

Abstract
Adenylation/adenylate-forming enzymes catalyze the activation of a carboxylic acid at the expense of ATP to form an acyl-adenylate intermediate and pyrophosphate (PPi). In a second half-reaction, adenylation enzymes catalyze the transfer of the acyl moiety of the acyl-adenylate onto an acceptor molecule, which can be either a protein or a small molecule. We describe the design, development, and validation of a coupled continuous spectrophotometric assay for adenylation enzymes that employs hydroxylamine as a surrogate acceptor molecule leading to the formation of a hydroxamate. The released pyrophosphate from the first half-reaction is measured using the pyrophosphatase–purine nucleoside phosphorylase coupling system with the chromogenic substrate 7-methylthioguanosine (MesG). The coupled hydroxamate–MesG assay is especially useful for characterizing the activity and inhibition of adenylation enzymes that acylate a protein substrate and/or fail to undergo rapid ATP-PPi exchange.
Keywords: adenylation, adenylate-forming, hydroxamate, MesG, enzyme assay
Introduction
Adenylation or adenylate-forming enzymes (AEs) are ubiquitous in all forms of life and are involved in numerous biological processes including protein synthesis1 and degradation,2 DNA synthesis,3 coenzyme biosynthesis,4–6 lipid metabolism,7 and biosynthesis of secondary metabolites including polyketides (PKs) and nonribosomal peptides (NRPs).8, 9 Adenylation enzymes catalyze a two-step adenylation–acylation reaction.9 In the initial adenylation half-reaction, the AE binds a substrate carboxylic acid and ATP, then catalyzes their condensation forming a tightly bound acyl-adenylate along with pyrophosphate (Figure 1A). In the subsequent acylation half-reaction, the AE binds the acceptor residue and catalyzes the transfer of the acyl moiety of the acyl-adenylate onto a nucleophilic oxygen, sulfur or nitrogen atom of an acceptor molecule leading to respective ester, thioester or amide products. In primary or intermediary metabolism, the acceptor is usually a diffusible small molecule (Figure 1B). The overall reaction can be easily followed by detection of the released pyrophosphate using pyrophosphatase and malachite green or purine nucleoside phosphorylase in conjunction with the chromogenic substrate 7-methylthioguanosine (MesG).10–12 Alternatively, the overall adenylation reaction can be followed through detection of AMP using the myokinase–pyruvate kinase–lactate dehydrogenase coupling system.13, 14 However, in secondary metabolism the acceptor molecule is a carrier protein (CP), which is either located in trans as a separate protein or more often is found in cis to an adenylation domain as part of a multifunctional protein.8, 15 In these latter cases, the overall coupled reaction requires stoichiometric protein and cannot be analyzed by steady-state kinetic methods, thus complicating assay development. A common solution is to decouple the enzymatic steps and measure only the adenylation activity using a pyrophosphate exchange assay that measures the adenylation reaction in reverse through incorporation of [32P]-pyrophosphate into ATP.16, 17 A primary drawback to the pyrophosphate exchange assay is the use of radioactive [32P]-pyrophosphate. In this latter regard, Bachmann and co-workers have recently described an elegant non-radioactive mass spectrometry based [18O]-ATP exchange assay.18
Figure 1.
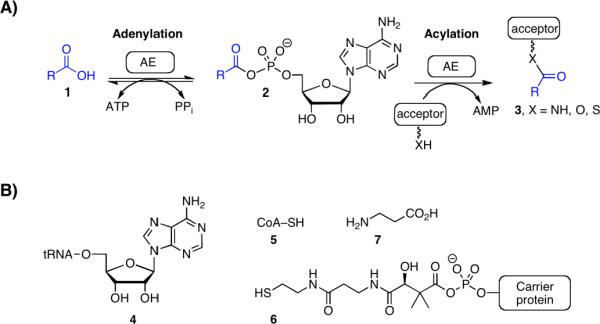
(A) Enzyme mechanism catalyzed by adenylation enzymes (AEs). (B) Representative acceptor molecules in primary and seconday metabolism (4–7). Ester formation: for aminoacyl tRNA synthetases, the acceptor residue is the 2' or 3' alcohol from the ribose sugar of the terminal adenosine residue of a cognate tRNA molecule (4). Thioester formation: For CoA ligases, the acceptor is the terminal sulfur atom of the coenzyme A molecule (5) and for carrier domains of polyketide synthases (PKSs) and nonribosomal peptide synthetases (NRPSs) the acceptor is the terminal sulfur atom of the phosphopantetheinyl (ppant) cofactor arm (6). Amide formation: For PanC involved in coenzyme A biosynthesis, the acceptor is the β-amino group of β-alanine.
Both [32P]-pyrophosphate and the [18O]-ATP de-coupled assays require rapid exchange of pyrophosphate with the acyl-adenylate. Challis and co-workers have reported that several adenylating enzymes involved in synthesis of bacterial siderophores (small molecule iron chelators) cannot release pyrophosphate and therefore could not be assayed through the conventional ATP-PPi exchange assays.19 Instead, a hydroxamate formation assay was used to characterize these AEs wherein hydroxylamine serves as a highly reactive surrogate acceptor molecule that leads to the formation of a hydroxamate, which in turn can be quantified spectrophotometrically through the addition of ferric iron (Figure 2).19 However, the reported hydroxamate formation assay suffers from substrate dependence, low sensitivity, and inability to monitor continuously. Herein we report the development of a coupled hydroxamate continuous assay that detects phosphate formation using the MesG assay system. The assay was validated against a panel of previously characterized adenylating enzymes. Next, we demonstrated the utility of this assay in the steady-state kinetic analysis of adenylation-carrier protein didomain in the overall forward direction and show this can also be used to characterize a fatty acid adenylating enzyme from M. tuberculosis that fails to undergo rapid ATP-PPi exchange. Finally, we show the coupled assay can be used to rapidly kinetically characterize a series of inhibitors against these adenylating enzymes.
Figure 2.

