

Abstract
The combination of irinotecan and erlotinib has been evaluated in clinical trials, although toxicity has been significant. We aimed to investigate the effect of erlotinib on SN-38 glucuronidation, and the association between UGT1A polymorphisms and SN-38 glucuronidation activity in the presence of erlotinib. The inhibitory effect of erlotinib on SN-38 glucuronidation was determined by measuring the formation rates for SN-38 glucuronide, using recombinant human UGT1A1, pooled human liver microsomes (HLM), and 52 Caucasian liver microsomes in the absence or presence of erlotinib. Inhibition kinetic studies were conducted. AUC ratios were used to predict the risk of potential drug-drug interactions (DDI) in vivo. Our data showed that erlotinib exhibited potent noncompetitive inhibition against SN-38 glucuronidation in pooled HLM and UGT1A1. Using the physiological and pharmacokinetic parameters obtained from the literature, we estimated the in vivo concentrations of unbound erlotinib available for UGT1A1 active site, and thus the AUC ratios of SN-38 were also quantitatively predicted. It is estimated that erlotinib administered at 50 mg/day or higher doses may result in at least a 24% increase in SN-38 AUC. Significant correlations were observed between SN-38 glucuronidation activity in the presence of erlotinib and UGT1A1*28 in 52 Caucasian liver microsomes. Our results suggest that erlotinib is a potent inhibitor of SN-38 glucuronidation via UGT1A1 inhibition. The coadministration of erlotinib with irinotecan may result in clinically significant DDI. UGT1A1*28 polymorphism correlates with erlotinib's effect on SN-38 glucuronidation. The present findings shed light on the development and optimization of combinations involving irinotecan and erlotinib.
Keywords: Erlotinib, Irinotecan, SN-38 glucuronidation, UGT1A1*28, Drug-drug interactions
1. Introduction
Irinotecan is one of the most commonly prescribed chemotherapy agents. Despite the extensive clinical experience with irinotecan, its usage has been limited by severe toxicity, including diarrhea and neutropenia. Fatal events (up to 5.3%) during single-agent irinotecan treatment have been reported (1). The risk of life-threatening neutropenia of irinotecan has been consistently associated with genetic variation in UGT1A1 (2, 3). Irinotecan requires metabolic activation by carboxylesterase 2 to form the active metabolite SN-38, which is further cleared via formation of SN-38 glucuronide (SN-38G) by UGT1A isoforms in human liver, including UGT1A1, 1A3, 1A6, and 1A9. Among these UGT isoforms, UGT1A1 shows the highest activity of SN-38 glucuronidation (4). There is accumulating evidence that plasma exposure to SN-38 is associated with increased risk of severe toxicity, primarily diarrhea and neutropenia (2, 5). The UGT1A1*28 polymorphism has been shown to be associated with reduced glucuronidation of SN-38 (6, 7), and increased clinical toxicity for patients treated with irinotecan (2, 8). We have showed that erlotinib is a potent inhibitor of UGT1A1-mediated 4-methylumbelliferone glucuronidation and bilirubin glucuronidation (9). These findings prompted us to assume that drug-drug interactions (DDI) might occur between erlotinib and irinotecan via the inhibition of UGT1A1-mediated SN-38 glucuronidation, and that genetic variation in UGT1A1 gene might be associated with the toxical outcome of the combination of both drugs.
Anti-epidermal growth factor receptor (EGFR) therapies have been utilized in combination with chemotherapy in multiple solid tumors, including colorectal and lung cancers (10). Targeting the EGFR pathway using a monoclonal antibody, such as cetuximab, has been demonstrated to be safe and effective when given alone or in combination with irinotecan (11).
Recently, the combination of irinotecan and erlotinib, an orally available, potent, and selective EGFR small molecule inhibitor approved for treatment of non-small cell lung and pancreatic cancer (12), has been evaluated in some studies. However, contradictory results have been reported with respect to the safety of the combination of irinotecan and erlotinib. It has been shown that erlotinib can enhance the antitumor activity of irinotecan, without enhanced toxicity, in the LoVo human colorectal tumor xenograft model (13). A clinical study of erlotinib with capecitabine and irinotecan in advanced colorectal cancer patients indicated that the combination has an acceptable safety profile and appears suitable for further Phase II studies (14). However, a phase I trial of irinotecan, infusional 5-fluorouracil, and leucovorin (FOLFIRI) with erlotinib was halted due to unexpectedly severe toxicities in several patients, including rash, diarrhea, and neutropenia, even at the lowest dose level (15).
