base for cleavage. Genetic and biochemical evidence suggests that eukaryotic cells utilize the Dna2 helicase/nuclease to cleave RPA-coated flaps to a length that will dissociate RPA and allow FEN1 entry.
The size of Dna2 varies from 172 kDa in yeast [53] to 120 kDa in humans [54-56]. Dna2 has two domains: one possesses ssDNA-dependent ATPase activity and ATP dependent 5′ – 3′ helicase activity and the other possesses 5′ – 3′ and a minor 3′ – 5′ ssDNA nuclease activity [57]. While the helicase activity of Dna2 is robust in S. cerevisiae, it was previously reported to be absent in X. laevis [58] and S. pombe [59] and extremely low in human cells [55,56]. However, robust helicase activity of xDna2 was recently detected using a nuclease deficient mutant [60]. Helicase activity is stimulated by high levels of ATP, which inhibits the nuclease, or is suppressed at high concentrations of Mg2+, which stimulate the nuclease. This has allowed us to alter the ratio of activities for biochemical studies. Acetylation of Dna2 in vitro by the transcriptional co-activator p300 simulates the ATPase, helicase, and nuclease activities of Dna2 [61]. Mutation or knockout of Dna2 in yeast induces a lethal phenotype and knockdown in human cells results in the formation of inter-nuclei chromatin bridges [54].
Design of Dna2 substrates
Dna2, similar to FEN1, is a tracking enzyme that requires a free 5′ end for cleavage activity. However, unlike FEN1, Dna2 does not cleave at the base of a flap, but instead cleaves at multiple sites in the single stranded region leaving a 5 – 6 nt flap as a terminal product. Similarly, Dna2 can track on ssRNA templates, but cannot cleave RNA as it does DNA. Flaps that are coated by RPA stimulate the 5′ – 3′ cleavage by Dna2 increasing the frequency of cuts on individual substrate molecules. However, the 3′ – 5′ cleavage is inhibited in the presence of RPA [62]. The 3′ – 5′ cleavage by Dna2 is thought to work on equilibrating flaps created during pol δ strand displacement synthesis [55]. The optimal substrate for Dna2 helicase and nuclease activity has a 5′ flap greater than 5 – 6 nt in length. Studies done in vitro have shown that Dna2 works in concert with RPA to remove flap impediments ultimately generating a short flap substrate for FEN1 [26].
Multiple activities of Dna2
ATPase Assay
ATPase activity of Dna2 is measured by incubating the purified protein with 1 μg of ssDNA oligonucleotide in the ATPase buffer (Table 1) with increasing concentrations of [ γ-32P] ATP at 37 °C for 20 mins. Reactions in the absence of protein and in the absence of oligonucleotide serve as controls. Each reaction is terminated using EDTA to a final concentration of 4 mM. Approximately 0.5 μl of this reaction is spotted on a polyethyleneimine cellulose plate and developed in 0.5 M LiCl/1 M formic acid. The results are analyzed using a PhosphorImager. Alternatively, ATPase activity can be measured using the Norit Protocol, wherein instead of just EDTA the reaction is stopped by 10 mM EDTA/10% acid-washed charcoal. Each reaction is allowed to sit on ice overnight and spun at 13,000 × g for 30 min the following day. Duplicate 100 μl supernatant samples are counted by the Cerenkov method [63]. ATP hydrolysis is determined by dividing the adjusted protein-dependent counts by the specific activity of ATP in each reaction.
Helicase Assay
Helicase activity of Dna2 is measured in a similar manner as described below in the Pif1 section. The helicase function of Dna2 can be improved by increasing the ATP concentration and decreasing the MgCl2 concentration in the helicase buffer (Table 1).
Nuclease activity
Nuclease activity of Dna2 is measured in a similar manner as described for FEN1. Initial cuts by Dna2 can be visualized using a 5′ labeled flap substrate and the final cuts can be visualized using a 3′ labeled flap substrate. Nuclease activity of Dna2 can be improved by increasing the MgCl2 and decreasing the ATP concentration in the nuclease buffer (Table 1).
Binding Assay
Binding activity of Dna2 to DNA can be measured in a similar manner as described for FEN1 and RPA. If MgCl2 and ATP are omitted from the EMSA buffer (Table 1), the helicase and nuclease activities of Dna2 will not be functional, allowing accurate assessment of binding. The dissociation constant of Dna2 on ssDNA is 5.5 + 0.77 nM [52] and can be measured in a similar manner as described in the RPA section.