Hydroxamate formation assay. In the absence of the native acceptor the tightly bound acyl adenylate can be released with hydroxylamine forming the acyl hydroxymate and releasing AMP. Product formation may then be monitored by coupling the release of PPi to the cleavage of the UV indicator MesG in a continuous format. Addition of Fe allows for end point monitoring of the hydroxamate.
Materials and Methods
Materials
Chemically competent E. coli Mach1 and BL21 STAR (DE3), plasmids pCR2.1-TOPO and pENTR/D-TOPO were purchased from Invitrogen (Carlsbad, CA, USA). Restriction enzymes were purchased from New England Biolabs (Ipswich, MA, USA). PrimeSTAR HS DNA polymerase was purchased from TAKARA Bio Inc (Otsu, Shiga, Japan). Primers for PCR were obtained from Integrated DNA Technologies (Coralville, IA, USA). Expression vectors were purchased from EMD biosciences (San Diego, CA, USA). Ni-NTA and DNA purification/isolation kits were obtained from Qiagen Sciences (Germantown, MD, USA). 7-Methylthioguanosine (MesG) was obtained from Berry and Associates (Dexter, MI). All other chemicals, biological buffers, and the coupling enzymes inorganic pyrophosphatase (I1643) and purine nucleoside phosphorylase (N8264) were purchased from Sigma–Aldrich (St. Louis, MO, USA). Enzymatic activity, kinetic parameters, and inhibition assays were performed on a Molecular Devices (Sunnyvale, CA, USA) M5e multi-mode plate reader. M. tuberculosis H37Rv genomic DNA was a gift of Dr. Clifton E. Barry 3rd (NIH). A. migulanus genomic DNA was purchased from ATCC (Manassas, VA).
Preparation of overexpression constructs
The genes vibE, basE, entE, and mbtA were cloned as previously described.20 The gene fadD28 was amplified by PCR from M. tuberculosis H37Rv genomic DNA using primers fadD28 F (CACCCATATGAGTGTGCGTTCCCTTC) and fadD28 R (GGGGATCCTCAGGCATCCAAGCGGGCGAATTG) and cloned into pENTR/TEV D-TOPO to produce plasmid pCDD079. Plasmid pCDD079 was digested with NdeI and BamHI and the gene was subcloned into pET28b to produce pCDD084, an expression vector for FadD28 with a hexahistidine appended to the N-terminus. The genes grsA(A) and grsA(AT) were cloned essentially as described.21 Primers grsA F (CACCATGGTAAACAGTTCTAAAAG) and either grsA R (CCTCGAGTAATACATCCTGCCAG) or grsAT R (CCTCGAGATTTGGTCTATACAA) were used to amplify the gene from A. migulanus genomic DNA (ATCC 9999). After capture of the PCR products using pENTR/SD D-TOPO the grsA(A) and grsA(AT) were digested with NcoI and XhoI and inserted into pET28b, creating the C-terminal His fusion expression vectors pCDD121 and pCDD122 respectively.
Overproduction and purification of adenylating enzymes
VibE, BasE, EntE, and MbtA were expressed and purified as previously described.20 Plasmid pCDD084 was transformed into BL21 STAR (DE3) (Invitrogen) for expression of FadD28. Overnight cultures (0.1 mL) were used to inoculate 100 mL ZYM-505222 containing kanamycin (50 μg/mL) and grown at 37 °C for 6 hours followed by 30 °C overnight. The culture was centrifuged and the cell pellets were frozen at −80 °C. Frozen pellets were resuspended in 20 mL lysis buffer (50 mM HEPES, 300 mM NaCl, 10 mM imidazole, pH 8.0). The solution was sonicated at 0 °C with three 2 min bursts (power 7, 30% duty cycle) with a 1 min break between each burst (Branson Sonifier 250). The lysate was centrifuged at 30,000 × g for 10 min at 4 °C then 2 mL of 50% Ni-NTA were added to the cleared supernatant. After incubating at 4 °C for 1 h, the lysate was loaded onto a column and the flow through was collected. The column was washed with 16 mL of wash buffer (50 mM HEPES, 300 mM NaCl, 20 mM imidazole, pH 8.0) and eluted with 3 mL of elution buffer (50 mM HEPES, 300 mM NaCl, 250 mM imidazole, pH 8.0) of which the first 0.5 mL was discarded. The protein solution was desalted on a PD-10 column into storage buffer (10 mM Tris·HCl, pH 8.0, 1.0 mM EDTA, 5% glycerol) and stored at −80 °C. Protein concentrations were determined by the Bradford method using BSA as a standard. The yield of FadD28 was approximately 90 mg/L.
For expression of GrsA(A) and GrsA(AT) plasmids pCDD121 and pCDD122 were transformed into BL21 STAR (DE3) and BL21 (DE3) pDHS4032 respectively. Plasmid pDHS4032 carries a chloramphenicol resistance gene and sfp, which expresses Sfp, a nonspecific phosphopantetheinyl transferase from Bacillus subtilis that converts the apo protein into the holo form.23 Both strains were expressed essentially as described.21 Overnight cultures (5 mL) were used to inoculate 500 mL LB supplemented with 10 mM MgCl2, 50 μg/mL kanamycin and 25 μg/mL chloramphenicol (GrsA(AT) only). Cultures were grown at 37 °C to an A600 of 0.6 andthen grown at 30 °C until an A600 of 1.5 and then induced with IPTG to 1.5 mM. Cells were grown for an additional 4 hours then centrifuged and frozen at −80 °C. GrsA(A) and GrsA(AT) were purified from the cell pellets as described for FadD28. The yield of GrsA(A) was approximately 23 mg/L while GrsA(AT) provided 5 mg/L.
