

Abstract
In order to proliferate and expand in an environment with limited nutrients, cancer cells co-opt cellular regulatory pathways that facilitate adaptation and thereby maintain tumor growth and survival potential. The endoplasmic reticulum (ER) is uniquely positioned to sense nutrient deprivation stress and subsequently engage signaling pathways that promote adaptive strategies. As such, components of the ER stress-signaling pathway represent potential anti-neoplastic targets. However, recent investigations into the role of the ER resident protein kinase PERK have paradoxically suggested both pro- and anti-tumorigenic properties. We have utilized animal models of mammary carcinoma to interrogate PERK contribution in the neoplastic process. The ablation of PERK in tumor cells resulted in impaired regeneration of intracellular antioxidants and accumulation of reactive oxygen species triggering oxidative DNA damage. Ultimately, PERK deficiency impeded progression through the cell cycle due to the activation of the DNA damage checkpoint. Our data reveal that PERK-dependent signaling is utilized during both tumor initiation and expansion to maintain redox homeostasis and thereby facilitates tumor growth.
Keywords: PERK, Nrf2, ROS, DNA damage, cell cycle checkpoints
INTRODUCTION
The initial growth of solid tumors is not well coordinated with the formation of new blood vessels necessary to support malignant expansion (Folkman et al., 1966). Thus, tumors rapidly outstrip the existing vasculature resulting in a microenvironment restricted for critical nutrients (glucose, oxygen). Limitation of glucose and oxygen is cytotoxic as production of ATP via glycolysis and reducing equivalents in the form of NADPH become hampered, while reactive oxygen species (ROS) are generated in the mitochondria (Brunelle et al., 2005; Guzy et al., 2005). Thus, nutrient-poor tumor microenvironment could have a profound impact on cancer cell energy metabolism and redox homeostasis (DeBerardinis et al., 2008; Spitz et al., 2000).
Cellular energy and redox homeostasis is critical for proper folding and modification of transmembrane and secreted proteins within the endoplasmic reticulum (ER). Perturbation of this homeostasis triggers a cellular checkpoint referred to as the Unfolded Protein Response (UPR). The capacity of the ER to respond to alterations in nutrient status makes it an effective early sensor of cellular stress associated with tumorigenesis. The UPR is mediated by three primary signal transducer molecules that span the ER membrane: PKR-like ER kinase, PERK/PEK/EIF2AK3 (Harding et al., 1999; Shi et al., 1998), inositol-requiring enzyme 1 (Ire 1α and Ire1β) (Tirasophon et al., 1998; Wang et al., 1998), and transmembrane transcription factor ATF6 (Haze et al., 1999; Li et al., 2000; Wang et al., 2000). Together, PERK, Ire1, and ATF6 alleviate ER stress by regulating expression of ER chaperones and components of ER-associated degradation system (Wu et al., 2007; Yamamoto et al., 2007), while PERK uniquely serves to reduce new protein synthesis. If stress is severe, UPR sensors trigger apoptotic program to eliminate damaged cells.
PERK is a transmembrane serine/threonine protein kinase that contains an N-terminal ER luminal domain, and a cytoplasmic C-terminal protein kinase domain (Harding et al., 1999; Shi et al., 1998). PERK phosphorylates two known substrates: the eukaryotic translation initiation factor 2 alpha (Harding et al., 1999) and the Nrf2 transcription factor (Cullinan et al., 2003). Phosphorylation of eIF2α attenuates translation initiation of most transcripts while concurrently increasing translation of select mRNAs such as the ATF4 transcription factor (Harding et al., 2000; Vattem and Wek, 2004). PERK-dependent attenuation of protein synthesis directly contributes to redox homeostasis by decreasing cellular consumption of reducing equivalents necessary for the disulfide bond formation during oxidative folding in the ER (Shimizu and Hendershot, 2009). PERK-dependent resistance to oxidative stress is further regulated by ATF4 (Harding et al., 2003). In contrast, PERK-dependent phosphorylation of Nrf2 releases it from an inhibitory E3 ligase complex containing Keap1 and cullin 3 (Cullinan et al., 2004) thereby permitting Nrf2 accumulation, nuclear import and induction of enzymes critical for the elimination of intracellular ROS (Alam et al., 1999; Hayes et al., 2000; Itoh et al., 1997; Wild et al., 1999). Thus, PERK function is critical for maintaining cellular redox homeostasis and reducing ROS-induced genotoxic stress.
Initial findings implicated PERK as anti-tumorigenic regulator as PERK deficiency inhibited the ability of Ras-transformed MEFs to grow as subcutaneous transplants (Bi et al., 2005; Blais et al., 2006). However, subsequent studies revealed that overexpression of a dominant negative PERK allele in MCF10A normal mammary epithelial cells rendered neoplastic growth characteristics (Sequeira et al., 2007). In addition, activation of Fv2E-PERK engineered to contain a drug-inducible dimerization domain reduced tumorigenic potential of squamous carcinoma T-HEp3 cells and SW620 colon carcinoma cells (Ranganathan et al., 2008). Finally, activation of PERK by overexpression of H-Ras in melanocytes was associated with a senescent phenotype (Denoyelle et al., 2006) suggesting that PERK may function as a barrier to malignant growth in certain contexts.
In this study, we interrogated the importance of PERK for tumorigenesis utilizing short hairpin RNA approach to reduce PERK levels in human breast and esophageal carcinoma cells. In addition, we generated a mammary gland-specific knockout of PERK in the mammary tumor-prone MMTV-Neu mouse strain. Our results reveal that loss of PERK renders tumor cells acutely susceptible to oxidative DNA damage thereby limiting tumor cell growth.
RESULTS
PERK is expressed in cancer cells wherein it potentiates tumor expansion
Markers of ER stress signaling, including phospho-eIF2α and GRP78 expression, are increased in a variety of tumor types (Daneshmand et al., 2007; Fernandez et al., 2000; Gazit et al., 1999; Lee et al., 2008). Because PERK mediates cell growth and survival under conditions of ER stress, we determined whether tumor-derived cells retain functional PERK. We assessed PERK expression in 4 breast and 3 esophageal human carcinoma-derived cell lines and compared it to the PERK levels in MCF10A cells, an immortalized, non-transformed breast epithelial cell line. PERK protein was readily detectable in all cell lines (Fig. 1A), and PERK function was preserved in cancer cells, as evidenced by their ability to activate PERK-dependent effectors (Fig. 1C).
Figure 1. PERK expression is maintained in cancer cells wherein it regulates tumor expansion in vivo.

(A) PERK protein levels were measured by immunoprecipitation (IP) followed by Western blot analysis in the following cell lines: MCF10A (1), MCF7 (2), T47D (3), MDA-MB231 (4), MDA-MB468 (5), TE3 (6), TE7 (7), KYSE 520 (8). (B) PERK protein levels following shRNA targeting of PERK. (C) Parental MDA-MB468 cell line, shPERK-transduced cells (shPERK), and shPERK-transduced cells reconstituted with mouse Myc-PERK (+mPERK) were treated with 2μg/ml tunicamycin for the indicated intervals. Western analysis for ATF4, CHOP, or β-actin. (D) Volume of orthotopic tumors formed from the mouse mammary tumor-derived cells transduced in vitro with empty vector virus (Neu/PERKloxP/loxP) or Cre-expressing retrovirus (Neu/PERKΔ/Δ) 28 days post-transplant (n=4). Representative image of tumors are provided. All p-values determined by Student t-test. (E) Western analysis of transgenic ErbB2 and PERK expression following infection of mouse mammary tumor-derived cells with control (Neu/PERKloxP/loxP) or Cre-expressing retrovirus (Neu/PERKΔ/Δ). Thapsigargin treatment (50nM, 1h) was used to demonstrate that PERK is functional.
To assess the role of PERK in human tumor cell growth and survival, we utilized lentivirus-delivered short hairpin RNAs (shRNA) to reduce endogenous levels of PERK (Fig. 1B). As expected, PERK knockdown resulted in the inhibition of downstream effectors, CHOP/ATF4, following challenge with tunicamycin (Fig. 1C).