In this study, we found that erlotinib exhibited a potent noncompetitive inhibition against SN-38 glucuronidation in pooled human liver microsomes (HLMs) and recombinant UGT1A1. The potential for drug interactions between erlotinib and SN-38 was estimated using the area under the plasma concentration–time curve (AUC) ratios. Significant correlations were observed between UGT1A1*28 and SN-38G formation rates in the absence and presence of erlotinib in 52 Caucasian liver microsomes.
2. Materials and methods
2.1. Chemicals
Erlotinib was purchased from Biaffin GmbH & Co KG (Kassel, Germany). SN-38 was obtained from Toronto Research Chemicals, Inc. (North York, Canada), SN-38G was gifts kindly provided by Dr. Kiyoshi Terada (Yakult Honsha Co. Ltd., Japan). β-glucuronidase (from Escherichia coli), alamethicin (from Trichoderma viride), Tris-HCl, ketoconazole, and UDPGA (trisodium salt) were purchased from Sigma-Aldrich (St. Louis, MO, USA). All other reagents were of HPLC grade or of the highest grade commercially available.
2.2. Human liver microsomes and liver tissue
Pooled HLMs and human UGT1A1 expressed in baculovirus-infected insect cells were purchased from BD Gentest Corp. (Woburn, MA, USA). Pooled HLMs were derived from 22 donors (90% Caucasian, 5% Hispanic and 5% African-American; 15 men and 7 women). The median age was 48 years with a range of 10 to 70.
Human liver tissue from 52 Caucasian donors was processed through Dr. Mary Relling's laboratory at St. Jude Children's Research Hospital (Memphis, TN, USA) and was provided by the Liver Tissue Procurement and Distribution System (funded by #NO1-DK-9-2310) and by the Cooperative Human Tissue Network. Samples were collected with human subjects approval.
2.3. SN-38 glucuronidation assay
Incubations were performed using conditions reported previously (16) with a slight modification. Incubation was performed at 37°C for 30 min. Microsomes were pre-incubated with 50 μg/mg protein alamethicin on ice for 15 min prior to incubation. SN-38 was dissolved in DMSO. The final concentration of DMSO in the incubation system was 1% (v/v). HPLC separation was achieved using a C18 column (3.9×300 mm I.D., 10μm) (Sigma-Aldrich, St. Louis, MO) at a flow rate of 1 ml/min and fluorescence detection at λex 355nm and λem 515 nm. A mobile phase consisted of 32/0.5/67.5 acetonitrile/ tetrahydrofuran/ 5 mM 1-heptanesulfonic acid in 50 mM potassium phosphate buffer (pH 2.7). The metabolites were quantified using the ratio of SN-38G to internal standard calibration curve. The precision was assessed by relative standard deviation (RSD = SD/Mean×100%), while the accuracy was calculated as relative mean error of calculated concentrations from nominal concentrations [RME = (calculated concentration-nominal concentration) / nominal concentration×100%]. The accuracy and precision of the back-calculated values for each concentration were less than 15% of the nominal values.
2.4. Kinetic study in pooled HLMs and UGT1A1
SN-38 (1-20 μM) was preincubated with pooled HLMs (0.5 mg /ml) or UGT1A1 (0.25 mg/ml) at 37°C for 5 min in a final volume of 200 μl of 25 mM Tris-HCl buffer (pH 7.4) containing 10 mM MgCl2, and 50 μg/mg protein alamethicin. After preincubation of the reaction mixture, the reaction was started by adding UDPGA for a final concentration of 5 mM. Incubation was performed at 37°C for 30 min. All experiments were performed in duplicate.
2.5. Inhibition of SN-38 glucuronidation activity assay
SN-38 was incubated in the presence of different concentrations of erlotinib (0, 0.05, 0.1, 0.2, 0.5, 1, 2, 10, and 50 μM). Ketoconazole, a known inhibitor of SN-38 glucuronidation (17), was used as a positive control. The reactions were continued for 30 min at 37°C in 0.5 mg/ml HLMs or 0.25 mg/ml UGT1A1. All experiments were separately performed in duplicate.