Hydroxamate–MesG assay
Reactions contained varying amounts of adenylation enzyme (0.005–5 μM) to maintain initial velocity conditions, 50 mM Tris pH 8.0, 2.5 mM ATP, 5 mM MgCl2, 0.5 mM DTT, 150 mM hydroxylamine pH 7, 0.1 U nucleoside phosphorylase, 0.04 U pyrophosphatase, 0.2 mM MesG, and varying concentrations of acid substrates (Table 1). Reactions (100 μL) were run in 96 well half-area UVStar plates (Greiner) and the cleavage of MesG was monitored at A360 on a Molecular Devices Spectra Max M5e. Working stocks of hydroxylamine were prepared fresh daily by combining 500 μL of 4 M hydroxylamine, 250 μL water, and 250 μL 7 M NaOH on ice. Hydroxylamine and NaOH stocks were stored at 4 °C for at least 1 month.
Table 1.
Kinetic parameters of EntEa
| substrate | KM (mM) | kcat (min−1) | Ki (mM) |
|---|---|---|---|
| NH2OH | 225 ± 51 | 3.2 ± 0.5 | 381 ± 113 |
ATP-[32P]-PPi exchange assay
Reactions were carried out as previously described, except for GrsA(A) and GrsA(AT), where the buffer was modified to 50 mM Tris pH 8.0, 2.5 mM ATP, 5 mM MgCl2, 0.5 mM DTT.20 This was done since GrsA(A) and GrsA(AT) have a reported KM for ATP of approximately 1 mM so the ATP in the hydroxamate–MesG assay (2.5 mM) was not saturating. All other enzymes have a lower KM and ATP was saturating in both assays.
Determination of kinetic parameters
Steady-state kinetic parameters of the substrate acids were determined for each enzyme using both the hydroxamate-MesG assay and the [32P]-pyrophosphate exchange assay using the standard assay conditions described above by measuring the initial velocity (v0) as a function of substrate acid concentration to provide a saturation curve, which was fit by nonlinear regression analysis to the Michaelis–Menten equation. The enzyme and substrate concentrations that were used in the hydroxamte-MesG assay are listed herein: EntE was used at 100 nM with salicylic acid (1.4–1000 μM), benzoic acid (0.2–10 mM) and 2,3-DHB (1–100 μM); VibE was used at 200 nM with salicylic acid (1.4–1000 μM) and 2,3-DHB (1–100 μM); BasE was used at 200 nM with salicylic acid (1.4–1000 μM) and 2,3-DHB (1–100 μM); MbtA was used at 1 μM with salicylic acid (0.4–300 μM); GrsA(A) and GrsA(AT) were used at 5 nM with D-phenylalanine (1.4–1000 μM); and FadD28 was used at 4.5 μM with tetradecanoic acid (0.14–100 μM). To determine the kinetic parameters of hydroxylamine, EntE and 2,3-DHB were fixed at 100 nM and 100 μM, respectively, while hydroxylamine was varied from 16.3–520 mM. The resulting data was fit to the substrate inhibition model (eq 1). For the pyrophosphate exchange assay, EntE was used at 2 nM with 2,3-DHB (0.4–50 μM) and 7 nM with salicylic acid (1.6–200 μM). For the other enzymes, VibE was used at 7 nM with 2,3-DHB (0.4–50 μM) or 30 nM with salicylic acid (3–500 μM); GrsA or GrsAT were used at 5 nM with D-phenylalanine (1.4–1000 μM).
Determination of values of inhibitors by the hydroxamate–MesG assay
determination was performed using our standard assay conditions. For EntE compound 15 was tested from 31–2000 nM using both salicylic acid (10 mM) and 2,3-DHB (100 μM) as the competing substrates. The enzyme was fixed at 50 nM for salicylic acid and 250 nM for 2,3-DHB. Compound 16 was varied from 0.3–200 nM while GrsA (5 nM) and D-phenylalanine (1 mM) were held constant. Compound 17 was varied from 0.4–50 μM while FadD28 and tetradecanoic acid were fixed at 4.25 μM and 33 μM, respectively. Initial velocities were fit to the Morrison equation (eq 2) using Prism 4 (GraphPad). Enzyme concentrations were constrained allowing the program to solve exclusively for . The for 15 with EntE using salicylic acid as the competitive ligand was used to solve the equation (eq 3) for Ki. This value was then used with the obtained using 2,3-DHB as the competitive ligand and the equation was solved for the KM of 2,3-DHB.
Results and Discussion
Rationale and Implementation
Adenylation enzymes catalyze the formation of an acyl-adenylate, which remains tightly bound in the enzyme active site as a result of the large interaction energy between the protein and this bisubstrate intermediate. Walsh and co-workers have shown dissociation of the acyl-adenylate occurs at a rate of more than 100-fold lower than the acylation rate of the native acceptor molecule.24 Consequently, in the absence of an acceptor molecule, catalytic turnover in the physiological relevant forward direction is very slow. The use of hydroxylamine, a potent nucleophile, as a surrogate acceptor molecule was expected to enhance turnover. As shown by Challis and co-workers, the hydroxamate product can be detected as an iron (III) hydroxamate complex through addition of ferric iron in an end-point assay.19 However, the absorbance of the hydroxamate product is weak, thus limiting sensitivity. Furthermore, not all hydroxamates form spectroscopically detectable complexes with ferric iron.
The overall adenylation–acylation reaction generates one molecule of pyrophosphate (PPi). Pyrophosphate has previously been measured using the continuous spectrophotometric assay available as the EnzChek Pyrophosphate Assay Kit (Invitrogen) wherein PPi is hydrolyzed to inorganic phosphate by inorganic pyrophosphatase.10, 11 Phosphate production is then coupled to phosphorolysis of 7-methylthioguanosine (MesG) catalyzed by the enzyme purine nucleoside phosphorylase (PNP) and the chromophoric product, 7-methylguanine, is monitored by absorbance at 360 nm. We envisioned that coupling of the hydroxamate formation assay with the pyrophosphate detection assay would provide a continuous spectroscopic assay if the large amounts of hydroxylamine do not interfere with inorganic pyrophosphatase or purine nucleoside phosphorylase.