Assay for Displacement of RPA by Dna2
In addition to cleaving long flaps that are displaced during Okazaki fragment maturation, one of the most important roles of Dna2 in the twonuclease pathway is to displace RPA from its ssDNA binding sites on the flaps. Removal ofRPA and flap cleavage are the means by which Dna2 prepares flaps for FEN1. Displacement of RPA can be measured using two different experimental protocols. One uses EMSA, in which a constant amount of RPA is prebound on the flap substrate for 5 mins in 20 μl EMSA buffer, followed by titrating in Dna2 and further incubating for 5 mins in the EMSA buffer at 37 °C. Four μl of EMSA loading dye is added to the reactions and they are loaded onto the native gel. Products are fractionated by electrophoresis, dried and then analyzed by PhosphorImager as previously described. As increasing concentrations of Dna2 displace RPA, a conversion should be observed from the RPA-substrate band to the Dna2-substrate band in the scanned gel. Control reactions having either RPA incubated with the substrate alone, or the highest concentration of Dna2 incubated with the substrate alone, will mark the expected positions of experimental bands. Additionally, these controls will determine whether the melting and helicase functions of the respective proteins act on the substrate. An example of the EMSA gel is depicted in Figure 5C.
Figure 5. Dna2 displaces RPA from ssDNA and cleaves at multiple sites on the ssDNA.

(A) Model for Dna2 5′ – 3′ helicase unwinding. (B) Model for the Dna2 5′ – 3′ nuclease activity. Note that helicase activity may displace into the downstream primer, but the terminal nuclease product is always a 5-6 nt flap. (C) EMSA gel showing Dna2E675A (nuclease mutant) displacement of RPA from the 5′ flap. (D) Denaturing gel showing RPA stimulation of Dna2 cleavage. The asterisk indicates the position of the radiolabel.
Figure 5C, D were originally published in Journal of Biological Chemistry by Stewart, J.A., et al., “Dynamic removal of replication protein A by Dna2 facilitates primer cleavage during Okazaki fragment processing in Saccharomyces cerevisiae”, J Biol. Chem., (2008); 283 (46):31356-65 © the American Society for Biochemistry and Molecular Biology
Stimulation of Dna2 by RPA, and displacement of RPA by Dna2, can also be measured using a cleavage assay. Stimulation of Dna2 by RPA is assessed by incubating Dna2 and increasing concentrations of RPA with the substrate in 20 μl of nuclease buffer (Table 1, adjust buffer as described in the nuclease assay section for Dna2) for 10 mins at 37 °C. Calculating the cleavage by Dna2 (described in Table 3) in the presence of RPA and comparing it to cleavage by Dna2 alone will yield the fold stimulation. Displacement of RPA by Dna2 can also be measured in a cleavage assay in which a fixed amount of RPA is preincubated with the flap substrate and then Dna2 is titrated into the reaction as described above. The cleavage pattern will delineate the footprint of Dna2 and provides an alternative assessment of displacement of RPA from the flap. An example of the EMSA gel is depicted in Figure 5D.
Properties of Pif1 In Vivo and In Vitro
Pif1 is an ATP-dependent 5′ – 3′ helicase [64] that localizes to both the mitochondria and nucleus based on alternative translation [65]. Human Pif1 has a predicted molecular weight of about 71 kDa [66]. Pif1 plays a significant role in telomere maintenance and mitochondrial DNA repair and recombination [67]. Post-translation phosphorylation of Pif1 regulates its function between two pathways: DNA double strand break (DSB) repair and telomere maintenance [68]. A role for Pif1 in Okazaki fragment maturation was implied by genetic experiments that indicated a functional association of Dna2 and Pif1 in vivo [69]. This report showed that the lethal phenotype of dna2 strains was partially rescued in dna2 pif1 strains, and completely rescued by deleting POL32 in dna2 pif1 strains. The deletion of POL32, the PCNA-binding subunit of pol δ, reduces the ability of pol δ to carry out strand displacement synthesis [70,71]. Taken together, these results suggest that POL32 and Pif1 are responsible for creating longer flaps and generate the need for Dna2 to process these long flaps.
Substrates for Pif1 Helicase Function
Pif1 cannot bind or track on RNA [72], implying that if it promotes flap lengthening, it begins unwinding when the DNA portion of the flap starts to be displaced. Pif1 can unwind approximately 20 – 30 nt before dissociating from the substrate [72-74]. Also, because of its directionality, Pif1 can only unwind 5′ flap substrates. The flap length requirement for Pif1 to load and unwind is 5 – 6 nt and longer. Pif1 demonstrates increasing helicase activity as the 5′ single stranded tail length increases until reaching a plateau [75]. Interestingly, a recent report suggests that Pif1 can bind to the template of a substrate containing a gap of either 20 or 30 nt between the upstream primer and downstream primer. On binding to such a substrate, it can not only track on the flap and unwind the downstream primer (Reaction1) but also it can bind to the template and unwind the upstream primer (Reaction 2). However, the frequency of occurrence of Reaction 2 during Okazaki fragment maturation may be minimal, since Pif1 would have to displace the protein components of the replisome in order to unwind the upstream primer [75].
Assessing Helicase Activity
The efficiency of Pif1 helicase activity on preferred substrates can be assessed by a helicase assay. Five fmol of 32P-labeled Pif1 substrate is incubated with increasing concentrations of Pif1 for 10 min at 37 °C in a 20 μl reaction containing helicase buffer (Table 1). Each reaction is terminated by addition of 4μ of helicase dye (Table 2). Reactions are then loaded on to 4 – 8% native gels and fractionated by electrophoresis at 150 V for 30 – 45 mins. After drying, the gel is exposed overnight to a phosphor screen. The image is analyzed on a PhosphorImager and the amount of substrate unwound is calculated as described in Table 3. Reports of the effect of RPA on the helicase activity of Pif1 have been contradictory, with one group showing RPA as having a stimulatory effect on Pif1 helicase function [72] and another group showing that it is inhibitory [76]. Possibly the observed stimulation was related to the ability of RPA to melt the DNA. An example of a helicase assay gel is shown in Figure 6B.