To determine whether PERK deficiency affects the ability of mammary carcinoma cells to form solid tumors in vivo, we utilized tumor-prone MMTV-Neu transgenic mice bearing PERKloxP/loxP allele (MMTV-Neu/PERKloxP/loxP). Primary tumors from MMTV-Neu/PERKloxP/loxP mice were isolated and transduced with empty vector retrovirus or retrovirus encoding Cre recombinase to excise perk (Fig. 1E). The primary tumor cells were then transplanted into mammary fat pads of 3-week old SCID mice. During a 28-day interval, PERK-deficient tumor cells generated tumors with a significantly reduced volume relative to PERK positive cells (Fig. 1D). Similar reduction in tumor volume was observed upon PERK knockdown in human MDA-MB468 cells (Fig. S1). These data collectively demonstrate a role for PERK as a critical regulator of mammary tumor expansion.
Loss of PERK in human cancer cells delays cell cycle progression through the G2/M phase
Gain and loss of PERK function can influence cell cycle progression of certain cells (Wei et al., 2008; Zhang et al., 2006b). Accordingly, subsequent to acute PERK knockdown in MDA-MB468 and T47D cells, we noted a 50% reduction in BrdU-incorporating S-phase cells with a concomitant increase in G2/M phase cells and a small increase in cell death (Fig. 2A and S2A). These data demonstrate that knockdown of PERK triggers a cell cycle delay at the G2/M boundary, resulting in a significant decrease in cancer cell proliferation in vitro.
Figure 2. PERK knockdown triggers a G2/M delay.

(A) MDA-MB468 cells were infected with control shRNA or anti-PERK shRNA for the indicated intervals. Cells were pulsed with BrdU 45 min prior to harvest for FACS analysis. (B) Kinetics of growth of the MDA-MB468 parental cell line, control shRNA-(shControl) or shPERK-transduced cells (shPERK), and shPERK-transduced cells reconstituted with mouse Myc-PERK (+mPERK). PERK protein levels following expression of shRNA targeting human PERK and reconstitution with mouse Myc-PERK are shown. (C) Proliferation rates in mammary gland sections from control (PERKloxP/loxP) and mammary gland-specific PERK knockout mice (PERKΔ/Δ) on pregnancy day 16 (P16) and lactation day 3 (L3) were determined by immunohistochemistry for BrdU (animals were injected with BrdU 1 h prior to being euthanized). (D) Quantification of BrdU-positive cells from (C) is shown; error bars indicate S.D. among 3 animals, 5 acini were counted per animal.
As independent assessment, we measured growth rates of parental versus stable PERK knockdown cells utilizing three different human carcinoma cell lines derived from two distinct cancer types. Strikingly, PERK knockdown resulted in a significant reduction in the growth rate of human breast carcinoma MDA-MB468 (Fig. 2B) and T47D (Fig. S2B) cells, as well as esophageal carcinoma TE3 (Fig. S2C) cells compared to control cell lines. To ensure that the reduced growth kinetics specifically reflected reduced levels of PERK, we restored PERK function by transducing cells with retrovirus encoding myc-tagged murine PERK (Fig. 2B). Murine PERK rescued tunicamycin-dependent induction of ATF4 and CHOP (Fig. 1C) and resulted in a significant rescue of attenuated cell growth (Fig. 2B). To determine whether attenuated proliferation of tumor cells following loss of PERK function is restricted to tumors, we determined whether excision of perk in normal mammary epithelium inhibits proliferation. Critically, perk excision in mammary epithelial cells did not influence their proliferative capacity (Fig. 2C–D).
Because a G2/M cell cycle delay/arrest is frequently associated with the activation of a double strand DNA break (DSB) checkpoint, we next tested for the evidence of DNA damage response pathway activation. Indeed, acute PERK knockdown coincided with accumulation of phospho-ATM and phospho-Chk2 positive foci in MDA-MB468 (Fig. 3A–B) and T47D cells (Fig. S3A–B). Coordinately, we noted increased phospho-Chk2 and pTyr-15 on CDK2 (Fig. 3C) as well as reduced CDK2 kinase activity, which could be restored by introduction of murine PERK (Fig. 3D). In addition, we noted a significant inhibition of CDK2 activity in a lysate prepared from tumor wherein PERK was excised (Fig. S3C). These data demonstrate that loss of PERK delays progression through the G2/M transition due to the activation of DNA damage checkpoint.
Figure 3. PERK knockdown triggers DNA damage response signaling pathway.

(A) Immunofluorescence staining for DNA damage-induced foci containing phospho-ATM and phospho-Chk2 following acute PERK knockdown (72 h after infection) in MDA-MB468 cells. (B) Quantification of phospho-ATM positive cells (>3 foci) is shown; error bars indicate S.D. from 3 slides, 5 fields were counted per slide. p-value was determined by Student t-test. (C) Western analysis of DNA damage response-associated markers following PERK knockdown. (D) IP/kinase assays assessing CDK2-dependent phosphorylation of histone H1 (bottom panel). CDK2 complexes were immunoprecipitated from MDA-MB 468 cells treated as indicated. PERK levels were assessed by IP/immunoblot and CDK2 recovery in precipitates was assessed by CDK2 immunoblot (middle panel).
Reactive Oxygen Species (ROS) accumulate in PERK deficient cells
Previous work revealed a role for PERK in the regulation of cellular redox homeostasis via direct phosphorylation of Nrf2 (Cullinan and Diehl, 2004; Cullinan et al., 2003) and translational regulation of ATF4 (Harding et al., 2003). We thus determined whether PERK loss contributed to increased cellular ROS in human breast carcinoma cells. Indeed, PERK knockdown led to significantly increased levels of ROS (Fig. 4A–B). Furthermore, growth curve analysis in the presence of ROS scavenger, N-acetyl cysteine (NAC), revealed that ROS accumulation contributed to reduced cell growth in PERK knockdown cells (Fig. 4C).
Figure 4. Increased levels of Reactive Oxygen Species (ROS) in PERK knockdown cells contribute to reduced kinetics of cell growth.
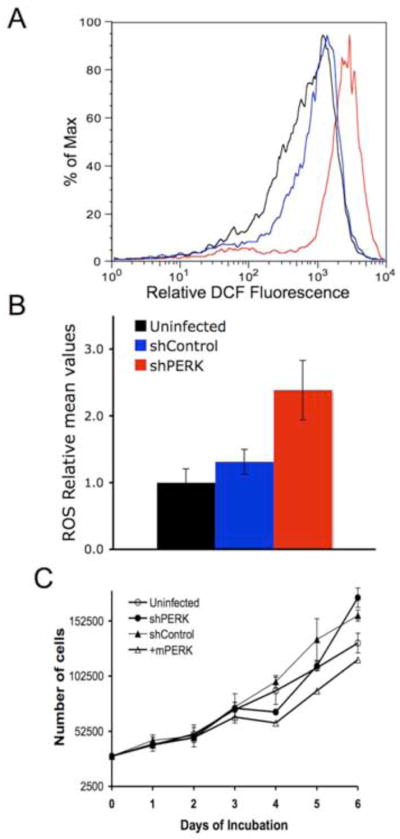
(A) DCF fluorescence measured by FACS using CM-H2DCFDA dye. (B) Mean values of ROS determined from three independent experiments. Uninfected (black), stably infected with empty vector (blue) or shPERK (red) in both (A) and (B). (C) Growth analysis of MDA-MB468 parental cell line, cells transduced with control shRNA, shPERK, or shPERK and reconstituted with mouse Myc-PERK in the presence of ROS scavenger N-acetylcysteine (NAC).