To initially evaluate the MesG coupled hydroxamate formation assay, we selected the prototypical stand-alone adenylating enzyme EntE from E. coli involved in biosynthesis of the siderophore enterobactin.25, 26 EntE catalyzes the acylation of the N-terminal carrier domain of EntB with 2,3-dihydroxybenzoic acid (2,3-DHB). Using the MesG coupled continuous assay, we showed that EntE, in the absence of acceptor, catalyzes the adenylation of 2,3-DHB and nonspecific release of the acyl adenylate with a rate of 0.177 min−1 with saturating concentrations of both ATP and 2,3-DHB. This rate corresponds to the leak rate of 2,3-DHB-AMP from EntE in the absence of an acceptor molecule (EntB or hydroxylamine).24 We further demonstrated the pyrophosphate formation is not a result of nonspecific ATP hydrolysis since both ATP and 2,3-DHB were required for activity. We then evaluated the initial velocity as a function of hydroxylamine concentration and observed the rate exhibited substrate inhibition kinetics with a KM of 225 ± 51 mM, Ki of 381 ± 113 mM and kcat of 3.2 ± 0.5 min−1 with saturating concentrations of both ATP and 2,3-DHB (Figure 3 and Table 1). We also confirmed that the acylation rate was linearly dependant on EntE concentration for this coupled assay system (Figure 4). The kcat was 18-fold higher than the leak rate, but 40-fold lower than the acylation rate of the native protein substrate EntB.24 Gulick and co-workers have shown that binding of carrier proteins to their respective adenylation enzymes induces a large ~140 ° rotation of the C-terminal domain that facilitates the acylation half-reaction and may explain the lower kcat value observed with hydroxylamine.9 Movement of the C-terminal domain provides space for the incoming nucleophile and ideally positions a catalytic lysine to deprotonate the incoming nucleophile. Additionally, this movement is expected to permit dissociation of the products.
Figure 3.
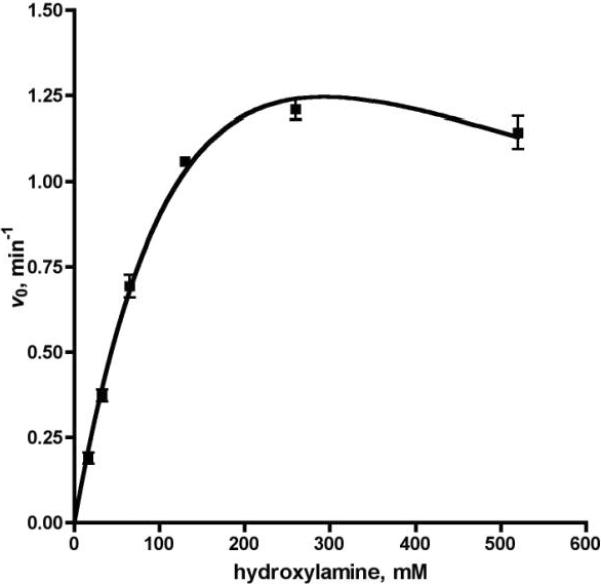
Steady-state kinetics of EntE: velocity vs. hydroxylamine. Each reaction contained EntE at 100 nM, 50 mM Tris pH 8.0, 2.5 mM ATP, 100 μM 2,3 DHBA, 5 mM MgCl2, 0.5 mM DTT, 0.1 U nucleoside phosphorylase, 0.04 U pyrophosphatase, and 0.2 mM MesG. Velocities were fit to the substrate inhibition equation.
Fig 4.

A) Enzyme dependant time course of pyrophosphate release. Each reaction contained 50 mM Tris pH 8.0, 2.5 mM ATP, 100 μM 2,3 DHBA, 5 mM MgCl2, 0.5 mM DTT, 150 mM hydroxylamine pH 7, 0.1 U nucleoside phosphorylase, 0.04 U pyrophosphatase, and 0.2 mM MesG. EntE was used at 0 (∎), 62.5 nM (▲), 125 nM (▼), 250 nM (◆), 500 nM (●), 1000 nM (□), 2000 nM (△). B) Secondary plot of linear relationship between EntE concentration and initial velocity.
Measurement of kinetic parameters
We evaluated the substrate specificity of EntE with 2,3-DHB, salicylic acid (SAL), and benzoic acid (BZ) with a saturating concentration of ATP and 150 mM hydroxylamine (not saturating due to substrate inhibition). Plots of the initial velocities, v0, versus substrate concentration provided saturation curves (see Figure 5), which were fit to the Michaelis–Menten equation to provide the apparent kinetic parameters shown in Table 2. The apparent kcat for 2,3-DHB was 1.1 min−1 while the KM was too low to evaluate. The KM for 2,3-DHB was determined indirectly as 0.018 μM using a bisubstrate inhibitor as detailed in the Materials and Methods. The KM values for the non-native substrates salicylic acid and benzoic acid were 19 μM and 2.1 mM, respectively. The apparent kcat values of salicylic acid and benzoic acid were 11.6 min−1 and 3.2 min−1, which are respectively 11-fold and 3-fold higher than the native substrate 2,3-DHB. To further investigate the origin of the increased kcat of salicylic acid and benzoic acid, we evaluated the initial velocities of reactions without hydroxylamine. Hydroxylamine did not alter the rate with benzoic acid indicating that 100% of the activity is due to leaking of the acyl-adenylate. Similar studies with salicylic acid demonstrated that 91% of the activity was attributable to leaking of the acyl-adenylate. Interestingly, in the absence of hydroxylamine the slope of the progress curves for both salicylic acid and benzoic acid rapidly began to plateau, which we believe is due to inhibition by the intermediate acyl-adenylates. This in turn suggests that the hydroxylamine reacts with the respective acyl-adenylates in solution resulting in cleavage of these tight-binding bisubstrate compounds. Leaking of the acyl-adenylates from the enzyme enables turnover and hence enhanced kcat values. The acyl adenylates of the non-native substrates SAL-AMP and BZ-AMP are bound more weakly owing to loss of key hydrogen bonds and hence more rapidly leak out of the active site, whereas 2,3-DHB-AMP is bound tightly and less than 10% of the observed activity is due to leaking of the acyl-adenylate.
Figure 5.