2.6. Genotyping and correlation studies in human liver microsomes
Fifty-two Caucasian liver microsomes were incubated with SN-38 with or without erlotinib as described above to determine the correlation of SN-38 glucuronidation activities in the absence or presence of erlotinib with genotypes of UGT1A1. Genotyping of UGT1A1*28 polymorphisms has been previously performed in the entire set of 52 Caucasian liver samples (18).
2.7. Kinetics analysis
Kinetic constants for SN-38 glucuronidation were obtained by fitting the experimental data to the Michaelis-Menten formula (eq. 1) (19),
| (2) |
where v is the rate of reaction, Vmax is the maximum velocity, Km is Michaelis constant (substrate concentration at half of Vmax of the reaction), and S is the substrate concentration.
The inhibition constant (Ki) values were determined using a range of concentrations of SN-38 (2-10 μM) and different concentrations of erlotinib (0-2 μM). Inhibition data from experiments were graphically represented by Dixon plots, and Ki values were calculated with nonlinear regression according to the equations for competitive (eq. 2), noncompetitive (eq. 3), (19),
| (2) |
| (3) |
where I is the inhibitor concentration, and Ki is the inhibition constant describing the affinity of the inhibitor for the enzyme. The type of inhibition was determined from the fitting of data to the enzyme inhibition models. Goodness of fit to kinetic and inhibition models was assessed from the F statistic, r2 values, parameter standard error estimates and 95% confidence intervals. Kinetic constants are reported as the value ± standard error of the parameter estimate. IC50 values (concentration of inhibitor that reduces enzyme activity by 50%) were determined by GraphPad Prism4 software (GraphPad Software Inc., La Jolla, CA).
2.8. Predicted concentrations of erlotinib at UGT catalytic sites
In view of the general assumption that only unbound drug is available for interaction with the enzyme active site, and in order to exclude the highest risk, we used the maximum unbound hepatic input concentration ([I]in,u) as the inhibitor concentration at UGT active site. The concentration was determined as described previously (9).
2.9. Calculation of AUCi/AUC ratio
The magnitude of inhibitory interactions of erlotinib was estimated as the ratio of the area under the plasma concentration–time curve of SN-38 in the presence and absence of erlotinib (AUCi/AUC). This ratio was determined as we described previously (9).
2.10. Data analysis and statistics
The t-test analysis was performed to assess the variance in SN-38G formation in the absence or presence of erlotinib in the whole population of 52 HLMs and between different genotypes, and spearman correlation analysis of each genotype versus SN-38G formation was performed by GraphPad Prism4 software (GraphPad Software Inc., La Jolla, CA). The threshold for statistical significance was set at P<0.05. All tests were two-sided.
3. Results
3.1. Kinetic studies in pooled HLMs and recombinant UGT1A
SN-38G formation rates were linear with microsomal protein concentration up to 1 mg/ml and time up to 1 h (data not shown). The kinetics of SN-38 glucuronidation in pooled HLMs (0.5 mg /ml) and recombinant UGT1A1 (0.25 mg/ml) were investigated. SN-38 (1-20 μM) was incubated with pooled HLMs or UGT1 A1 at 37°C for 30 min. The values of apparent Km in HLMs and UGT1A1 were 4.1 ± 0.3 and 2.5 ± 0.3 μM, respectively. The values of apparent Vmax in HLMs and UGT1A1 were 256 ± 8 and 2505 ± 103 pmol/min/mg protein, respectively.
3.2. Inhibition of SN-38 glucuronidation activity by erlotinib in pooled HLMs
SN-38 glucuronidation rates in the presence of various concentrations of potential inhibitors were investigated in pooled HLMs. The concentration of substrate, SN-38, was 5μM, which was close to its Km value. The IC50 value of ketoconazole in HLMs was calculated as 8.0 ± 0.3 μM, which is comparable with previously published data (17).
Erlotinib exhibited a UDPGA- and concentration- dependent inhibition against SN-38 glucuronidation activity in HLMs. The IC50 value was calculated as 0.58 ± 0.05 μM. No glucuronidation of erlotinib was found in the course of the incubations.