ROS accumulation triggers oxidative DNA damage
To determine whether PERK loss and subsequent accumulation of ROS triggers oxidative DNA damage, we quantified 8-oxoguanine adducts, an oxidation product of guanine. PERK knockdown resulted in a significant increase in 8-oxoguanine adducts (Fig. 5A–B). We also noted a significant increase in 8-oxoguanine adducts in both human and mouse PERK-deficient tumors (Fig. 5C–F) consistent with the idea that ROS-mediated cellular damage could also contribute to reduced growth of PERK deficient tumors in vivo.
Figure 5. ROS accumulation triggers oxidative DNA lesions in PERK-deficient breast cancer cells and tumors.

(A) Detection of 8-oxoguanine oxidized DNA adduct (8-OxoG) using a FITC conjugated 8-OxoG binding peptide in parental MDA-MB468 cells, MDA-MB468 cells transduced with control shRNA, shPERK, or shPERK and reconstituted with mouse Myc-PERK. (B) Quantification of 8-oxoG positive cells from 3 independent experiments. (C) 8-OxoG in paraffin sections from tumors formed by parental MDA-MB468 cells, MDA-MB468 cells transduced with control shRNA, or shPERK. (D) Quantification of 8-OxoG positive cells shown in (C) is provided and error bars indicate S.D. from 4 animals. (E) Detection of 8-OxoG in paraffin sections from orthotopic tumors formed by mouse mammary tumor-derived cells transduced with empty vector (Neu/PERKloxP/loxP) or retrovirus expressing Cre recombinase (Neu/PERKΔ/Δ). (F) Quantification of 8-OxoG positive cells from (E); error bars indicate S.D. from 4 animals. All p-values determined by Student t-test.
Following DNA damage, H2AX, a histone H2 variant, is phosphorylated around regions of double strand breaks (DSBs) by the PI3K family members ATM and ATR, and it accumulates in DNA repair foci wherein it contributes to the recruitment of DNA repair proteins (Bassing and Alt, 2004). We thus measured the accumulation of phosphorylated H2AX, γ-H2AX, as a surrogate marker for DSBs formation. A 2-fold increase in γ-H2AX was noted in PERK knockdown cells relative to parental cells and/or cells infected with an empty vector consistent with increased DNA breaks (Fig. 6A–B). As γ-H2AX is also thought to accumulate under conditions not associated with DSBs, we assessed the accumulation of damaged DNA in single cells through the use of a COMET assay. Indeed, PERK knockdown triggered accumulation of cells with pronounced tail moment (Fig. 6C). Critically, we also noted increased levels of γ-H2AX-positive cells in PERK deficient tumors in vivo (Fig. S4A–B).
Figure 6. ROS accumulation triggers DNA double strand breaks in PERK-deficient breast cancer cells and tumors.

(A) Immunofluorescent staining of γ-H2AX foci in control or PERK knockdown MDA-MB468 cells. (B) Quantification of γ-H2AX positive (>5 foci) cells under standard tissue culture conditions. Error bars represent S.D. from 3 independent experiments performed in triplicate. (C) Quantification of the COMET tail moment in control or PERK knockdown cells under standard tissue culture conditions. Error bars indicate S.D. from 3 experiments.
While activation of a DSB checkpoint typically results in a transient arrest and cell cycle restart following repair, it is also associated with cellular senescence when triggered by oncogene induction. However, increased accumulation of p19Arf and tri-methylated H3K9 was not observed in PERK deficient tumors suggesting that loss of PERK does not induce a senescent phenotype (Fig. S4C).
Reduced activity of Nrf2 leads to increased oxidative stress in PERK knockdown cells
Nrf2, a direct PERK substrate (Cullinan et al., 2003), contributes to the transcriptional regulation of genes whose protein products mediate cellular redox homeostasis (Buetler et al., 1995; Hayes et al., 2000). Consistent with impaired Nrf2 activation in PERK knockdown cells, expression of two distinct Nrf2 target genes, NQO1 (Itoh et al., 1997) and GCLC (Wild et al., 1999) was decreased compared to the uninfected or control cells (Fig. 7A).
Figure 7. Reduced activity of Nrf2 causes increased oxidative stress in PERK knockdown cells.

(A) Quantitative real time PCR analysis of Nrf2 target genes NQO1 and GCLC in the indicated cell lines asynchronously proliferating under standard conditions. (B) Purified recombinant Nrf2-Neh2 domain of WT, T80A, S40A or T80A/S40A, was incubated with purified recombinant ΔN-PERK in the in vitro kinase assay. Phosphorylated Nrf2-Neh2 was detected by autoradiography (upper panel). (C) 293T cells were transfected with WT Nrf2 or Nrf2-T80A. 24 hours after transfection, cells were left untreated (C) or treated with tunicamycin (Tu) for 2 hours followed by immunoprecipitation with anti-Nrf2 antibody. Threonine phosphorylation was detected using a phospho-Thr reactive antibody. Nrf2 in the IP and the whole cell lysate (WCL) was detected with Nrf2 specific antibody. (D) Proliferation of the indicated cell lines was assessed by a 6-day growth curve under standard tissue culture conditions as described in materials and methods. PERK levels were detected by IP/Western blot analysis. (E) Oxidized guanine in damaged DNA was detected by a FITC-conjugated 8-OxoG binding peptide in PERK knockdown cells infected with pBabe control vector (shPERK) or Nrf2-HA (shPERK Nrf2-HA). Quantification of 8-OxoG positive cells is provided. Error bars represent S.D. from 3 experiments. (F) 8-OxoG was detected in PERK knockdown cells transfected with scramble siRNA (Scrm), or keap1 siRNA (sikeap1). Error bars in graphs represent S.D. from 3 experiments. Western blot panels demonstrate levels of Nrf2-HA and Keap1 in PERK knockdown cells.
To address the role of Nrf2 downstream of PERK, we sought to define the site of PERK phosphorylation in Nrf2. Because PERK-dependent phosphorylation disrupts Nrf2-Keap1 binding, we focused on the Neh2 domain of Nrf2 that binds directly to Keap1 (Lo et al., 2006). Indeed, PKC can phosphorylate serine 40 in this domain (Huang et al., 2002). Purified recombinant PERK phosphorylated wild type Nrf2-Neh2 and a serine 40 to alanine mutant; however, mutation of threonine 80 to alanine abrogated phosphorylation (Fig. 7B). We confirmed stress-dependent phosphorylation of this residue in Nrf2 using a phospho-threonine reactive antibody (Fig. 7C). The threonine 80 is essential for Keap1-Nrf2 binding (Lo et al., 2006), providing a biochemical basis for phosphorylation-dependent disruption of the Keap1-Nrf2 interaction.
We subsequently wished to determine whether cellular phenotypes resulting from loss of PERK could be rescued through enforced Nrf2 function. Nrf2 activity is restricted via its association with an E3 ligase wherein Keap1 functions as Nrf2-specific adaptor thereby targeting Nrf2 to cullin 3 (Cullinan et al., 2004; Furukawa et al., 2003; Furukawa and Xiong, 2005; Kobayashi et al., 2004; Zhang et al., 2004). We previously demonstrated that basal levels of active Nrf2 can be elevated via either overexpression of Nrf2 or knockdown of Keap1 (Cullinan and Diehl, 2004). Accordingly, expression of HA-Nrf2 restored normal growth to MDA-MB468 cells wherein PERK was ablated (Fig. 7D). Consistent with its role in PERK-dependent regulation of redox homeostasis, introduction of HA-Nrf2 significantly attenuated oxidative DNA damage (Fig. 7E). To independently confirm that increased Nrf2 activity can compensate for loss of PERK function and reduce oxidative DNA damage, we knocked down Keap1 in cells wherein PERK was stably reduced. This resulted in a significant reduction in oxidative DNA damage (Fig. 7F).