Fig 6. Pif1 can elongate the pol dd d displaced flap.

(A) Model of the Pif1 5′ – 3′ helicase activity. (B) Native gel assessing Pif1 helicase activity on a 30 and 53 nt flap substrate. The asterisk indicates the position of the radiolabel.
Helicase activity of Dna2 and strand melting activity of RPA can be measured using assays similar to those described above using appropriate substrates.
Strand Annealing Assay
To assess the strand annealing functions of Pif1 and RPA, labeled, annealed flap substrates are heated to 95 °C. Five fmol of the heated substrate are incubated with increasing concentrations of Pif1 and RPA in EMSA buffer for 30 mins at 37 °C. The reaction is terminated with 4 μl of EMSA dye and fractionated by electrophoresis as described above (Pif1 helicase section). The substrate alone lane will serve as a control for the percentage of increase in annealing that is observed in the presence of increasing concentration of Pif1 or RPA. Percentage of stand annealing is calculated as described in Table 3.
Full Reconstitution
Reconstituting the Okazaki processing pathway using the minimal purified proteins allows for the assessment of the dynamic function of each Okazaki protein component as it interacts in concert with all of the others. Optimal protein and buffer concentrations obtained from the action of individual proteins on their preferred substrates were used to guide the conditions chosen for the reconstitution system. The substrate design involves two adjacent primers annealed to a template, simulating successive Okazaki fragments. The key readouts ofthe reconstitution experiments are strand displacement synthesis from the upstream primer, cleavage of the downstream primer, and formation of the final ligation product.
To measure synthesis, a 5′ labeled upstream primer (44 nt) and a 60 nt downstream primer are annealed to an 110 nt template, as described in the pol δ section. All substrates used in the reconstitution reactions are adducted with biotin and conjugated with streptavidin at both ends of the template prior to starting the reaction. Each of the protein components (i.e. pol δ, PCNA, RFC, Pif1, RPA, Dna2 and FEN1) is incubated at optimal concentration with 5 fmol of labeled substrate in 20 μl reaction volume containing the reconstitution buffer (Table 1). The reactions are terminated, fractionated by electrophoresis, and analyzed as previously described. Comparing primer extension by the elongation complex alone (pol δ, PCNA, RFC) or with additional Okazaki fragment processing proteins (Pif1, RPA, Dna2 and FEN1) provides information about the stimulatory or inhibitory roles of the other proteins on the efficiency of strand displacement synthesis.
Assessment of downstream primer cleavage can be done with Dna2, FEN1 or the combination of Dna2 and FEN1. Use of a 5′ labeled downstream primer allows assessment of the first cleavages, and use of a 3′ labeled downstream primer displays the distribution of the farthest cleavages. Dna2 and FEN1 cleavages allow for inference of the extent of strand displacement synthesis by the elongation complex. Greater strand displacement synthesis usually increases the length of flap cut off by the first cleavage and decreases the length of primer left after the farthest cleavage. The presence of Pif1 allows for increased strand displacement synthesis in vitro [7]. Additionally, RPA inhibits the loading of FEN1, creating a requirement for Dna2 to activate flaps for cleavage [24]. The coordinated effort of Dna2 and FEN1 creates a nicked substrate, which is suitable for Lig I activity [24].
The final readout in the reconstitution system is ligation efficiency. For the maturation of an Okazaki fragment into a full length functional DNA, the fragments need to be efficiently sealed together. In the presence of minimum reconstitution proteins, ligation efficiency can be measured using an Okazaki substrate containing either a 5′ labeled upstream primer or a 3′labeled downstream primer. During the reconstitution reaction, the proper functioning of each of the components will create a nick that can be sealed by Lig I. An example of an Okazaki fragment maturation reconstitution assay is shown in Figure 7B. However, there is a caveat based on the structure of the substrates, since we use a substrate with a relatively short 60 nt downstream primer, it is possible that the elongation complex, with aid from the nucleases, helicases and RPA, would lead to full displacement of the downstream primer. With a labeled upstream primer, the full-length extension product would be indistinguishable from the ligation product on a denaturing gel so that measurement of the ligation efficiency would not be accurate. If the downstream primer were labeled, strand displacement would result in a loss of the radiolabel on the formation of a full-length synthesis product, so the consequences of making that product could not be determined. Neither substrate is adequate for measuring ligation efficiency.
Fig 7. Full Okazaki fragment processing reconstitution system.