Steady-state kinetics of EntE. A) initial velocity vs. salicylic acid. B) initial velocity vs. benzoic acid. Each reaction contained EntE at 100 nM and standard assay buffer (50 mM Tris pH 8.0, 2.5 mM ATP, 5 mM MgCl2, 0.5 mM DTT, 150 mM hydroxylamine pH 7, 0.1 U nucleoside phosphorylase, 0.04 U pyrophosphatase, and 0.2 mM MesG).
Table 2.
Kinetic parameters of adenylating enzymes.
| Hydroxamate–MesG Assay | [32P]-PPi-ATP Assay | ||||||
|---|---|---|---|---|---|---|---|
| Enzyme | Substratea | kcat (min−1) | KM (μM) | kcat/KM (min−1μM−1) | kcat (min−1) | KM (μM) | kcat/KM (min−1μM−1) |
| EntE | SAL | 18.0 ± 0.6 | 24.02 ± 3.77 | 0.75 ± 0.12 | 89.8 ± 1.9 | 118 ± 8 | 0.76 ± 0.05 |
| 2,3-DHB | 1.1 ± 0.1 | 0.018 | 58.3 | 416 ± 25 | 7.5 ± 1.3 | 55.5 ± 10.2 | |
| BZ | 3.2 ± 0.2 | 2131 ± 294 | 0.0010 ± 0.0002 | < 1 | NDb | NDb | |
| VibE | SAL | 4.5 ± 0.2 | 17.6 ± 3.5 | 0.25 ± 0.05 | 40.0 ± 1.5 | 57.3 ± 6.8 | 0.7 ± 0.1 |
| 2,3-DHB | 0.23 ± 0.01 | NDb | NDb | 138 ± 1 | 0.46 ± 0.01 | 300 ± 7 | |
| BasE | SAL | 13.6 ± 0.4 | 3.5 ± 0.6 | 3.9 ± 0.7 | 75 ± 3c | 13.2 ± 1.9c | 5.7 ± 0.8c |
| 2,3-DHB | 0.98 ± 0.03 | NDb | NDb | 207 ± 9c | 1.51 ± 0.30c | 137 ± 28c | |
| MbtA | SAL | 0.283 ± 0.001 | 0.31 ± 0.01 | 0.91 ± 0.03 | 222 ± 3 | 2.79 ± 0.16 | 79.5 ± 4.7 |
| GrsA | D-Phe | 283.9 ± 8.2 | 69.4 ± 7.4 | 4.1 ± 0.5 | 479.7 ± 23.9 | 106.0 ± 18.5 | 4.5 ± 0.8 |
| GrsAT | D-Phe | 93.9 ± 4.6 | 99.7 ± 16.8 | 0.9 ± 0.2 | 238.8 ± 9.9 | 156.4 ± 20.4 | 1.5 ± 0.2 |
| FadD28 | C14 | 0.21 ± 0.01 | 5.3 ± 0.9 | 0.04 ± 0.01 | NDb | NDb | NDb |
SAL = salicylic acid, 2,3-DHB = 2,3-dihydroxybenzoic acid, BZ = benzoic acid, D-Phe = D-phenylalanine, C14 = tetradecanoic acid.
not determined.
see ref. 20.
For comparative purposes, the kinetic parameters were also determined using the conventional ATP-PPi exchange assay, which has been the most commonly employed assay for evaluating stand alone aryl acid adenylating enzymes and NRPS adenylation domains. Although direct comparision of the substrate dependence of exchange kinetics to steady state turnover kinetics is not meaningful, we did observe that the hydroxamate-MesG coupled assay successfully reproduced the specificity constants (kcat/KM) determined with the ATP-PPi exchange assay. Thus, the specificity constant for 2,3-DHB with EntE was 58.3 min−μM−1 using the hydroxamate-MesG assay and 55.9 min−1μM−1 using the ATP-PPi exchange assay. Excellent correlation was also observed for salicylic acid and benzoic acid as shown in Table 2. Furthermore the results with the hydroxamate-MesG assay recapitulated the known trends in KM as observed with the ATP-PPi exchange assay while the trend in kcat values were opposite to the results using the ATP-PPi exchange assay.25
To verify the generality of the hydroxamate-MesG assay, we next evaluated the substrate specificity of three other aryl acid adenylating enzymes involved in synthesis of bacterial siderophores including BasE from Acinetobacter baumannii, VibE from Vibrio cholerae, and MbtA from Mycobacterium tuberculosis.20, 27, 28 The native substrate for BasE and VibE is 2,3-DHB, while the native substrate for MbtA is salicylic acid. These activated acids are transferred onto cognate carrier proteins in analogy to the EntE–EntB protein pair. The kinetic parameters for each of these enzymes with 2,3-DHB and salicylic acid are shown in Table 2. Overall, the trend in KM values paralleled those determined with the ATP-PPi exchange assay while the trend in kcat's was directly opposite. However, as observed with the model enzyme EntE, the specificity constants (kcat/KM) of VibE and BasE were in close agreement to the values obtained with the ATP-PPi exchange assay.
Next, we investigated the prototypical nonribosomal peptide synthetase GrsA involved in biosynthesis of gramicidin that contains the adenylation domain and thiolation domain on the same polypeptide.21, 29 Previous attempts to functionally characterize the adenylation activity of the phenylalanine adenylation domain have relied on the ATP-PPi exchange assay, since the carrier domain is located in cis precluding steady-state kinetic analysis in the overall forward direction. To simplify analysis we studied the excised phenylalanine adenylation domain GrsA(A) as well as the excised adenylation-thiolation didomain construct GrsA(AT).21 We evaluated the substrate D-phenylalanine using both the hydroxamate-MesG and ATP-PPi exchange assays. Unlike the stand-alone adenylating enzymes (i.e. EntE, VibE, BasE, MbtA) both GrsA(A) and GrsA(AT) were highly active in the hydroxamate-MesG assay and the kcat and KM values were within approximately 2-fold of the values obtained with the ATP-PPi exchange assay (Table 2).