Dual role for PERK in tumorigenesis in vivo
Using mouse models, we addressed two additional issues. First, we determined whether deletion of PERK attenuated MMTV-Neu initiated tumorigenesis. MMTV-Neu transgenic mice were crossed with PERKloxP/loxP/MMTV-Cre mice (Bobrovnikova-Marjon et al., 2008) generating MMTV-Neu/PERKΔ/Δ. Mice that did not inherit the MMTV-Cre transgene were used as a control (MMTV-Neu/PERKloxP/loxP). Analysis of tumor-free survival revealed that PERK loss delayed MMTV-Neu-induced tumor formation (Fig. 8A). Histological signature of tumors was characteristic of the MMTV-Neu mouse model (Fig. 8B). PERK excision in mammary epithelium was assessed by immunoblot (Fig. 8C; Bobrovnikova-Marjon et al., 2008) and RT-PCR (Fig. S5).
Figure 8. PERK loss attenuates MMTV-Neu-driven mammary tumorigenesis in mice, but promotes spontaneous mammary tumor formation in aged mammary gland-specific PERK knockout mice.

(A) Kaplan-Meier analysis of tumor-free survival for cohorts of MMTV-Neu/PERKloxP/loxP (n=21) and MMTV-Neu/PERKΔ/Δ Δ/Δ (n=27) mice. (B) Hematoxylin and eosin staining demonstrating histology of control (Neu/PERKloxP/loxP) and PERK knockout (Neu PERKΔ/Δ) mammary gland tumors. (C) Western analysis for PERK, ErbB2, and eIF4E levels on whole protein extracts from control (Neu/PERKloxP/loxP) and PERK knockout (Neu/PERKΔ/Δ) mammary gland tumors or mammary gland from lactating PERKloxP/loxP dam (L10). (D) Nrf2 was precipitated from tumor lysates prepared from MMTV-Neu/PERKΔ/Δ or control mice and blotted for phospho-Thr and Nrf2. PERK expression was determined by immunoblot. (E) PERK excision delays development of Neu-driven hyperplastic lesions. Representative mammary glands from 9- to 14-months old control (Neu/PERKloxP/loxP) and PERK knockout (Neu/PERKΔ/Δ) mice revealing pre-malignant lesions are shown. (F) Hematoxylin and eosin staining on lungs from control (Neu/PERKloxP/loxP, n=24) and PERK knockout (Neu/PERKΔ/Δ, n=27) mice revealing metastatic lesions. (G) Troma-1 (cytokeratin-8) staining on lung specimens containing metastatic foci. LT=lung tissue; Met=metastasis; OL=overlay. (H) Hematoxylin and eosin staining for tumor histology and whole mount of hyperplastic lesions in mammary glands of PERKΔ/Δ aged females. (I) qRT-PCR for ErbB2 on genomic DNA from PERKΔ/Δ tumors and FISH analysis on paraffin sections from the same animals. Levels of ErbB2 in tumors were compared to matched spleen tissues.
Tumor formation was not due to outgrowth of cells exhibiting inefficient PERK excision as only two tumors retained detectable PERK protein (Fig. 8C). To determine whether Thr-80 phosphorylation of Nrf2 was dependent upon PERK in vivo, we immunoprecipitated Nrf2 from tumor lysates and assessed p-Thr levels. Reduced levels of p-Thr were observed in immunocomplexes from all 4 MMTV-Neu/PERKΔ/Δ tumor lysates analyzed (Fig. 8D), suggesting that phosphorylation of Nrf2 in the tumor environment is a non-redundant function of PERK.
While use of the MMTV promoter to drive both PERK excision and oncogene expression permits targeting of the same cell population, it does not allow us to control the timing of perk excision with tumor onset. Our previous work revealed that perk is efficiently excised in the mammary gland of virgin mice by 4 months of age (Bobrovnikova-Marjon et al., 2008). Because this is substantially prior to MMTV-Neu-induced tumor onset, we presumed that perk excision occurs prior to tumor initiation. We inferred from this that loss of PERK delays tumor onset. To further address this possibility, we collected mammary glands from 9 through 14-months old MMTV-Neu/PERKloxP/loxP and MMTV-Neu/PERKΔ/Δ mice to assess the onset of pre-malignant lesions. No pre-malignant lesions were identified in 9-months old MMTV-Neu/PERKΔ/Δ mice (n=4), and 1 out of 4 mice exhibited a pre-neoplastic lesion in 12–14 months-old group (Fig. 8E). In contrast, hyperplastic lesions were apparent in MMTV-Neu/PERKloxP/loxP mice at 9 and 14 months (Fig. 8E). Although tumor histology did not suggest that loss of PERK resulted in a less aggressive tumor, we also noted a 2-fold reduction in the incidence of lung metastases in PERK knockout mice (Fig. 8F). Mammary origin of the metastases was consistent with positive cytokeratin-8 staining (Fig. 8G). Collectively, our data demonstrate two critical points. First, acute PERK loss of function compromises tumor growth and expansion. Second, deletion of PERK delays Neu-dependent tumor onset and significantly reduces lung metastases.
DNA damage and activation of a DNA damage checkpoint may serve as a tumor barrier, while long-term genotoxic stress accompanied by mutational inactivation of DNA damage response mechanisms is pro-tumorigenic (Bartkova J, 2005; Gorgoulis VG, 2005; Stracker et al., 2008). Thus, we considered whether loss of PERK might contribute to increased spontaneous mammary tumorigenesis. We utilized MMTV-Cre/PERKloxP/loxP mice, which do not exhibit detectable proliferative defects during postnatal mammary gland development (Bobrovnikova-Marjon et al., 2008), and aged these animals for up to 24 months. During this interval, 6 of 29 animals developed overt mammary adenocarcinoma; in addition, pre-malignant adenomas were observed in several aged mice analyzed (Fig. 8H), while only 2 of 19 control PERKloxP/loxP mice developed carcinomas over this same interval. We noted amplification of the ErbB2 allele in 2 of 6 MMTV-Cre/PERKloxP/loxP animals by FISH and confirmed the amplification in one of these tumors by qRT-PCR (Fig. 8I). These findings demonstrate that long-term PERK inactivation could increase susceptibility to spontaneous tumor formation due to increased genomic instability.
DISCUSSION
PERK signaling facilitates cellular adaptation to reduced glucose and oxygen availability, as well as challenges imposed by chemicals that inhibit protein folding. Because cancer cells acquire the ability to survive and proliferate in an environment with restricted glucose and oxygen availability, we investigated whether PERK signaling provides tumor-derived cells with a growth or survival advantage. Strikingly, our data reveal that loss of PERK triggers a significant attenuation of tumor cell proliferation that results from increased oxidative DNA damage, which subsequently leads to the G2/M cell cycle checkpoint activation. Reduced proliferation correlates with compromised signaling via Nrf2 (Cullinan and Diehl, 2004), a PERK responsive transcription factor. Collectively, our data suggest that PERK/Nrf2 signaling protects cancer cells from oxidative stress triggered by growth within a harsh tumor microenvironment, thereby promoting tumorigenesis. The data also opens the possibility that PERK activity could be protective from chemotherapeutics that function in part through the generation of reactive oxygen species. Thus, PERK inhibitors could be potentially used as a chemo- and radiosensitization treatment.
PERK substrates include eIF2α (Harding et al., 2000; Shi et al., 1998) and the Nrf2 transcription factor (Cullinan et al., 2003). Inhibition of PERK is associated with increased oxidative stress, particularly in response to the agents that perturb the ER function. Previous work suggests that PERK regulates the cellular redox homeostasis via both eIF2α–dependent translational upregulation of ATF4 (Harding et al., 2003) and direct phosphorylation of Nrf2 by PERK (Cullinan and Diehl, 2004). Nrf2 then regulates expression of enzymes necessary for the production of major cellular antioxidant, glutathione (Shenvi et al., 2009; Wild et al., 1999; Wild and Mulcahy, 2000). That Nrf2 can contribute to tumor growth and survival stems from recent work demonstrating that tumorigenesis is associated with constitutive activation of the Nrf2 axis via inactivating mutations in Keap1 in breast (Nioi and Nguyen, 2007) and lung cancer (Ohta et al., 2008), as well as mutations in Nrf2 leading to its aberrant accumulation (Shibata et al., 2008). Indeed, recent work revealed that deletion of Nrf2 in alveolar epithelial cells results in accumulation of oxidized 8-oxoguanine DNA adducts triggering DNA damage response involving ATM, Chk1 and Chk2, and leading to G2/M cell cycle checkpoint activation (Reddy et al., 2008). Furthermore, RNAi-mediated inactivation of Nrf2 reduced tumorigenic potential of lung cancer cells (Singh et al., 2008). However, this work contrasts with studies utilizing the TRAMP mouse model of prostate cancer, which revealed that loss of Nrf2 is associated with increased cellular transformation due to elevated ROS levels and oxidative DNA damage (Frohlich et al., 2008).