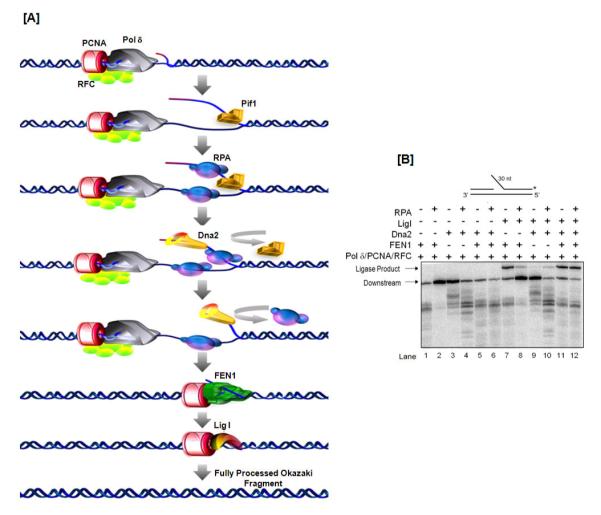
(A) Model of the two-nuclease pathway (see text for description of individual steps). (B) Denaturing gel assessing ligation efficiency of an Okazaki substrate in a reconstituted system. The asterisk indicates the position of the radiolabel.
Figure 7B was originally published in Journal of Biological Chemistry by Pike, J.E., et al., “Pif1 helicase lengthens some Okazaki fragment flaps necessitating Dna2 nuclease/helicase action in the two-nuclease processing pathway”, J Biol. Chem., (2009); 284(37):25170-80 © the American Society for Biochemistry and Molecular Biology
In order to properly measure percentage ligation, the downstream primer is modified such that it extends five nts beyond the annealed template. The 5′ end of the upstream primer is labeled. If the elongation complex fully displaces the downstream primer, the extended product would run at 110 nt, corresponding to the template length, on a denaturing gel. However, if the downstream primer were partially displaced, cleaved by FEN1, and then ligated to the upstream primer, the ligated strand would run at 115 nt on a denaturing gel as the downstream primer extends five nt beyond the template. The five nt difference in length is readily distinguished by electrophoresis.
We have also previously described a system that detects and quantifies only the ligation that occurs after FEN1 cleavage of a flap made by strand displacement synthesis. It excludes any substrates on which FEN1 acts before the upstream primer is extended to make a nick with the downstream primer. Therefore, only the dynamic, coordinated action of displacement, cleavage and ligation is measured. The design of this system is beyond the scope of this review and the reader can refer to the manuscript by Pike et al [21] for further information.
CONCLUDING REMARKS
The central purpose of this review is to summarize the functions of proteins involved in Okazaki fragment maturation and their activities on specific substrates, including some important information on the actual methodology of activity measurements. Two decades of information from both biochemical and genetic investigations have provided us with the capability to reconstitute the Okazaki fragment maturation process in vitro using the minimal protein complement required for effective processing. Use of substrates and techniques described in this review should equip the reader to design activity assays and reconstitute the replication system. Many structural and biochemical functions of components of the Okazaki processing complex still need to be identified. Additionally, helicases such as Bloom’s syndrome protein and Werner’s syndrome protein, might have significant roles in the maturation process. Post-translational modification of protein components of the Okazaki processing complex also alters the functional characteristics of these proteins in vitro. Using techniques described in this review, it should be possible to elucidate the roles of these proteins and modifications in the effective processing of the Okazaki fragments to form a functional DNA.
ACKNOWLEDGEMENTS
We would like to thank present and former members of the Bambara research group for sharing their published results for this review. We also thank Dr. Judith L. Campbell and members of the Bambara laboratory for critically reading the manuscript. This work was supported by National Institutes of Health (NIH) Grant GM024441 to R.A.B. J.W.G was supported by training grant by NIH T32-GM068411.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].Waga S, Stillman B. The DNA replication fork in eukaryotic cells. Annu Rev Biochem. 1998;67:721–51. doi: 10.1146/annurev.biochem.67.1.721. [DOI] [PubMed] [Google Scholar]
- [2].Pursell ZF, Isoz I, Lundstrom EB, Johansson E, Kunkel TA. Yeast DNA polymerase epsilon participates in leading-strand DNA replication. Science. 2007;317:127–30. doi: 10.1126/science.1144067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Arezi B, Kuchta RD. Eukaryotic DNA primase. Trends Biochem Sci. 2000;25:572–6. doi: 10.1016/s0968-0004(00)01680-7. [DOI] [PubMed] [Google Scholar]
- [4].Tsurimoto T, Stillman B. Replication factors required for SV40 DNA replication in vitro. I. DNA structure-specific recognition of a primer-template junction by eukaryotic DNA polymerases and their accessory proteins. J Biol Chem. 1991;266:1950–60. [PubMed] [Google Scholar]