Lastly, we examined the fatty acid adenylating enzyme FadD28 involved in biosynthesis of the phthiocerol dimycocerosate lipids, which are virulence factors in M. tuberculosis.30′32 FadD28 has been shown to activate and transfer long-chain fatty acids to a N-terminal carrier protein domain of the mycobacterial polyketide synthase enzyme Mas thereby linking fatty acid and polyketide biosynthesis.33 Our attempts to characterize FadD28 using the established ATP-PPi assay or the recently reported [18O]-ATP exchange assay failed, which we believe is due to tight binding of the pyrophosphate in analogy to the adenylate-forming enzymes disclosed by Challis and co-workers. The hydroxamate-formation assay was also unsuccessful due to the lack of absorbance of the ferric-hydroxamate complex, likely due to the long lipid chain, which prevents formation of a spectroscopically observable tris-hydroxamate complex. However, the coupled hydroxamate–MesG assay was successful and provided the kinetic parameters of tetradecanoic acid activation catalyzed by FadD28 (Table 2). The apparent kcat values were dependent on hydroxylamine concentration and approximately 50% of the activity was due to leaking of the acyl-adenylate.
Evaluation of bisubstrate inhibitors of adenylating enzymes
One of the primary motivations in the development of the coupled hydroxamate–MesG assay was the ability to rapidly screen inhibitors. Previously, we have utilized the ATP-PPi exchange end-point assay, which is extremely robust and very sensitive.34–36 However, this low-throughput radioassay is relatively tedious involving several liquid handling steps. The assay is also limited to adenylating enzymes that undergo rapid exchange between ATP and PPi. To demonstrate the utility of the hydroxamate-MesG assay for evaluation of adenylation inhibitors, we investigated several bisubstrate inhibitors including salicyl-AMS 15,37 phenylalanyl-AMS 16,35 and tetradecanoyl-AMS 1738 against EntE, GrsA, and FadD28, respectively (Figure 6). The continuous, non-radioactive, hydroxamate-MesG assay performed extremely well and enabled rapid measurement of the apparent inhibition constants for each inhibitor–enzyme pair. Since 15–17 displayed tight-binding inhibition under the assay conditions (apparent Ki ≤ 10 × [E]), the fractional initial velocities (vi/v0) and [I] were fit to the Morrison equation (eq 2, see Experimental Section) by nonlinear regression analysis, where vi is the initial velocity with inhibitor and v0 represents the initial velocity for a DMSO control to obtain apparent Ki values for 15–17 (Table 3).
Figure 6.
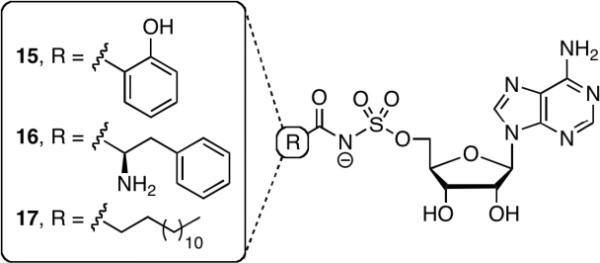
Bisubstrate Inhibitors 15-17 designed for inhibition of salicylic acid, phenylalanine, and long chain fatty acid adenylating enzymes, respectively.
Table 3.
Inhibition constants for 15–17.
| Protein | Inhibitor | Substratea | , nM |
|---|---|---|---|
| EntE | 15, Sal-AMS | SAL | 8.2 ± 1.3 |
| 15, Sal-AMS | 2,3-DHB | 207.2 ± 9.7 | |
| GrsA | 16, Phe-AMS | D-Phe | 8.0 ± 0.8 |
| FadD28 | 17, tetradecyl-AMS | C14 | 2482 ± 203 |
Substrate employed for the respective adenylating enzyme. SAL = salicylic acid, 2,3-DHB = 2,3-dihydroxybenzoic acid, D-Phe = D-phenylalanine, C14 = tetradecanoic acid
In summary, we have described the development and application of an experimentally straightforward continuous coupled hydroxamate–MesG assay to study adenylating enzymes that acylate a protein substrate. The assay was validated against a panel of previously characterized aryl acid adenylating enzymes involved in siderophore biosynthesis and shown to accurately reproduce the specificity constants (kcat/KM) as determined by the conventional ATP-PPi exchange assay. Next, we demonstrated the utility of this assay in the first steady-state kinetic analysis of Grs(AT), an adenylation-carrier protein didomain, in the overall forward direction. In this case the hydroxamate-MesG assay provided kinetic parameters commensurate with the ATP-PPi exchange assay. We highlighted the value of the hydroxamate–MesG assay to characterize adenylating enzymes that fail to undergo rapid ATP-PPi exchange in the study of FadD28, a fatty acid adenylating enzyme from M. tuberculosis. Finally, we showed the hydroxamate–MesG assay is extremely useful for the characterization of adenylation inhibitors.
Supplementary Material
Figure 7.

A) Inhibition of EntE by Sal-AMS in the presence of salicylic acid. Reactions contained 50 nM EntE, 10 mM salicylic acid, and standard assay buffer (50 mM Tris pH 8.0, 2.5 mM ATP, 5 mM MgCl2, 0.5 mM DTT, 150 mM hydroxylamine pH 7, 0.1 U nucleoside phosphorylase, 0.04 U pyrophosphatase, and 0.2 mM MesG). B) Inhibition of GrsA by D-Phenylalanyl-AMS. Reactions contained 5 nM GrsA, 1 mM D-phenylalanine, and standard assay buffer. C) Inhibition of FadD28 by Myristoyl-AMS. Reactions contained 4.25 μM FadD28, 33 μM myristic acid, and standard assay buffer.
Acknowledgements
We thank Dr. Benjamin Duckworth for carefully proofreading this manuscript. C.C.A. acknowledge membership in and support from the Region V `Great Lakes' Regional Center of Excellence in Biodefense and Emerging Infectious Diseases Consortium (NIH award 1-U54-AI-057153).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary Materials Available Figure of % activity due to leaking of acyl-adenylate for all substrate/enzyme combinations evaluated are available free of charge via the Internet at http://www.sciencedirect.com/science/journal/00032697.
