

Abstract
Recombinant proteins are important therapeutics due to potent, highly specific, and nontoxic actions in vivo. However, they are expensive medicines to manufacture, chemically unstable, and difficult to administer with low patient uptake and compliance. Small molecule drugs are cheaper and more bioavailable, but less target-specific in vivo and often have associated side effects. Here we combine some advantages of proteins and small molecules by taking short amino acid sequences that confer potency and selectivity to proteins, and fixing them as small constrained molecules that are chemically and structurally stable and easy to make. Proteins often use short α-helices of just 1–4 helical turns (4–15 amino acids) to interact with biological targets, but peptides this short usually have negligible α-helicity in water. Here we show that short peptides, corresponding to helical epitopes from viral, bacterial, or human proteins, can be strategically fixed in highly α-helical structures in water. These helix-constrained compounds have similar biological potencies as proteins that bear the same helical sequences. Examples are (i) a picomolar inhibitor of Respiratory Syncytial Virus F protein mediated fusion with host cells, (ii) a nanomolar inhibitor of RNA binding to the transporter protein HIV-Rev, (iii) a submicromolar inhibitor of Streptococcus pneumoniae growth induced by quorum sensing pheromone Competence Stimulating Peptide, and (iv) a picomolar agonist of the GPCR pain receptor opioid receptor like receptor ORL-1. This approach can be generally applicable to downsizing helical regions of proteins with broad applications to biology and medicine.
Keywords: helix, inhibitor, structure, mimetic, anti-infective
Proteins are key functional components that define life, aging, disease, and death. Their uses in medicine, science, and industry are however limited by their complexity, high costs, chemical instability, and low bioavailability. Consequently, new chemical technology is needed to create simpler, smaller, cheaper, more stable, and bioavailable molecules that can execute or inhibit selected functions of proteins. If generic approaches could be developed to stabilize or recreate new molecular shapes that mimic structural components of proteins (e.g., helices, turns, strands, and combinations), the resulting molecules could potentially be extremely valuable for interrogating biological systems and for exploring prospective applications such as new pharmaceuticals, diagnostics, vaccines, and nanomaterials.
Some proteins elicit biological function through a single short α-helical segment (usually 4–15 amino acids in 1–4 helical turns) that interacts with nucleic acids or proteins (1–3). However, short synthetic peptides of this length are usually not thermodynamically stable helices in water and adopt only random structures (4). Techniques developed as generalized strategies for stabilising or mimicking short peptide α-helices have been limited by lack of helical structure in water, by sequence or context dependent helicity, by low biological potency or by poor target specificity (5–10). Either these techniques have failed to induce α-helicity through an entire peptide, had complex synthetic routes that limited broad utility, or have not been applicable across a wide range of biological sequences and receptor types. Nonpeptide compounds developed to mimic just the sidechains of protein α-helices have suffered from selectivity and potency issues and lack generic applicability. A truly generic technology for mimicking short protein helices of < 15 residues of different functions and origins would be valuable for creating biological probes and biopharmaceuticals.
Here we set out to downsize viral [respiratory syncytial virus (RSV) F protein, HIV rev], bacterial [Streptococcus pneumoniae Competence Stimulating Peptide (CSP)] and human (nociceptin) proteins to short synthetic peptides with strategically enforced water-stable α-helical structures. Helix-constrained and unconstrained peptides are compared for a correlation between α-helicity and function, and to determine if this approach to helix preorganization for receptor binding is generic in producing the same properties exhibited by native proteins bearing the same helical sequences. We report here circular dichroism (CD) and NMR data for helix-constrained peptides in water, with NMR-derived solution structures and biological activities for four constrained peptides that have well defined α-helical structures in contrast to their unconstrained analogues. This structure induction correlates with biological activities that are superior to those of unconstrained peptides, approaching and in some cases surpassing those of the proteins from which the peptide sequences were originally derived.
Results
Cyclic Pentapeptide Ac-(1,5-cyclo)-[KAAAD]-NH2 Is the Smallest α-Helical Structure.