3.3. Inhibition kinetic analysis in pooled HLMs and recombinant UGT1A1
Erlotinib was found to strongly inhibit SN-38G formation in pooled HLMs. The representative Lineweaver-Burk plots (Fig. 1A) and analysis of the parameters of the enzyme inhibition model suggested that the inhibition type was noncompetitive. Based on analysis of nonlinear regression of inhibition data and Dixon plots presented in Fig. 1B, erlotinib showed noncompetitive inhibition against SN-38 glucuronidation in pooled HLMs with a Ki of 0.68 ± 0.04 μM. Erlotinib also exerted noncompetitive inhibition against SN-38 glucuronidation in recombinant UGT1A1 with a Ki of 0.81 ±0.05 μM (Fig. 1C & 1D).
Fig 1.
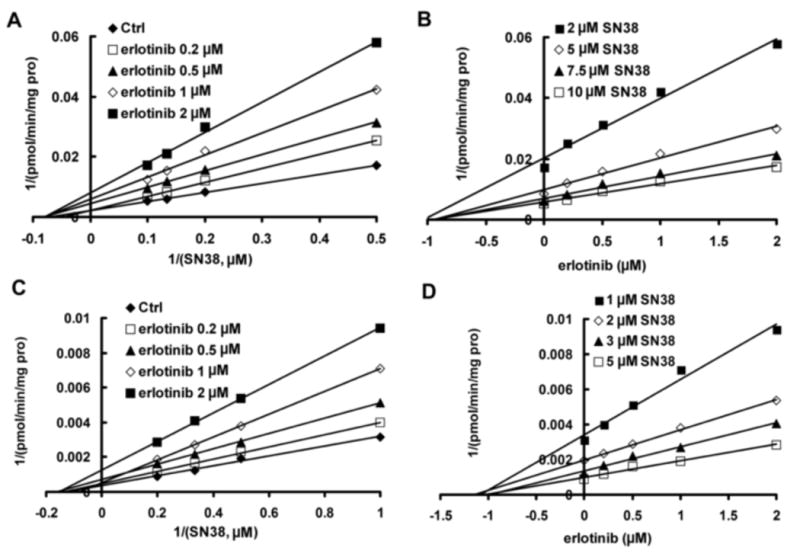
Representative Lineweaver-Burk plots (A,C) and Dixon plots (B,D) of the effects of erlotinib on SN-38 glucuronide formation in pooled human liver microsomes (A,B) and recombinant UGT1A1 (C,D). Reactions were performed in the presence of SN-38 (2, 5, 7.5, and 10 μM) and various concentrations of erlotinib (0, 0.1, 0.5, 1, and 2 μM) in pooled human liver microsomes (0.5 mg/ml) or recombinant UGT1A1 (0.25 mg/ml), 50 μg/mg protein alamethicin, 5 mM UDPGA, and 10 mM MgCl2 in a 25 mM Tri-HCl buffer (pH 7.4) in a final volume of 200 μl at 37°C for 30 min. All data points shown represent the mean of duplicate measurements.
3.4. Calculated AUC ratio of SN-38 in the presence and absence of erlotinib
The values of [I]max after oral administration of 50, 100, 150 mg/day erlotinib were calculated as 1.42, 2.64, and 3.72 μM, respectively. The calculated values are comparable to the reported actual values (1.29, 2.53, and 3.65 μM), and fall inside the interval of variability (20, 21). The values of [I]in,u after oral administration of 50, 100, 150 mg/day erlotinib were calculated as 0.16, 0.18, and 1.46 μM, respectively.
After oral administration of erlotinib of 50, 100, or 150 mg/day, the AUC of coadministered SN-38 is predicted to increase by 24%, 26%, and 46%, respectively.
3.5. Genotype-phenotype correlation study
We tested the hypothesis that UGT1A1*28 is associated with erlotinib's effects on SN-38 glucuronidation activity by incubating HLMs with SN-38 in the absence or presence of erlotinib. Our previously reported genotyping analysis of this entire set of 52 Caucasian liver samples revealed 25 heterozygote (6/7) and 7 homozygote (7/7) for (TA)7TAA allele (UGT1A1*28). The remaining 20 samples were homozygote for (TA)6TAA allele (6/6, wild type) (18).