References
- 1.Ibba M, Soll D. Aminoacyl-tRNA synthesis. Annu. Rev. Biochem. 2000;69:617–650. doi: 10.1146/annurev.biochem.69.1.617. [DOI] [PubMed] [Google Scholar]
- 2.Schulman BA, Harper JW. Ubiquitin-like protein activation by E1 enzymes: the apex for downstream signalling pathways. Nat. Rev. Mol. Cell Biol. 2009;10:319–331. doi: 10.1038/nrm2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shuman S. DNA ligases: progress and prospects. J. Biol. Chem. 2009;284:17365–17369. doi: 10.1074/jbc.R900017200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chapman-Smith A, Cronan JE., Jr. The enzymatic biotinylation of proteins: a post-translational modification of exceptional specificity. Trends Biochem. Sci. 1999;24:359–363. doi: 10.1016/s0968-0004(99)01438-3. [DOI] [PubMed] [Google Scholar]
- 5.Webb ME, Smith AG, Abell C. Biosynthesis of pantothenate. Nat. Prod. Rep. 2004;21:695–721. doi: 10.1039/b316419p. [DOI] [PubMed] [Google Scholar]
- 6.Chen L, Petrelli R, Felczak K, Gao G, Bonnac L, Yu JS, Bennett EM, Pankiewicz KW. Nicotinamide adenine dinucleotide based therapeutics. Curr. Med. Chem. 2008;15:650–670. doi: 10.2174/092986708783885282. [DOI] [PubMed] [Google Scholar]
- 7.Watkins PA. Fatty acid activation. Prog. Lipid Res. 1997;36:55–83. doi: 10.1016/s0163-7827(97)00004-0. [DOI] [PubMed] [Google Scholar]
- 8.Sieber SA, Marahiel MA. Molecular mechanisms underlying nonribosomal peptide synthesis: approaches to new antibiotics. Chem. Rev. 2005;105:715–738. doi: 10.1021/cr0301191. [DOI] [PubMed] [Google Scholar]
- 9.Gulick AM. Conformational Dynamics in the Acyl-CoA Synthetases, Adenylation Domains of Non-ribosomal Peptide Synthetases, and Firefly Luciferase. ACS Chem. Biol. 2009 doi: 10.1021/cb900156h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Webb MR. A continuous spectrophotometric assay for inorganic phosphate and for measuring phosphate release kinetics in biological systems. Proc. Natl. Acad. Sci. USA. 1992;89:4884–4887. doi: 10.1073/pnas.89.11.4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Upson RH, Haugland RP, Malekzadeh MN. A spectrophotometric method to measure enzymatic activity in reactions that generate inorganic pyrophosphate. Anal. Biochem. 1996;243:41–45. doi: 10.1006/abio.1996.0479. [DOI] [PubMed] [Google Scholar]
- 12.McQuade TJ, Shallop AD, Sheoran A, Delproposto JE, Tsodikov OV, Garneau-Tsodikova S. A nonradioactive high-throughput assay for screening and characterization of adenylation domains for nonribosomal peptide combinatorial biosynthesis. Anal. Biochem. 2009;386:244–250. doi: 10.1016/j.ab.2008.12.014. [DOI] [PubMed] [Google Scholar]
- 13.Pfleiderer G, Kreiling A, Wieldan T. On pantothenic acid synthetase from E. coli. I. Concentration with the aid of an optical test. Biochem. Z. 1960;333:302–307. [PubMed] [Google Scholar]
- 14.Zheng R, Blanchard JS. Steady-state and pre-steady-state kinetic analysis of Mycobacterium tuberculosis pantothenate synthetase. Biochemistry. 2001;40:12904–12912. doi: 10.1021/bi011522+. [DOI] [PubMed] [Google Scholar]
- 15.Mercer AC, Burkart MD. The ubiquitous carrier protein--a window to metabolite biosynthesis. Nat. Prod. Rep. 2007;24:750–773. doi: 10.1039/b603921a. [DOI] [PubMed] [Google Scholar]
- 16.Linne U, Marahiel MA. Reactions catalyzed by mature and recombinant nonribosomal peptide synthetases. Methods Enzymol. 2004;388:293–315. doi: 10.1016/S0076-6879(04)88024-8. [DOI] [PubMed] [Google Scholar]
- 17.Otten LG, Schaffer ML, Villiers BR, Stachelhaus T, Hollfelder F. An optimized ATP/PP(i)-exchange assay in 96-well format for screening of adenylation domains for applications in combinatorial biosynthesis. Biotechnol. J. 2007;2:232–240. doi: 10.1002/biot.200600220. [DOI] [PubMed] [Google Scholar]
- 18.Phelan VV, Du Y, McLean JA, Bachmann BO. Adenylation enzyme characterization using gamma -(18)O(4)-ATP pyrophosphate exchange. Chem. Biol. 2009;16:473–478. doi: 10.1016/j.chembiol.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kadi N, Challis GL. Chapter 17. Siderophore biosynthesis a substrate specificity assay for nonribosomal peptide synthetase-independent siderophore synthetases involving trapping of acyl-adenylate intermediates with hydroxylamine. Methods Enzymol. 2009;458:431–457. doi: 10.1016/S0076-6879(09)04817-4. [DOI] [PubMed] [Google Scholar]
- 20.Neres J, Wilson DJ, Celia L, Beck BJ, Aldrich CC. Aryl acid adenylating enzymes involved in siderophore biosynthesis: fluorescence polarization assay, ligand specificity, and discovery of non-nucleoside inhibitors via high-throughput screening. Biochemistry. 2008;47:11735–11749. doi: 10.1021/bi801625b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stachelhaus T, Marahiel MA. Modular structure of peptide synthetases revealed by dissection of the multifunctional enzyme GrsA. J. Biol. Chem. 1995;270:6163–6169. doi: 10.1074/jbc.270.11.6163. [DOI] [PubMed] [Google Scholar]