- [5].Burgers PM. Saccharomyces cerevisiae replication factor C. II. Formation and activity of complexes with the proliferating cell nuclear antigen and with DNA polymerases delta and epsilon. J Biol Chem. 1991;266:22698–706. [PubMed] [Google Scholar]
- [6].Ayyagari R, Gomes XV, Gordenin DA, Burgers PM. Okazaki fragment maturation in yeast. I. Distribution of functions between FEN1 AND DNA2. J Biol Chem. 2003;278:1618–25. doi: 10.1074/jbc.M209801200. [DOI] [PubMed] [Google Scholar]
- [7].Rossi ML, Pike JE, Wang W, Burgers PM, Campbell JL, Bambara RA. Pif1 helicase directs eukaryotic Okazaki fragments toward the two-nuclease cleavage pathway for primer removal. J Biol Chem. 2008;283:27483–93. doi: 10.1074/jbc.M804550200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Chu WK, Hickson ID. RecQ helicases: multifunctional genome caretakers. Nat Rev Cancer. 2009;9:644–54. doi: 10.1038/nrc2682. [DOI] [PubMed] [Google Scholar]
- [9].Bartos JD, Wang W, Pike JE, Bambara RA. Mechanisms by which Bloom protein can disrupt recombination intermediates of Okazaki fragment maturation. J Biol Chem. 2006;281:32227–39. doi: 10.1074/jbc.M606310200. [DOI] [PubMed] [Google Scholar]
- [10].Sharma S, Sommers JA, Wu L, Bohr VA, Hickson ID, Brosh RM., Jr. Stimulation of flap endonuclease-1 by the Bloom’s syndrome protein. J Biol Chem. 2004;279:9847–56. doi: 10.1074/jbc.M309898200. [DOI] [PubMed] [Google Scholar]
- [11].Wang W, Bambara RA. Human Bloom protein stimulates flap endonuclease 1 activity by resolving DNA secondary structure. J Biol Chem. 2005;280:5391–9. doi: 10.1074/jbc.M412359200. [DOI] [PubMed] [Google Scholar]
- [12].Brosh RM, Jr., von Kobbe C, Sommers JA, Karmakar P, Opresko PL, Piotrowski J, Dianova I, Dianov GL, Bohr VA. Werner syndrome protein interacts with human flap endonuclease 1 and stimulates its cleavage activity. EMBO J. 2001;20:5791–801. doi: 10.1093/emboj/20.20.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Bohr VA. Rising from the RecQ-age: the role of human RecQ helicases in genome maintenance. Trends Biochem Sci. 2008;33:609–20. doi: 10.1016/j.tibs.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Ouyang KJ, Woo LL, Ellis NA. Homologous recombination and maintenance of genome integrity: cancer and aging through the prism of human RecQ helicases. Mech Ageing Dev. 2008;129:425–40. doi: 10.1016/j.mad.2008.03.003. [DOI] [PubMed] [Google Scholar]
- [15].Li JJ, Kelly TJ. Simian virus 40 DNA replication in vitro. Proc Natl Acad Sci U S A. 1984;81:6973–7. doi: 10.1073/pnas.81.22.6973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Burgers PM. Polymerase dynamics at the eukaryotic DNA replication fork. J Biol Chem. 2009;284:4041–5. doi: 10.1074/jbc.R800062200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Gerik KJ, Li X, Pautz A, Burgers PM. Characterization of the two small subunits of Saccharomyces cerevisiae DNA polymerase delta. J Biol Chem. 1998;273:19747–55. doi: 10.1074/jbc.273.31.19747. [DOI] [PubMed] [Google Scholar]
- [18].Liu L, Mo J, Rodriguez-Belmonte EM, Lee MY. Identification of a fourth subunit of mammalian DNA polymerase delta. J Biol Chem. 2000;275:18739–44. doi: 10.1074/jbc.M001217200. [DOI] [PubMed] [Google Scholar]
- [19].Podust VN, Chang LS, Ott R, Dianov GL, Fanning E. Reconstitution of human DNA polymerase delta using recombinant baculoviruses: the p12 subunit potentiates DNA polymerizing activity of the four-subunit enzyme. J Biol Chem. 2002;277:3894–901. doi: 10.1074/jbc.M109684200. [DOI] [PubMed] [Google Scholar]
- [20].Zuo S, Gibbs E, Kelman Z, Wang TS, O’Donnell M, MacNeill SA, Hurwitz J. DNA polymerase delta isolated from Schizosaccharomyces pombe contains five subunits. Proc Natl Acad Sci U S A. 1997;94:11244–9. doi: 10.1073/pnas.94.21.11244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Pike JE, Burgers PM, Campbell JL, Bambara RA. Pif1 helicase lengthens some Okazaki fragment flaps necessitating Dna2 nuclease/helicase action in the two-nuclease processing pathway. J Biol Chem. 2009;284:25170–80. doi: 10.1074/jbc.M109.023325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Tom S, Henricksen LA, Bambara RA. Mechanism whereby proliferating cell nuclear antigen stimulates flap endonuclease 1. J Biol Chem. 2000;275:10498–505. doi: 10.1074/jbc.275.14.10498. [DOI] [PubMed] [Google Scholar]