As shown in Fig. 2A, t-test analysis indicated that the SN-38 glucuronidation activities in the presence of erlotinib both in the whole 52 samples and among the three genotypes were significantly lower than those in the absence of erlotinib (P<0.01). In addition, the SN-38 glucuronidation activities in the absence or presence of erlotinib in samples with 7/7 genotype were significantly lower than those with 6/6 (P<0.01) and 6/7 genotypes (P<0.05). Spearman correlation analysis revealed strong negative correlations between UGT1A1*28 in the 52 samples and SN-38G formation activity either in the absence of erlotinib (r=-0.60, P<0.0001) or in the presence of erlotinib (r=-0.55, P<0.0001) (Fig. 2A). No significant correlations were observed between residual activity percentage and UGT1A1*28 (P>0.05) (Fig. 2B).
Fig 2.
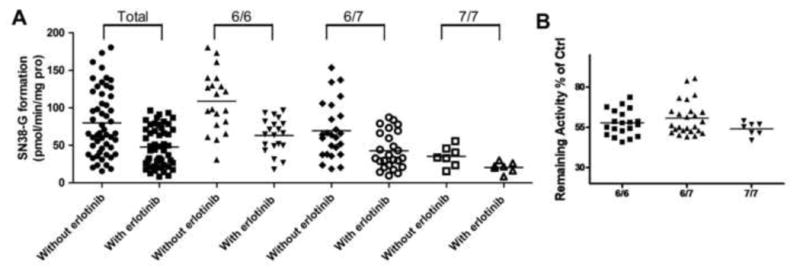
The variance in SN-38 glucuronide formation in the absence or presence of 0.5 μM erlotinib in the whole 52 Caucasian liver microsomes and among different UGT1A1*28 genotypes (A), as well as inhibition potential of erlotinib (B) in 52 Caucasian liver microsomes. Incubations were performed as described under Materials and Methods. Data represent means of duplicate determinations.
To investigate the influence of erlotinib concentrations on this relationship, further study was conducted in 9 liver samples (3 in each UGT1A1*28 genotype 6/6, 6/7 and 7/7). The samples were randomly selected from the samples whose volume was sufficient for further experiments. As shown in Fig. 3A-D, the SN-38 glucuronidation activities in the presence of 0, 0.15, 0.5, or 2 μM erlotinib in samples with 7/7 genotype were significantly lower than those with other genotypes (6/6 and 6/7) (P<0.05). UGT1A1*28 was associated with SN-38G formation in the presence of 0, 0.15, 0.5, or 2 μM erlotinib in nine samples (Spearman r=-0.95, P<0.0005; r=-0.98, P<0.0005; r=-0.90, P<0.001; r=-0.95, P<0.0005, respectively) (Fig. 3 A-D). No significant correlations were observed between the residual activity percentage and UGT1A1*28 in the same set of samples.
Fig 3.
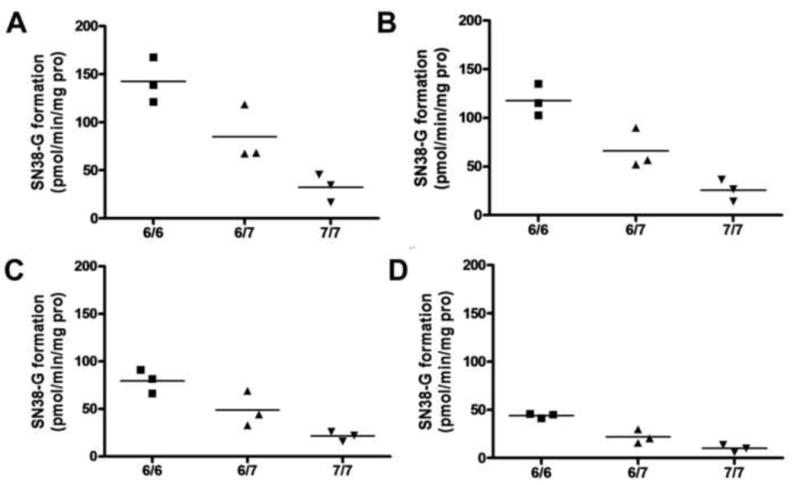
Correlation of UGT1A1*28 genotype with SN-38 glucuronide formation rates in the presence of 0 (A), 0.15 (B), 0.5 (C), or 2 μM erlotinib (D) on SN-38 glucuronidation in 9 Caucasian liver microsomes. Incubations were performed as described under Materials and Methods. Data represent means of duplicate determinations.