Our work supports a model wherein targeted deletion of PERK in human cancer cells results in inactivation of Nrf2 axis leading to significant attenuation of tumor growth due to accumulation of oxidized DNA adducts and DNA damage checkpoint activation. However, the potential contribution of impaired translational control via PERK-dependent regulation of eIF2α can not be excluded (Back et al., 2009). In our analysis, we did not always detect reduced levels of phospho-eIF2α in PERK-deficient tumors (Fig. 8D), suggesting compensatory regulation. Because phosphorylation of Nrf2 was impaired (Fig. 8D), we suggest that loss of Nrf2 activation in PERK deficient cells largely contributes to the observed phenotype.
Recent investigation of PERK function during tumorigenesis has been paradoxical. Initial findings suggested that PERK potentiates the ability of transformed MEFs to grow as subcutaneous transplants (Bi et al., 2005; Blais et al., 2006). In contrast, expression of a dominant negative PERK allele in immortalized MCF10A normal mammary epithelial cells contributed to neoplastic growth that was associated with increased levels of ErbB2 expression (Sequeira et al., 2007). In view of this paradox, we sought to address the role of endogenous PERK in the regulation of tumor progression in a more physiological setting. We utilized a short hairpin RNA approach to reduce PERK levels in human cancer cells and Cre-loxP system to excise perk in murine mammary epithelium and mammary tumors. Both approaches revealed that PERK deficiency significantly compromises growth of established tumors in vivo. We found that loss of PERK significantly impaired cellular ROS buffering mechanisms resulting in oxidative DNA damage and subsequent engagement of the DNA damage response (DDR) checkpoint.
Activation of the DDR checkpoint was associated with decreased growth, cell cycle arrest and apoptosis and thus has been correlated with tumor suppression (Bartkova J, 2005; Gorgoulis VG, 2005; Stracker et al., 2008). Ultimately, unabated DNA damage can override the DNA damage checkpoint, resulting in fortuitous mutations that promote neoplastic growth. Our findings provide evidence for both. Transformation due to oncogene activation (Behrend et al., 2003) is generally associated with increased levels of protein synthesis as well as depletion of cellular antioxidant defenses. Importantly, this same mechanism would affect growing tumors that outstrip their blood vessel supply and experience hypoxia resulting in hypoxia-induced ROS accumulation (Clanton, 2007; Guzy and Schumacker, 2006; Liu et al., 2008). Indeed, we observed that PERK loss affects both tumor onset and expansion. These findings suggest that tumor cells co-opt PERK signaling pathway to promote their growth and survival in a cytotoxic tumor microenvironment. In normal tissue, loss of PERK function does not have a dramatic consequence. This may reflect conditions of normal homeostatic proliferation, which does not induce generation of significant mitochondrial ROS. However, over the lifetime of an organism, PERK inactivation does contribute to genomic instability thereby increasing tumor susceptibility as noted by the increased frequency of spontaneous mammary carcinomas occurring in aged PERK null mammary glands.
If loss of PERK triggers a DNA damage checkpoint, how then do tumor cells escape permitting restoration of cell proliferation in vitro and in vivo, albeit at a reduce rate? Checkpoint activation can be averted via mutagenesis and/or inactivation of the DNA damage signaling pathway components, such as Chk2, levels of which can be reduced in cancer (Wu et al., 2001), or CDC25 family of phosphatases that are upregulated by tumor cells (Evans, 2000; Wu et al., 1998). Analysis of PERK knockdown cells or Cre-excised PERK lines that had been cultured in vitro for extended periods revealed a down-regulation of phospho-ATM and phospho-Chk2 levels and reduced pTyr15 on CDK2 (data not shown). Finally, analysis of DNA damage signaling components in MMTV-Neu/PERKΔ/Δ and control MMTV-Neu/PERKloxP/loxP mice revealed that pTyr15 CDK2 levels and CDC25A levels were generally higher in the PERK-null background (Fig. S6). Collectively, this reveals that DNA damage checkpoint mechanisms can be averted over time due to the gradual increase in CDC25A protein levels (Fig. S6) or through down-regulation of ATM/Chk2 signaling. Of note, the difference in CDC25A phosphatase protein levels between control and PERK-deficient Neu-transgenic mice tumors was particularly striking in the late latency tumors, suggesting that this event might be necessary for tumor formation in a PERK-null background (Fig. S6).
In summary, PERK regulates cancer cell redox homeostasis within the tumor microenvironment via buffering ROS accumulation and thereby preventing activation of oxidative DNA damage checkpoint. Loss of PERK in cancer cells suppresses tumor onset and expansion, while it increases genomic instability in normal cells and may result in tumor development. In the context of therapeutic intervention, simple inhibition of PERK may provide some short-term benefit. However, when combined with agents that increase DNA damage burden, short-term inhibition of PERK may result in a highly effective novel therapeutic approach.
Methods
Animals and tissue
Experiments were conducted in accordance with the Animal Welfare Act and the Department of Health and Human Services Guide. Mammary gland-specific perk knockout animals (Bobrovnikova-Marjon et al., 2008) were mated to mice bearing the Neu transgene under the control of MMTV-LTR promoter (Jackson laboratories; Guy et al., 1992). The Neu and Cre transgene bearing offspring were bred to homozygocity for the LoxP allele of perk thus generating mammary gland-specific perk ‘null’. Littermates bearing Neu but not the Cre transgene were used as controls. The No. 4 inguinal gland was extracted and processed for whole-mount analysis as previously described (Lin et al., 2008).
Cell culture and Transfection
MDA-MB468 and T47D cells were maintained in DMEM (GIBCO) supplemented with 10% fetal bovine serum (FBS), 0.01mg/ml insulin and antibiotics. TE3, TE7, and KYSE-520 were grown in RPMI1640 supplemented with 2mM L-glutamine, 10% FBS, and antibiotics. Transfections were performed using Lipofectamin Plus (Invitrogen) according to manufacturer’s instructions. Tumor-derived mammary epithelial cell lines were derived and cultured as described (Lin et al., 2008).
Immunoprecipitation and immunobloting
Cells were lysed in EBC buffer (50mM Tris pH 8.0; 120mM NaCl; 0.5% NP-40) supplemented with protease and phosphatase inhibitors. Antibodies used for immunobloting analysis, immunofluorescence and IHC included PERK (Rockland Immunochemicals); human ATF4, histone H3 (tri methyl K9), phospho-Chk2 (Thr68) (Abcam); human CHOP (Affinity Bioreagents), β-actin (Sigma, AC-15), Nrf2, Keap 1, CDK2, and p19ARF (Santa Cruz Biotechnology); γ-H2AX (Ser139), phospho-eIF2α, eIF4E, Cdc25A, phospho-Tyr15 CDK2, phospho-Thr160 CDK2, phospho-Thr (Cell signaling); troma-1 (Developmental Studies Hybridoma Bank, University of Iowa), ErbB2 (Calbiochem), Chk2 (BD Pharmingen), eIF2α (BioSource), phospho-ATM (Millipore).