- [23].Cho IT, Kim DH, Kang YH, Lee CH, Amangyelid T, Nguyen TA, Hurwitz J, Seo YS. Human replication factor C stimulates flap endonuclease 1. J Biol Chem. 2009;284:10387–99. doi: 10.1074/jbc.M808893200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Bae SH, Bae KH, Kim JA, Seo YS. RPA governs endonuclease switching during processing of Okazaki fragments in eukaryotes. Nature. 2001;412:456–61. doi: 10.1038/35086609. [DOI] [PubMed] [Google Scholar]
- [25].Biswas EE, Zhu FX, Biswas SB. Stimulation of RTH1 nuclease of the yeast Saccharomyces cerevisiae by replication protein A. Biochemistry. 1997;36:5955–62. doi: 10.1021/bi962890u. [DOI] [PubMed] [Google Scholar]
- [26].Stewart JA, Campbell JL, Bambara RA. Significance of the dissociation of Dna2 by flap endonuclease 1 to Okazaki fragment processing in Saccharomyces cerevisiae. J Biol Chem. 2009;284:8283–91. doi: 10.1074/jbc.M809189200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Hasan S, Stucki M, Hassa PO, Imhof R, Gehrig P, Hunziker P, Hubscher U, Hottiger MO. Regulation of human flap endonuclease-1 activity by acetylation through the transcriptional coactivator p300. Mol Cell. 2001;7:1221–31. doi: 10.1016/s1097-2765(01)00272-6. [DOI] [PubMed] [Google Scholar]
- [28].Henneke G, Koundrioukoff S, Hubscher U. Phosphorylation of human Fen1 by cyclin-dependent kinase modulates its role in replication fork regulation. Oncogene. 2003;22:4301–13. doi: 10.1038/sj.onc.1206606. [DOI] [PubMed] [Google Scholar]
- [29].Kucherlapati M, Yang K, Kuraguchi M, Zhao J, Lia M, Heyer J, Kane MF, Fan K, Russell R, Brown AM, Kneitz B, Edelmann W, Kolodner RD, Lipkin M, Kucherlapati R. Haploinsufficiency of Flap endonuclease (Fen1) leads to rapid tumor progression. Proc Natl Acad Sci U S A. 2002;99:9924–9. doi: 10.1073/pnas.152321699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Zheng L, Dai H, Zhou M, Li M, Singh P, Qiu J, Tsark W, Huang Q, Kernstine K, Zhang X, Lin D, Shen B. Fen1 mutations result in autoimmunity, chronic inflammation and cancers. Nat Med. 2007;13:812–9. doi: 10.1038/nm1599. [DOI] [PubMed] [Google Scholar]
- [31].Liu Y, Kao HI, Bambara RA. Flap endonuclease 1: a central component of DNA metabolism. Annu Rev Biochem. 2004;73:589–615. doi: 10.1146/annurev.biochem.73.012803.092453. [DOI] [PubMed] [Google Scholar]
- [32].Harrington JJ, Lieber MR. DNA structural elements required for FEN-1 binding. J Biol Chem. 1995;270:4503–8. doi: 10.1074/jbc.270.9.4503. [DOI] [PubMed] [Google Scholar]
- [33].Kao HI, Henricksen LA, Liu Y, Bambara RA. Cleavage specificity of Saccharomyces cerevisiae flap endonuclease 1 suggests a double-flap structure as the cellular substrate. J Biol Chem. 2002;277:14379–89. doi: 10.1074/jbc.M110662200. [DOI] [PubMed] [Google Scholar]
- [34].Harrington JJ, Lieber MR. The characterization of a mammalian DNA structure-specific endonuclease. EMBO J. 1994;13:1235–46. doi: 10.1002/j.1460-2075.1994.tb06373.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Murante RS, Rust L, Bambara RA. Calf 5′ to 3′ exo/endonuclease must slide from a 5′ end of the substrate to perform structure-specific cleavage. J Biol Chem. 1995;270:30377–83. doi: 10.1074/jbc.270.51.30377. [DOI] [PubMed] [Google Scholar]
- [36].Hohl M, Dunand-Sauthier I, Staresincic L, Jaquier-Gubler P, Thorel F, Modesti M, Clarkson SG, Scharer OD. Domain swapping between FEN-1 and XPG defines regions in XPG that mediate nucleotide excision repair activity and substrate specificity. Nucleic Acids Res. 2007;35:3053–63. doi: 10.1093/nar/gkm092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Bornarth CJ, Ranalli TA, Henricksen LA, Wahl AF, Bambara RA. Effect of flap modifications on human FEN1 cleavage. Biochemistry. 1999;38:13347–54. doi: 10.1021/bi991321u. [DOI] [PubMed] [Google Scholar]
- [38].Nolan JP, Shen B, Park MS, Sklar LA. Kinetic analysis of human flap endonuclease-1 by flow cytometry. Biochemistry. 1996;35:11668–76. doi: 10.1021/bi952840+. [DOI] [PubMed] [Google Scholar]
- [39].Lehman IR. DNA ligase: structure, mechanism, and function. Science. 1974;186:790–7. doi: 10.1126/science.186.4166.790. [DOI] [PubMed] [Google Scholar]
- [40].Shuman S. DNA ligases: progress and prospects. J Biol Chem. 2009;284:17365–9. doi: 10.1074/jbc.R900017200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Levin DS, Vijayakumar S, Liu X, Bermudez VP, Hurwitz J, Tomkinson AE. A conserved interaction between the replicative clamp loader and DNA ligase in eukaryotes: implications for Okazaki fragment joining. J Biol Chem. 2004;279:55196–201. doi: 10.1074/jbc.M409250200. [DOI] [PubMed] [Google Scholar]