4. Discussion
We demonstrated that erlotinib exhibited a UDPGA- and concentration- dependent potent inhibition against SN-38 glucuronidation activity. The inhibition type was noncompetitive. This occurs when the inhibitor binds at a site away from the substrate binding site, causing a reduction in the catalytic rate. The fractional inhibition is identical at all substrate concentrations and cannot be overcome by increasing substrate concentration due to the reduction in Vmax (19). There are several noncompetitive inhibitors of UGTs enzymes that have been reported, such as phenylbutazone and quinine, which may suggest distinct binding sites for substrate and inhibitor in these enzymes (22, 23). Nevertheless, the molecular basis of these UGTs noncompetitive inhibitions was unknown.
The quantitative prediction of drug-drug interaction risk indicated that the coadministration of erlotinib at clinically available dose could significantly increase the AUC of SN-38, the active metabolite of irinotecan, implying a clinically significant interaction might occur between erlotinib and irinotecan.
It is noteworthy that in vitro data tend to underestimate inhibition of drug glucuronidation in vivo (22), and our calculation is based only on hepatic SN-38 glucuronidation, whereas the extent of extrahepatic glucuronidation is unknown. In addition, the bioavailability of erlotinib can be increased to almost 100% with the intake of food (24). Furthermore, CYP3A4 is involved in the metabolism of both erlotinib and irinotecan, and both irinotecan and SN-38 have been suggested to be mechanism-based inactivators of CYP3A4, making their interactions in vivo more complex (25, 26).
High intraluminal levels of SN-38 are believed to be the direct cause of the severe diarrhea occurring after irinotecan therapy, and high plasma levels of SN-38 are associated with neutropenia (2, 5). Glucuronidation of SN-38 to the inactive SN-38G decreases the toxicities of irinotecan (27). Therefore, our results may explain the unexpectedly severe toxicities found in clinical trials of the combinations of irinotecan with erlotinib.
Erlotinib also exhibited potent noncompetitive inhibition against SN-38 glucuronidation in recombinant UGT1A1 with a Ki similar to that in HLMs. In view of the fact that UGT1A1, 1A3, 1A6, and 1A9 are involved in SN-38 glucuronidation and UGT1A1 shows the highest activity (4), the inhibition of erlotinib against SN-38G formation should be attributed to the predominant inhibitory effect of erlotinib on UGT1A1 activity.
Significant correlations were observed between SN-38G formation rates in the presence of erlotinib and UGT1A1*28. Due to limitation of samples, only one concentration of erlotinib was evaluated in the whole set of human liver samples, but more detailed evaluation of nine samples in the presence of various concentrations of erlotinib lent further support to this finding. This suggests that adverse effects of the combination of irinotecan with erlotinib would be associated with the UGT1A1*28 polymorphism.
The present findings shed light on the development and optimization of the combinations involving irinotecan and erlotinib, and also provide the basis for further clinical studies investigating adverse effects of the combination of irinotecan with erlotinib.
Acknowledgments
This work was supported by Pharmacogenetics of Anticancer Agents Research Group, National Institutes of Health (NIH)/National Institute of General Medical Sciences grant U01GM61393.
Grant support: This work was supported by Pharmacogenetics of Anticancer Agents Research Group, National Institutes of Health (NIH)/National Institute of General Medical Sciences grant U01GM61393.