Lentivirus, Retrovirus shRNA/siRNA
293T cells were transfected with PMDL, VSVG, REV and pLKO.1 containing shRNA against PERK (IDTRCN0000001401, Open Biosystem) or pLKO.1 empty vector as control using Lipofectamine Plus (Invitrogene) for stable knockdown or FuGene (Roche) for acute knockdown experiment. Viral supernatants were harvested 48h after transfection and concentrated using SW-28 rotor for stable knockdown infections. Concentrated virus was used to infect human cell lines in the presence of 10μg/ml polybrene. Selection to create stably knocked down cell lines was conducted with puromycin at 5μg/ml. Retroviruses were produced as previously described (Brewer and Diehl, 2000). MDA-MB468 PERK knockdown cells were transfected with siRNA by using HiPerfect (Qiagen). Scrambled (Scrm) and keap1-specific siRNA Smartpool were from Dharmacon. Experiments were conducted 72 h after transfection.
Growth curves
3×104 cells were plated in 6 cm dish. Cells were counted every 24h for 5 days using hemocytometer. ROS scavenger N-acetylcysteine (NAC) was used at 5 mM where indicated. Culture media was changed every 3 days. Each experiment was done in triplicate.
Quantitative and semiquantitative RT-PCR
RNA was prepared from cultured cells or frozen tissues using TRIzol (Invitrogen), followed by isopropanol precipitation. Genomic DNA (gDNA) was isolated using Qiagen DNeasy kit. Quantitative RT-PCR reactions were performed using SYBR Green (SuperArray). All primer sequences are available upon request. Semiquantitative RT-PCR for PERK excision efficiency was performed as described (Zhang et al., 2006a).
Immunofluorescence
Cells were permeabilized with ice cold MeOH:acetone (1:1) for 10min at −20°C, allowed to air dry, and rehydrated for 10min with PBS. Blocking was performed with 10% FBS/PBS for 40 minutes at room temperature. Primary and secondary antibodies were diluted in 10% FBS/PBS and incubated for 2h or 30 minutes at RT, respectively.
FISH
Fluorescent in situ hybridization for ErbB2 was performed on paraffin sections following treatment with proteinase K. Biotin-labeled probe was generated by random priming method with ErbB2 full-length cDNA (ID 5356166, Open Biosystems) and visualized with streptavidin-Texas Red.
ROS measurement
Cells were incubated with 3ml PBS (with Calcium and Magnesiun) containing 5mM 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate, acetyl ester (CM-H2DCFDA, Invitrogen) for 30min in the dark at 37°C. Cells were washed with PBS, trypsinized, washed, resuspended in PBS and analyzed by FACS.
8-oxyguanine staining
4×105 cells were plated on glass coverslips. 8-oxyguanine was detected using OxyDNA test (Biotrin). Briefly, cells were permeabilized with MeOH:Acetone (1:1), washed with wash solution, and incubated with protein-FITC for 1h. For tumor sections, antigen retrieval was performed by heating in 50mM Tris pH 9.5 for 12min. Slides were immersed in ice cold MeOH for 10min at −20°C. Sections were blocked with 10% FBS in PBS and incubated overnight at 4°C with FITC-conjugate in wash solution.
Comet assay
DNA fragmentation was tested by alkaline electrophoresis comet assay (Trevigen) according to manufacturer’s instructions. Data were analyzed using Comet assay IV software (Perceptive Instruments).
Orthotopic injections
5×106 cells were mixed with 30μl matrigel:media (1:1) and were injected into the mammary fat pad of 3 months old female SCID mice (Charles River). Animals were sacrificed after 37 (human cells) or 28 (mouse cells) days, tumor size was measured using a caliper, and tumor volume was calculated using the formula: Volume (cc) = p×(lenght)×(width)2/6 (Bruns et al., 2004).
Immunohistochemistry
Antigen retrieval was performed in 10mM citrate buffer, pH 6.0 (Biogenex). Endogenous peroxidase activity was blocked with 3% peroxide in MeOH. Sections were blocked with 1X Power Block Reagent (Biogenex) followed by incubation with primary antibody. Detection was performed with biotinylated secondary antibodies and ABC-HRP reagent followed by DAB substrate (Vector laboratories).
In vitro kinase assay
For the detection of CDK2 kinase activity, cells or tissues were solubilized in EBC buffer. Complexes were isolated by precipitation with a CDK2 reactive antibody from 200μg total protein. The kinase assay was performed using recombinant histone H1 with 10μCi of γ-32P-ATP for 10 min at 30°C. Reactions were resolved by SDS-PAGE, transferred to PVDF membrane, and visualized by autoradiography. Total Histone H1 was visualized by ponceau stain.
Cell cycle analysis
Cells were pulsed with 10μM BrdU 45 min prior to being harvested. Cells were washed with PBS, fixed with ethanol, and stained with anti-BrdU (BD Pharmingen) and FITC-conjugated secondary antibody (BD Pharmingen) and then with propidium iodide (10 μg/ml) for 30 min prior to FACS analysis. Cell cycle profiles based on DNA content and BrdU incorporation were assessed using FlowJo software, and the sub-G1 population of cells served as a readout for apoptotic cells.
Supplementary Material
Acknowledgments
The authors wish to thank Serge Fuchs for critical reading of the manuscript, Margarita Romero for outstanding technical assistance and the AFCRI histology core. This work was supported by National Institutes of Health grants F32CA1238252 (EBM), P01 CA104838 and a Leukemia & Lymphoma Scholar award (JAD).
References
- Alam J, Stewart D, Touchard C, Boinapally S, Choi AM, Cook JL. Nrf2, a Cap‘n’Collar transcription factor, regulates induction of the heme oxygenase-1 gene. J Biol Chem. 1999;274:26071–8. doi: 10.1074/jbc.274.37.26071. [DOI] [PubMed] [Google Scholar]
- Back SH, Scheuner D, Han J, Song B, Ribick M, Wang J, et al. Translation attenuation through eIF2alpha phosphorylation prevents oxidative stress and maintains the differentiated state in beta cells. Cell Metab. 2009;10:13–26. doi: 10.1016/j.cmet.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartkova JHZ, Koed K, Kramer A, Tort F, Zieger K, Guldberg P, Sehested M, Nesland JM, Lukas C, Orntoft T, Lukas J, Bartek J. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–70. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- Bassing CH, Alt FW. H2AX may function as an anchor to hold broken chromosomal DNA ends in close proximity. Cell Cycle. 2004;3:149–53. doi: 10.4161/cc.3.2.689. [DOI] [PubMed] [Google Scholar]
- Behrend L, Henderson G, Zwacka RM. Reactive oxygen species in oncogenic transformation. Biochem Soc Trans. 2003;31:1441–4. doi: 10.1042/bst0311441. [DOI] [PubMed] [Google Scholar]
- Bi M, Naczki C, Koritzinsky M, Fels D, Blais J, Hu N, et al. ER stress-regulated translation increases tolerance to extreme hypoxia and promotes tumor growth. Embo J. 2005;24:3470–81. doi: 10.1038/sj.emboj.7600777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blais JD, Addison CL, Edge R, Falls T, Zhao H, Wary K, et al. Perk-dependent translational regulation promotes tumor cell adaptation and angiogenesis in response to hypoxic stress. Mol Cell Biol. 2006;26:9517–32. doi: 10.1128/MCB.01145-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobrovnikova-Marjon E, Hatzivassiliou G, Grigoriadou C, Romero M, Cavener DR, Thompson CB, et al. PERK-dependent regulation of lipogenesis during mouse mammary gland development and adipocyte differentiation. Proc Natl Acad Sci U S A. 2008;105:16314–9. doi: 10.1073/pnas.0808517105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer JW, Diehl JA. PERK mediates cell-cycle exit during the mammalian unfolded protein response. Proc Natl Acad Sci U S A. 2000;97:12625–30. doi: 10.1073/pnas.220247197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunelle JK, Bell EL, Quesada NM, Vercauteren K, Tiranti V, Zeviani M, et al. Oxygen sensing requires mitochondrial ROS but not oxidative phosphorylation. Cell Metab. 2005;1:409–14. doi: 10.1016/j.cmet.2005.05.002. [DOI] [PubMed] [Google Scholar]