- [42].Vijayakumar S, Dziegielewska B, Levin DS, Song W, Yin J, Yang A, Matsumoto Y, Bermudez VP, Hurwitz J, Tomkinson AE. Phosphorylation of human DNA ligase I regulates its interaction with replication factor C and its participation in DNA replication and DNA repair. Mol Cell Biol. 2009;29:2042–52. doi: 10.1128/MCB.01732-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Pascal JM, O’Brien PJ, Tomkinson AE, Ellenberger T. Human DNA ligase I completely encircles and partially unwinds nicked DNA. Nature. 2004;432:473–8. doi: 10.1038/nature03082. [DOI] [PubMed] [Google Scholar]
- [44].Tom S, Henricksen LA, Park MS, Bambara RA. DNA ligase I and proliferating cell nuclear antigen form a functional complex. J Biol Chem. 2001;276:24817–25. doi: 10.1074/jbc.M101673200. [DOI] [PubMed] [Google Scholar]
- [45].Jonsson ZO, Hindges R, Hubscher U. Regulation of DNA replication and repair proteins through interaction with the front side of proliferating cell nuclear antigen. Embo J. 1998;17:2412–25. doi: 10.1093/emboj/17.8.2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Levin DS, Bai W, Yao N, O’Donnell M, Tomkinson AE. An interaction between DNA ligase I and proliferating cell nuclear antigen: implications for Okazaki fragment synthesis and joining. Proc Natl Acad Sci U S A. 1997;94:12863–8. doi: 10.1073/pnas.94.24.12863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Maga G, Frouin I, Spadari S, Hubscher U. Replication protein A as a “fidelity clamp” for DNA polymerase alpha. J Biol Chem. 2001;276:18235–42. doi: 10.1074/jbc.M009599200. [DOI] [PubMed] [Google Scholar]
- [48].Yuzhakov A, Kelman Z, Hurwitz J, O’Donnell M. Multiple competition reactions for RPA order the assembly of the DNA polymerase delta holoenzyme. Embo J. 1999;18:6189–99. doi: 10.1093/emboj/18.21.6189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Fanning E, Klimovich V, Nager AR. A dynamic model for replication protein A (RPA) function in DNA processing pathways. Nucleic Acids Res. 2006;34:4126–37. doi: 10.1093/nar/gkl550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Bartos JD, Willmott LJ, Binz SK, Wold MS, Bambara RA. Catalysis of strand annealing by replication protein A derives from its strand melting properties. J Biol Chem. 2008;283:21758–68. doi: 10.1074/jbc.M800856200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Hellman LM, Fried MG. Electrophoretic mobility shift assay (EMSA) for detecting protein-nucleic acid interactions. Nat Protoc. 2007;2:1849–61. doi: 10.1038/nprot.2007.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Stewart JA, Miller AS, Campbell JL, Bambara RA. Dynamic removal of replication protein A by Dna2 facilitates primer cleavage during Okazaki fragment processing in Saccharomyces cerevisiae. J Biol Chem. 2008;283:31356–65. doi: 10.1074/jbc.M805965200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Budd ME, Campbell JL. A yeast replicative helicase, Dna2 helicase, interacts with yeast FEN-1 nuclease in carrying out its essential function. Mol Cell Biol. 1997;17:2136–42. doi: 10.1128/mcb.17.4.2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Duxin JP, Dao B, Martinsson P, Rajala N, Guittat L, Campbell JL, Spelbrink JN, Stewart SA. Human dna2 is a nuclear and mitochondrial DNA maintenance protein. Mol Cell Biol. 2009;29:4274–82. doi: 10.1128/MCB.01834-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Kim JH, Kim HD, Ryu GH, Kim DH, Hurwitz J, Seo YS. Isolation of human Dna2 endonuclease and characterization of its enzymatic properties. Nucleic Acids Res. 2006;34:1854–64. doi: 10.1093/nar/gkl102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Masuda-Sasa T, Imamura O, Campbell JL. Biochemical analysis of human Dna2. Nucleic Acids Res. 2006;34:1865–75. doi: 10.1093/nar/gkl070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Bae SH, Kim JA, Choi E, Lee KH, Kang HY, Kim HD, Kim JH, Bae KH, Cho Y, Park C, Seo YS. Tripartite structure of Saccharomyces cerevisiae Dna2 helicase/endonuclease. Nucleic Acids Res. 2001;29:3069–79. doi: 10.1093/nar/29.14.3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Liu Q, Choe W, Campbell JL. Identification of the Xenopus laevis homolog of Saccharomyces cerevisiae DNA2 and its role in DNA replication. J Biol Chem. 2000;275:1615–24. doi: 10.1074/jbc.275.3.1615. [DOI] [PubMed] [Google Scholar]