Footnotes
Conflict of Interest Statement: Dr. Mark J. Ratain is a consultant for Ambit, Hoffman-LaRoche, OSI Pharmaceuticals and Genentech.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Fuchs CS, Moore MR, Harker G, Villa L, Rinaldi D, Hecht JR. Phase III comparison of two irinotecan dosing regimens in second-line therapy of metastatic colorectal cancer. J Clin Oncol. 2003;21:807–14. doi: 10.1200/JCO.2003.08.058. [DOI] [PubMed] [Google Scholar]
- 2.Innocenti F, Undevia SD, Iyer L, Chen PX, Das S, Kocherginsky M, et al. Genetic variants in the UDP-glucuronosyltransferase 1A1 gene predict the risk of severe neutropenia of irinotecan. J Clin Oncol. 2004;22:1382–8. doi: 10.1200/JCO.2004.07.173. [DOI] [PubMed] [Google Scholar]
- 3.Hoskins JM, Goldberg RM, Qu P, Ibrahim JG, McLeod HL. UGT1A1*28 genotype and irinotecan-induced neutropenia: dose matters. J Natl Cancer Inst. 2007;99:1290–5. doi: 10.1093/jnci/djm115. [DOI] [PubMed] [Google Scholar]
- 4.Hanioka N, Ozawa S, Jinno H, Ando M, Saito Y, Sawada J. Human liver UDP-glucuronosyltransferase isoforms involved in the glucuronidation of 7-ethyl-10-hydroxycamptothecin. Xenobiotica. 2001;31:687–99. doi: 10.1080/00498250110057341. [DOI] [PubMed] [Google Scholar]
- 5.Ramchandani RP, Wang Y, Booth BP, Ibrahim A, Johnson JR, Rahman A, et al. The role of SN-38 exposure, UGT1A1*28 polymorphism, and baseline bilirubin level in predicting severe irinotecan toxicity. J Clin Pharmacol. 2007;47:78–86. doi: 10.1177/0091270006295060. [DOI] [PubMed] [Google Scholar]
- 6.Iyer L, Hall D, Das S, Mortell MA, Ramirez J, Kim S, et al. Phenotype-genotype correlation of in vitro SN-38 (active metabolite of irinotecan) and bilirubin glucuronidation in human liver tissue with UGT1A1 promoter polymorphism. Clin Pharmacol Ther. 1999;65:576–82. doi: 10.1016/S0009-9236(99)70078-0. [DOI] [PubMed] [Google Scholar]
- 7.Ando Y, Saka H, Asai G, Sugiura S, Shimokata K, Kamataki T. UGT1A1 genotypes and glucuronidation of SN-38, the active metabolite of irinotecan. Ann Oncol. 1998;9:845–7. doi: 10.1023/a:1008438109725. [DOI] [PubMed] [Google Scholar]
- 8.Iyer L, Das S, Janisch L, Wen M, Ramirez J, Karrison T, et al. UGT1A1*28 polymorphism as a determinant of irinotecan disposition and toxicity. Pharmacogenomics J. 2002;2:43–7. doi: 10.1038/sj.tpj.6500072. [DOI] [PubMed] [Google Scholar]
- 9.Liu Y, Ramirez J, House L, Ratain MJ. Comparison of the drug-drug interactions potential of erlotinib and gefitinib via inhibition of UDP-glucuronosyltransferases. Drug Metab Dispos. 2010;38:32–9. doi: 10.1124/dmd.109.029660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vokes EE, Chu E. Anti-EGFR therapies: clinical experience in colorectal, lung, and head and neck cancers. Oncology (Williston Park) 2006;20 l 2:15–25. [PubMed] [Google Scholar]
- 11.Cunningham D, Humblet Y, Siena S, Khayat D, Bleiberg H, Santoro A, et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med. 2004;351:337–45. doi: 10.1056/NEJMoa033025. [DOI] [PubMed] [Google Scholar]
- 12.Arora A, Scholar EM. Role of tyrosine kinase inhibitors in cancer therapy. J Pharmacol Exp Ther. 2005;315:971–9. doi: 10.1124/jpet.105.084145. [DOI] [PubMed] [Google Scholar]
- 13.Chen J, Smith M, Kolinsky K, Adames V, Mehta N, Fritzky L, et al. Antitumor activity of HER1/EGFR tyrosine kinase inhibitor erlotinib, alone and in combination with CPT-11 (irinotecan) in human colorectal cancer xenograft models. Cancer Chemother Pharmacol. 2007;59:651–9. doi: 10.1007/s00280-006-0320-8. [DOI] [PubMed] [Google Scholar]
- 14.Bajetta E, Di Bartolomeo M, Buzzoni R, Ferrario E, Dotti KF, Mariani L, et al. Dose finding study of erlotinib combined to capecitabine and irinotecan in pretreated advanced colorectal cancer patients. Cancer Chemother Pharmacol. 2009;64:67–72. doi: 10.1007/s00280-008-0852-1. [DOI] [PubMed] [Google Scholar]