- Bruns CJ, Koehl GE, Guba M, Yezhelyev M, Steinbauer M, Seeliger H, et al. Rapamycin-induced endothelial cell death and tumor vessel thrombosis potentiate cytotoxic therapy against pancreatic cancer. Clin Cancer Res. 2004;10:2109–19. doi: 10.1158/1078-0432.ccr-03-0502. [DOI] [PubMed] [Google Scholar]
- Buetler TM, Gallagher EP, Wang C, Stahl DL, Hayes JD, Eaton DL. Induction of phase I and phase II drug-metabolizing enzyme mRNA, protein, and activity by BHA, ethoxyquin, and oltipraz. Toxicol Appl Pharmacol. 1995;135:45–57. doi: 10.1006/taap.1995.1207. [DOI] [PubMed] [Google Scholar]
- Clanton TL. Hypoxia-induced reactive oxygen species formation in skeletal muscle. J Appl Physiol. 2007;102:2379–88. doi: 10.1152/japplphysiol.01298.2006. [DOI] [PubMed] [Google Scholar]
- Cullinan SB, Diehl JA. PERK-dependent activation of Nrf2 contributes to redox homeostasis and cell survival following ER stress. J Biol Chem. 2004;279:20076–20087. doi: 10.1074/jbc.M314219200. [DOI] [PubMed] [Google Scholar]
- Cullinan SB, Gordan JD, Jin J, Harper JW, Diehl JA. The Keap1-BTB protein is an adaptor that bridges Nrf2 to a Cul3-based E3 ligase: oxidative stress sensing by a Cul3-Keap1 ligase. Mol Cell Biol. 2004;24:8477–86. doi: 10.1128/MCB.24.19.8477-8486.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullinan SB, Zhang D, Hannink M, Arvisais E, Kaufman RJ, Diehl JA. Nrf2 Is a Direct PERK Substrate and Effector of PERK-Dependent Cell Survival. Mol Cell Biol. 2003;23:7198–7209. doi: 10.1128/MCB.23.20.7198-7209.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daneshmand S, Quek ML, Lin E, Lee C, Cote RJ, Hawes D, et al. Glucose-regulated protein GRP78 is up-regulated in prostate cancer and correlates with recurrence and survival. Hum Pathol. 2007;38:1547–52. doi: 10.1016/j.humpath.2007.03.014. [DOI] [PubMed] [Google Scholar]
- DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;7:11–20. doi: 10.1016/j.cmet.2007.10.002. [DOI] [PubMed] [Google Scholar]
- Denoyelle C, Abou-Rjaily G, Bezrookove V, Verhaegen M, Johnson TM, Fullen DR, et al. Anti-oncogenic role of the endoplasmic reticulum differentially activated by mutations in the MAPK pathway. Nat Cell Biol. 2006;8:1053–63. doi: 10.1038/ncb1471. [DOI] [PubMed] [Google Scholar]
- Evans KL. Overexpression of CDC25A associated with poor prognosis in breast cancer. Mol Med Today. 2000;6:459. doi: 10.1016/s1357-4310(00)01824-4. [DOI] [PubMed] [Google Scholar]
- Fernandez PM, Tabbara SO, Jacobs LK, Manning FC, Tsangaris TN, Schwartz AM, et al. Overexpression of the glucose-regulated stress gene GRP78 in malignant but not benign human breast lesions. Breast Cancer Res Treat. 2000;59:15–26. doi: 10.1023/a:1006332011207. [DOI] [PubMed] [Google Scholar]
- Folkman J, Cole P, Zimmerman S. Tumor behavior in isolated perfused organs: in vitro growth and metastases of biopsy material in rabbit thyroid and canine intestinal segment. Ann Surg. 1966;164:491–502. doi: 10.1097/00000658-196609000-00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frohlich DA, McCabe MT, Arnold RS, Day ML. The role of Nrf2 in increased reactive oxygen species and DNA damage in prostate tumorigenesis. Oncogene. 2008;27:4353–62. doi: 10.1038/onc.2008.79. [DOI] [PubMed] [Google Scholar]
- Furukawa M, He YJ, Borchers C, Xiong Y. Targeting of protein ubiquitination by BTB-Cullin 3-Roc1 ubiquitin ligases. Nat Cell Biol. 2003;5:1001–7. doi: 10.1038/ncb1056. [DOI] [PubMed] [Google Scholar]
- Furukawa M, Xiong Y. BTB protein Keap1 targets antioxidant transcription factor Nrf2 for ubiquitination by the Cullin 3-Roc1 ligase. Mol Cell Biol. 2005;25:162–71. doi: 10.1128/MCB.25.1.162-171.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazit G, Lu J, Lee AS. De-regulation of GRP stress protein expression in human breast cancer cell lines. Breast Cancer Res Treat. 1999;54:135–46. doi: 10.1023/a:1006102411439. [DOI] [PubMed] [Google Scholar]
- Gorgoulis VGVL, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, Venere M, Ditullio RA, Jr, Kastrinakas NG, Levy B, Kletsas D, Yoneta A, Herlyn M, Kittas C, Halazonetis TD. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434:907–13. doi: 10.1038/nature03485. [DOI] [PubMed] [Google Scholar]
- Guy CT, Webster MA, Schaller M, Parsons TJ, Cardiff RD, Muller WJ. Expression of the neu protooncogene in the mammary epithelium of transgenic mice induces metastatic disease. Proc Natl Acad Sci U S A. 1992;89:10578–82. doi: 10.1073/pnas.89.22.10578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzy RD, Hoyos B, Robin E, Chen H, Liu L, Mansfield KD, et al. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 2005;1:401–8. doi: 10.1016/j.cmet.2005.05.001. [DOI] [PubMed] [Google Scholar]
- Guzy RD, Schumacker PT. Oxygen sensing by mitochondria at complex III: the paradox of increased reactive oxygen species during hypoxia. Exp Physiol. 2006;91:807–19. doi: 10.1113/expphysiol.2006.033506. [DOI] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell. 2000;5:897–904. doi: 10.1016/s1097-2765(00)80330-5. [DOI] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397:271–4. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell. 2003;11:619–33. doi: 10.1016/s1097-2765(03)00105-9. [DOI] [PubMed] [Google Scholar]
- Hayes JD, Chanas SA, Henderson CJ, McMahon M, Sun C, Moffat GJ, et al. The Nrf2 transcription factor contributes both to the basal expression of glutathione S-transferases in mouse liver and to their induction by the chemopreventive synthetic antioxidants, butylated hydroxyanisole and ethoxyquin. Biochem Soc Trans. 2000;28:33–41. doi: 10.1042/bst0280033. [DOI] [PubMed] [Google Scholar]
- Haze K, Yoshida H, Yanagi H, Yura T, Mori K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol Biol Cell. 1999;10:3787–99. doi: 10.1091/mbc.10.11.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang HC, Nguyen T, Pickett CB. Phosphorylation of Nrf2 at Ser-40 by protein kinase C regulates antioxidant response element-mediated transcription. J Biol Chem. 2002;277:42769–74. doi: 10.1074/jbc.M206911200. [DOI] [PubMed] [Google Scholar]
- Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun. 1997;236:313–22. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- Kobayashi A, Kang MI, Okawa H, Ohtsuji M, Zenke Y, Chiba T, et al. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol. 2004;24:7130–9. doi: 10.1128/MCB.24.16.7130-7139.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HK, Xiang C, Cazacu S, Finniss S, Kazimirsky G, Lemke N, et al. GRP78 is overexpressed in glioblastomas and regulates glioma cell growth and apoptosis. Neuro Oncol. 2008;10:236–43. doi: 10.1215/15228517-2008-006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Baumeister P, Roy B, Phan T, Foti D, Luo S, et al. ATF6 as a transcription activator of the endoplasmic reticulum stress element: thapsigargin stress-induced changes and synergistic interactions with NF-Y and YY1. Mol Cell Biol. 2000;20:5096–106. doi: 10.1128/mcb.20.14.5096-5106.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin DI, Lessie MD, Gladden AB, Bassing CH, Wagner KU, Diehl JA. Disruption of cyclin D1 nuclear export and proteolysis accelerates mammary carcinogenesis. Oncogene. 2008;27:1231–42. doi: 10.1038/sj.onc.1210738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Wise DR, Diehl JA, Simon MC. Hypoxic reactive oxygen species regulate the integrated stress response and cell survival. J Biol Chem. 2008;283:31153–62. doi: 10.1074/jbc.M805056200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo SC, Li X, Henzl MT, Beamer LJ, Hannink M. Structure of the Keap1:Nrf2 interface provides mechanistic insight into Nrf2 signaling. Embo J. 2006;25:3605–17. doi: 10.1038/sj.emboj.7601243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nioi P, Nguyen T. A mutation of Keap1 found in breast cancer impairs its ability to repress Nrf2 activity. Biochem Biophys Res Commun. 2007;362:816–21. doi: 10.1016/j.bbrc.2007.08.051. [DOI] [PubMed] [Google Scholar]
- Ohta T, Iijima K, Miyamoto M, Nakahara I, Tanaka H, Ohtsuji M, et al. Loss of Keap1 function activates Nrf2 and provides advantages for lung cancer cell growth. Cancer Res. 2008;68:1303–9. doi: 10.1158/0008-5472.CAN-07-5003. [DOI] [PubMed] [Google Scholar]
- Ranganathan AC, Ojha S, Kourtidis A, Conklin DS, Aguirre-Ghiso JA. Dual function of pancreatic endoplasmic reticulum kinase in tumor cell growth arrest and survival. Cancer Res. 2008;68:3260–8. doi: 10.1158/0008-5472.CAN-07-6215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy NM, Kleeberger SR, Bream JH, Fallon PG, Kensler TW, Yamamoto M, et al. Genetic disruption of the Nrf2 compromises cell-cycle progression by impairing GSH-induced redox signaling. Oncogene. 2008;27:5821–32. doi: 10.1038/onc.2008.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sequeira SJ, Ranganathan AC, Adam AP, Iglesias BV, Farias EF, Aguirre-Ghiso JA. Inhibition of proliferation by PERK regulates mammary acinar morphogenesis and tumor formation. PLoS ONE. 2007;2:e615. doi: 10.1371/journal.pone.0000615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenvi SV, Smith EJ, Hagen TM. Transcriptional regulation of rat gamma-glutamate cysteine ligase catalytic subunit gene is mediated through a distal antioxidant response element. Pharmacol Res. 2009 doi: 10.1016/j.phrs.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Vattem KM, Sood R, An J, Liang J, Stramm L, et al. Identification and characterization of pancreatic eukaryotic initiation factor 2 alpha-subunit kinase, PEK, involved in translational control. Mol Cell Biol. 1998;18:7499–509. doi: 10.1128/mcb.18.12.7499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata T, Ohta T, Tong KI, Kokubu A, Odogawa R, Tsuta K, et al. Cancer related mutations in NRF2 impair its recognition by Keap1-Cul3 E3 ligase and promote malignancy. Proc Natl Acad Sci U S A. 2008;105:13568–73. doi: 10.1073/pnas.0806268105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu Y, Hendershot LM. Oxidative folding: cellular strategies for dealing with the resultant equimolar production of reactive oxygen species. Antioxid Redox Signal. 2009 doi: 10.1089/ars.2009.2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A, Boldin-Adamsky S, Thimmulappa RK, Rath SK, Ashush H, Coulter J, et al. RNAi-mediated silencing of nuclear factor erythroid-2-related factor 2 gene expression in non-small cell lung cancer inhibits tumor growth and increases efficacy of chemotherapy. Cancer Res. 2008;68:7975–84. doi: 10.1158/0008-5472.CAN-08-1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitz DR, Sim JE, Ridnour LA, Galoforo SS, Lee YJ. Glucose deprivation-induced oxidative stress in human tumor cells. A fundamental defect in metabolism? Ann N Y Acad Sci. 2000;899:349–62. doi: 10.1111/j.1749-6632.2000.tb06199.x. [DOI] [PubMed] [Google Scholar]
- Stracker TH, Couto SS, Cordon-Cardo C, Matos T, Petrini JH. Chk2 suppresses the oncogenic potential of DNA replication-associated DNA damage. Mol Cell. 2008;31:21–32. doi: 10.1016/j.molcel.2008.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirasophon W, Welihinda AA, Kaufman RJ. A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes Dev. 1998;12:1812–24. doi: 10.1101/gad.12.12.1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vattem KM, Wek RC. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc Natl Acad Sci U S A. 2004;101:11269–74. doi: 10.1073/pnas.0400541101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XZ, Harding HP, Zhang Y, Jolicoeur EM, Kuroda M, Ron D. Cloning of mammalian Ire1 reveals diversity in the ER stress responses. Embo J. 1998;17:5708–17. doi: 10.1093/emboj/17.19.5708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Shen J, Arenzana N, Tirasophon W, Kaufman RJ, Prywes R. Activation of ATF6 and an ATF6 DNA binding site by the endoplasmic reticulum stress response. J Biol Chem. 2000;275:27013–20. doi: 10.1074/jbc.M003322200. [DOI] [PubMed] [Google Scholar]
- Wei J, Sheng X, Feng D, McGrath B, Cavener DR. PERK is essential for neonatal skeletal development to regulate osteoblast proliferation and differentiation. J Cell Physiol. 2008;217:693–707. doi: 10.1002/jcp.21543. [DOI] [PubMed] [Google Scholar]
- Wild AC, Moinova HR, Mulcahy RT. Regulation of gamma-glutamylcysteine synthetase subunit gene expression by the transcription factor Nrf2. J Biol Chem. 1999;274:33627–36. doi: 10.1074/jbc.274.47.33627. [DOI] [PubMed] [Google Scholar]
- Wild AC, Mulcahy RT. Regulation of gamma-glutamylcysteine synthetase subunit gene expression: insights into transcriptional control of antioxidant defenses. Free Radic Res. 2000;32:281–301. doi: 10.1080/10715760000300291. [DOI] [PubMed] [Google Scholar]
- Wu J, Rutkowski DT, Dubois M, Swathirajan J, Saunders T, Wang J, et al. ATF6alpha optimizes long-term endoplasmic reticulum function to protect cells from chronic stress. Dev Cell. 2007;13:351–64. doi: 10.1016/j.devcel.2007.07.005. [DOI] [PubMed] [Google Scholar]
- Wu W, Fan YH, Kemp BL, Walsh G, Mao L. Overexpression of cdc25A and cdc25B is frequent in primary non-small cell lung cancer but is not associated with overexpression of c-myc. Cancer Res. 1998;58:4082–5. [PubMed] [Google Scholar]
- Wu X, Webster SR, Chen J. Characterization of tumor-associated Chk2 mutations. J Biol Chem. 2001;276:2971–4. doi: 10.1074/jbc.M009727200. [DOI] [PubMed] [Google Scholar]
- Yamamoto K, Sato T, Matsui T, Sato M, Okada T, Yoshida H, et al. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Dev Cell. 2007;13:365–76. doi: 10.1016/j.devcel.2007.07.018. [DOI] [PubMed] [Google Scholar]
- Zhang DD, Lo SC, Cross JV, Templeton DJ, Hannink M. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol Cell Biol. 2004;24:10941–53. doi: 10.1128/MCB.24.24.10941-10953.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Hamanaka RB, Bobrovnikova-Marjon E, Gordan JD, Dai MS, Lu H, et al. Ribosomal stress couples the unfolded protein response to p53-dependent cell cycle arrest. J Biol Chem. 2006a;281:30036–45. doi: 10.1074/jbc.M604674200. [DOI] [PubMed] [Google Scholar]
- Zhang W, Feng D, Li Y, Iida K, McGrath B, Cavener DR. PERK EIF2AK3 control of pancreatic beta cell differentiation and proliferation is required for postnatal glucose homeostasis. Cell Metab. 2006b;4:491–7. doi: 10.1016/j.cmet.2006.11.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.