- [59].Kang HY, Choi E, Bae SH, Lee KH, Gim BS, Kim HD, Park C, MacNeill SA, Seo YS. Genetic analyses of Schizosaccharomyces pombe dna2(+) reveal that dna2 plays an essential role in Okazaki fragment metabolism. Genetics. 2000;155:1055–67. doi: 10.1093/genetics/155.3.1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Wawrousek K, Fortini B, Polaczek P, Chen L, Liu Q, Dunphy B, Campbell J. Xenopus DNA2 is a helicase/nuclease that is found in complexes with replication proteins And-1/Ctf4 and Mcm10 and DSB response proteins Nbs1 and ATM. Cell Cycle. 2010 doi: 10.4161/cc.9.6.11049. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Balakrishnan L, Stewart J, Polaczek P, Campbell JL, Bambara RA. Acetylation of Dna2 and FEN1 by p300 promotes DNA stability by creating long Flap intermediates. J Biol Chem. 2009 doi: 10.1074/jbc.M109.086397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Masuda-Sasa T, Polaczek P, Peng XP, Chen L, Campbell JL. Processing of G4 DNA by Dna2 helicase/nuclease and replication protein A (RPA) provides insights into the mechanism of Dna2/RPA substrate recognition. J Biol Chem. 2008;283:24359–73. doi: 10.1074/jbc.M802244200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Cherenkov PA. Visible emission of clean liquids by action of radiation. Dokl.A kad.Nauk SSSR. 1934;2:451–54. [Google Scholar]
- [64].Lahaye A, Stahl H, Thines-Sempoux D, Foury F. PIF1: a DNA helicase in yeast mitochondria. Embo J. 1991;10:997–1007. doi: 10.1002/j.1460-2075.1991.tb08034.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Pinter SF, Aubert SD, Zakian VA. The Schizosaccharomyces pombe Pfh1p DNA helicase is essential for the maintenance of nuclear and mitochondrial DNA. Mol Cell Biol. 2008;28:6594–608. doi: 10.1128/MCB.00191-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Zhang DH, Zhou B, Huang Y, Xu LX, Zhou JQ. The human Pif1 helicase, a potential Escherichia coli RecD homologue, inhibits telomerase activity. Nucleic Acids Res. 2006;34:1393–404. doi: 10.1093/nar/gkl029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Schulz VP, Zakian VA. The saccharomyces PIF1 DNA helicase inhibits telomere elongation and de novo telomere formation. Cell. 1994;76:145–55. doi: 10.1016/0092-8674(94)90179-1. [DOI] [PubMed] [Google Scholar]
- [68].Makovets S, Blackburn EH. DNA damage signalling prevents deleterious telomere addition at DNA breaks. Nat Cell Biol. 2009;11:1383–6. doi: 10.1038/ncb1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Budd ME, Reis CC, Smith S, Myung K, Campbell JL. Evidence suggesting that Pif1 helicase functions in DNA replication with the Dna2 helicase/nuclease and DNA polymerase delta. Mol Cell Biol. 2006;26:2490–500. doi: 10.1128/MCB.26.7.2490-2500.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Burgers PM, Gerik KJ. Structure and processivity of two forms of Saccharomyces cerevisiae DNA polymerase delta. J Biol Chem. 1998;273:19756–62. doi: 10.1074/jbc.273.31.19756. [DOI] [PubMed] [Google Scholar]
- [71].Stith CM, Sterling J, Resnick MA, Gordenin DA, Burgers PM. Flexibility of eukaryotic Okazaki fragment maturation through regulated strand displacement synthesis. J Biol Chem. 2008;283:34129–40. doi: 10.1074/jbc.M806668200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Boule JB, Zakian VA. The yeast Pif1p DNA helicase preferentially unwinds RNA DNA substrates. Nucleic Acids Res. 2007;35:5809–18. doi: 10.1093/nar/gkm613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Lahaye A, Leterme S, Foury F. PIF1 DNA helicase from Saccharomyces cerevisiae. Biochemical characterization of the enzyme. J Biol Chem. 1993;268:26155–61. [PubMed] [Google Scholar]
- [74].Zhou JQ, Qi H, Schulz VP, Mateyak MK, Monson EK, Zakian VA. Schizosaccharomyces pombe pfh1+ encodes an essential 5′ to 3′ DNA helicase that is a member of the PIF1 subfamily of DNA helicases. Mol Biol Cell. 2002;13:2180–91. doi: 10.1091/mbc.02-02-0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].George T, Wen Q, Griffiths R, Ganesh A, Meuth M, Sanders CM. Human Pif1 helicase unwinds synthetic DNA structures resembling stalled DNA replication forks. Nucleic Acids Res. 2009;37:6491–502. doi: 10.1093/nar/gkp671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Gu Y, Masuda Y, Kamiya K. Biochemical analysis of human PIF1 helicase and functions of its N-terminal domain. Nucleic Acids Res. 2008;36:6295–308. doi: 10.1093/nar/gkn609. [DOI] [PMC free article] [PubMed] [Google Scholar]