- 15.Messersmith WA, Laheru DA, Senzer NN, Donehower RC, Grouleff P, Rogers T, et al. Phase I trial of irinotecan, infusional 5-fluorouracil, and leucovorin (FOLFIRI) with erlotinib (OSI-774): early termination due to increased toxicities. Clin Cancer Res. 2004;10:6522–7. doi: 10.1158/1078-0432.CCR-04-0746. [DOI] [PubMed] [Google Scholar]
- 16.Iyer L, Hall D, Das S, Mortell MA, Ramirez J, Kim S, et al. Phenotype-genotype correlation of in vitro SN-38 (active metabolite of irinotecan) and bilirubin glucuronidation in human liver tissue with UGT1A1 promoter polymorphism. Clin Pharmacol Ther. 1999;65:576–582. doi: 10.1016/S0009-9236(99)70078-0. [DOI] [PubMed] [Google Scholar]
- 17.Yong WP, Ramirez J, Innocenti F, Ratain MJ. Effects of ketoconazole on glucuronidation by UDP-glucuronosyltransferase enzymes. Clin Cancer Res. 2005;11:6699–704. doi: 10.1158/1078-0432.CCR-05-0703. [DOI] [PubMed] [Google Scholar]
- 18.Innocenti F, Liu W, Chen P, Desai AA, Das S, Ratain MJ. Haplotypes of variants in the UDP-glucuronosyltransferase1A9 and 1A1 genes. Pharmacogenet Genomics. 2005;15:295–301. doi: 10.1097/01213011-200505000-00004. [DOI] [PubMed] [Google Scholar]
- 19.Copeland RA. Enzymes: A practical introduction to structure, mechanism, and data analysis. New York: Wiley-VCH; 2000. [Google Scholar]
- 20.Frohna P, Lu J, Eppler S, Hamilton M, Wolf J, Rakhit A, et al. Evaluation of the absolute oral bioavailability and bioequivalence of erlotinib, an inhibitor of the epidermal growth factor receptor tyrosine kinase, in a randomized, crossover study in healthy subjects. J Clin Pharmacol. 2006;46:282–90. doi: 10.1177/0091270005284193. [DOI] [PubMed] [Google Scholar]
- 21.Yamamoto N, Horiike A, Fujisaka Y, Murakami H, Shimoyama T, Yamada Y, et al. Phase I dose-finding and pharmacokinetic study of the oral epidermal growth factor receptor tyrosine kinase inhibitor Ro50-8231 (erlotinib) in Japanese patients with solid tumors. Cancer Chemother Pharmacol. 2008;61:489–96. doi: 10.1007/s00280-007-0494-8. [DOI] [PubMed] [Google Scholar]
- 22.Uchaipichat V, Mackenzie PI, Elliot DJ, Miners JO. Selectivity of substrate (trifluoperazine) and inhibitor (amitriptyline, androsterone, canrenoic acid, hecogenin, phenylbutazone, quinidine, quinine, and sulfinpyrazone) “probes” for human udp-glucuronosyltransferases. Drug Metab Dispos. 2006;34:449–56. doi: 10.1124/dmd.105.007369. [DOI] [PubMed] [Google Scholar]
- 23.Luukkanen L, Taskinen J, Kurkela M, Kostiainen R, Hirvonen J, Finel M. Kinetic characterization of the 1A subfamily of recombinant human UDP-glucuronosyltransferases. Drug Metab Dispos. 2005;33:1017–26. doi: 10.1124/dmd.105.004093. [DOI] [PubMed] [Google Scholar]
- 24.Smith J. Erlotinib: small-molecule targeted therapy in the treatment of non-small-cell lung cancer. Clin Ther. 2005;27:1513–34. doi: 10.1016/j.clinthera.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 25.Li J, Zhao M, He P, Hidalgo M, Baker SD. Differential metabolism of gefitinib and erlotinib by human cytochrome P450 enzymes. Clin Cancer Res. 2007;13:3731–7. doi: 10.1158/1078-0432.CCR-07-0088. [DOI] [PubMed] [Google Scholar]
- 26.Hanioka N, Ozawa S, Jinno H, Tanaka-Kagawa T, Nishimura T, Ando M, et al. Interaction of irinotecan (CPT-11) and its active metabolite 7-ethyl-10-hydroxycamptothecin (SN-38) with human cytochrome P450 enzymes. Drug Metab Dispos. 2002;30:391–6. doi: 10.1124/dmd.30.4.391. [DOI] [PubMed] [Google Scholar]
- 27.Gupta E, Lestingi TM, Mick R, Ramirez J, Vokes EE, Ratain MJ. Metabolic fate of irinotecan in humans: correlation of glucuronidation with diarrhea. Cancer Res. 1994;54:3723–5. [PubMed] [Google Scholar]