- 22.Studier FW. Protein production by auto-induction in high density shaking cultures. Protein. Expr. Purif. 2005;41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 23.Quadri LE, Weinreb PH, Lei M, Nakano MM, Zuber P, Walsh CT. Characterization of Sfp, a Bacillus subtilis phosphopantetheinyl transferase for peptidyl carrier protein domains in peptide synthetases. Biochemistry. 1998;37:1585–1595. doi: 10.1021/bi9719861. [DOI] [PubMed] [Google Scholar]
- 24.Ehmann DE, Shaw-Reid CA, Losey HC, Walsh CT. The EntF and EntE adenylation domains of Escherichia coli enterobactin synthetase: sequestration and selectivity in acyl-AMP transfers to thiolation domain cosubstrates. Proc. Natl. Acad. Sci. USA. 2000;97:2509–2514. doi: 10.1073/pnas.040572897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rusnak F, Faraci WS, Walsh CT. Subcloning, expression, and purification of the enterobactin biosynthetic enzyme 2,3-dihydroxybenzoate-AMP ligase: demonstration of enzyme-bound (2,3-dihydroxybenzoyl)adenylate product. Biochemistry. 1989;28:6827–6835. doi: 10.1021/bi00443a008. [DOI] [PubMed] [Google Scholar]
- 26.Drake EJ, Nicolai DA, Gulick AM. Structure of the EntB multidomain nonribosomal peptide synthetase and functional analysis of its interaction with the EntE adenylation domain. Chem. Biol. 2006;13:409–419. doi: 10.1016/j.chembiol.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 27.Keating TA, Marshall CG, Walsh CT. Vibriobactin biosynthesis in Vibrio cholerae: VibH is an amide synthase homologous to nonribosomal peptide synthetase condensation domains. Biochemistry. 2000;39:15513–15521. doi: 10.1021/bi001651a. [DOI] [PubMed] [Google Scholar]
- 28.Quadri LE, Sello J, Keating TA, Weinreb PH, Walsh CT. Identification of a Mycobacterium tuberculosis gene cluster encoding the biosynthetic enzymes for assembly of the virulence-conferring siderophore mycobactin. Chem. Biol. 1998;5:631–645. doi: 10.1016/s1074-5521(98)90291-5. [DOI] [PubMed] [Google Scholar]
- 29.Luo L, Walsh CT. Kinetic analysis of three activated phenylalanyl intermediates generated by the initiation module PheATE of gramicidin S synthetase. Biochemistry. 2001;40:5329–5337. doi: 10.1021/bi015518+. [DOI] [PubMed] [Google Scholar]
- 30.Camacho LR, Constant P, Raynaud C, Laneelle MA, Triccas JA, Gicquel B, Daffe M, Guilhot C. Analysis of the phthiocerol dimycocerosate locus of Mycobacterium tuberculosis. Evidence that this lipid is involved in the cell wall permeability barrier. J. Biol. Chem. 2001;276:19845–19854. doi: 10.1074/jbc.M100662200. [DOI] [PubMed] [Google Scholar]
- 31.Sirakova TD, Fitzmaurice AM, Kolattukudy P. Regulation of expression of mas and fadD28, two genes involved in production of dimycocerosyl phthiocerol, a virulence factor of Mycobacterium tuberculosis. J. Bacteriol. 2002;184:6796–6802. doi: 10.1128/JB.184.24.6796-6802.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Trivedi OA, Arora P, Vats A, Ansari MZ, Tickoo R, Sridharan V, Mohanty D, Gokhale RS. Dissecting the mechanism and assembly of a complex virulence mycobacterial lipid. Mol. Cell. 2005;17:631–643. doi: 10.1016/j.molcel.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 33.Trivedi OA, Arora P, Sridharan V, Tickoo R, Mohanty D, Gokhale RS. Enzymic activation and transfer of fatty acids as acyl-adenylates in mycobacteria. Nature. 2004;428:441–445. doi: 10.1038/nature02384. [DOI] [PubMed] [Google Scholar]
- 34.Somu RV, Wilson DJ, Bennett EM, Boshoff HI, Celia L, Beck BJ, Barry CE, 3rd, Aldrich CC. Antitubercular nucleosides that inhibit siderophore biosynthesis: SAR of the glycosyl domain. J. Med. Chem. 2006;49:7623–7635. doi: 10.1021/jm061068d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Qiao CH, Gupte A, Boshoff HI, Wilson DJ, Bennett EM, Somu RV, Barry CE, Aldrich CC. 5'-O-[(N-Acyl)sulfamoyl]adenosines as antitubercular agents that inhibit MbtA: An adenylation enzyme required for siderophore biosynthesis of the mycobactins. J. Med. Chem. 2007;50:6080–6094. doi: 10.1021/jm070905o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Neres J, Labello NP, Somu RV, Boshoff HI, Wilson DJ, Vannada J, Chen L, Barry CE, 3rd, Bennett EM, Aldrich CC. Inhibition of siderophore biosynthesis in Mycobacterium tuberculosis with nucleoside bisubstrate analogues: structure-activity relationships of the nucleobase domain of 5'-O-[N-(salicyl)sulfamoyl]adenosine. J. Med. Chem. 2008;51:5349–5370. doi: 10.1021/jm800567v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Somu RV, Boshoff H, Qiao C, Bennett EM, Barry CE, 3rd, Aldrich CC. Rationally designed nucleoside antibiotics that inhibit siderophore biosynthesis of Mycobacterium tuberculosis. J. Med. Chem. 2006;49:31–34. doi: 10.1021/jm051060o. [DOI] [PubMed] [Google Scholar]
- 38.Arora P, Goyal A, Natarajan VT, Rajakumara E, Verma P, Gupta R, Yousuf M, Trivedi OA, Mohanty D, Tyagi A, Sankaranarayanan R, Gokhale RS. Mechanistic and functional insights into fatty acid activation in Mycobacterium tuberculosis. Nat. Chem. Biol. 2009;5:166–173. doi: 10.1038/nchembio.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.