We previously demonstrated that arginine-containing pentapeptides Ac-(1,5-cyclo)-[KXRXD]-NH2, and related hexapeptides featuring an amide bond linkage between lysine and aspartate side chains spaced three residues apart, were α-helical by CD spectroscopy (11). However, CD spectra are notoriously unreliable for characterising α-helicity in short peptides (11–13). Here we report the NMR solution structure of Ac-(1,5-cyclo)-[KAAAD]-NH2 (1), containing three alanines, that is unambiguously the smallest three dimensional α-helical structure ever reported in water. Relative intensities of αN(i,i + 2), αN(i,i + 3), αN(i,i + 4) rotating-frame Overhauser enhancements (ROEs) together with other NMR parameters (Table S1, Table S2, Table S3, Table S4, Fig. S1, Fig. S2, Fig. S3, Fig. S4, and Fig. S5) definitively characterize the α-helical structure of 1 in water (Fig. 1A). It exhibits all the key features that define α-helical turns in proteins, such as specific dihedral angles (ϕ -58°, ψ -47°), three intramolecular hydrogen bonds, 3.6 residues per helical turn, a rise of 1.5 Å per residue, and a helix radius of 2.3 Å. This is an important finding because such a short α-helical structure lacks the internal structural stabilization afforded to protein helices through interaction with other parts of the protein. In agreement with our earlier study of small model peptides (11), CD spectral fingerprints show that the α-helical pentapeptide (1) is stable to aqueous phosphate buffer, acid, base, trypsin degradation, does not unfold in a denaturing environment (e.g., 8 M guanidine), and the CD spectral molar ellipticity does not increase at 222 nm in 50% 2,2,2-trifluoroethanol (TFE) indicating that maximum helicity is enforced by the constraint (Fig. 1B and Table S5). This exceptional structural and chemical stability, achieved without incorporating any unnatural components, offers significant advantages over other methods being trialled for protein helix mimicry (5–10).
Fig. 1.

A stable α-helical turn tolerates amino acids from protein helices. (A) Compound 1, Ac-(1,5-cyclo)-[KAAAD]-NH2, is α-helical. Seventeen lowest energy NMR-derived structures for 1 in 90% H2O∶10% D2O at 298 K (backbone heavy atom pairwise rmsd 0.32 Å) showing α-helix (green), linker (purple), and three hydrogen bonds (dashes) vs. idealized α-helix (gray ribbon). (B) Compound 1 was stable (by circular dichroism changes at 222 nm after 30 min, 298 K) in aq. phosphate buffer (black), 1 M TFA (orange), 0.1 M KOH (red), 200 nM trypsin in 10 mM PBS at pH 7.4 (blue), 1∶1 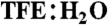 (purple), and 8 M guanidine (green). (C) Helicity depended on amino acid side chains at i and i + 4 positions that are covalently linked in cyclic peptides (2–9) Ac-(cyclo-1,5)-[(ith residue)-ARA-(i + 4 residue)]-NH2. The most α-helical had i = Lys, i + 4 = Asp. (D) % helicity in Ac-(2,6-cyclo)-R1[K2X3X4X5D6]-NH2 (10–18, Fig. S6) correlated both with the free energy (ΔΔGα, black) of each amino acid in stabilizing a protein α-helix (14) and with the probability (Pα, red) of an amino acid being in a protein α-helix (15). ΔΔGα and Pα values were summed for X3, X4, and X5 amino acids in each compound using literature data for each amino acid (14, 15). Helicity (%), ΔΔGα (kcal/mol), Pα (relative units) values are respectively 91, -2.31, 4.80 (10, A3A4A5); 100, -2.16, 4.70 (11, A3L4A5); 80, -2.04, 4.64 (12, A3M4A5); 84, -1.87, 4.42 (13, A3Q4A5); 76, -1.95, 4.65 (14, A3F4A5); 32, -1.54, 3.20 (15, A3G4A5); 32, -1.12, 2.04 (16, G3S4A5); 30, -1.05, 1.32 (17, S3S4S5); 14, 0.0, 1.41 (18, G3G4G5). Data for C and D were collected in 10 mM phosphate buffer (pH 7.4) at 25 °C (11).
(purple), and 8 M guanidine (green). (C) Helicity depended on amino acid side chains at i and i + 4 positions that are covalently linked in cyclic peptides (2–9) Ac-(cyclo-1,5)-[(ith residue)-ARA-(i + 4 residue)]-NH2. The most α-helical had i = Lys, i + 4 = Asp. (D) % helicity in Ac-(2,6-cyclo)-R1[K2X3X4X5D6]-NH2 (10–18, Fig. S6) correlated both with the free energy (ΔΔGα, black) of each amino acid in stabilizing a protein α-helix (14) and with the probability (Pα, red) of an amino acid being in a protein α-helix (15). ΔΔGα and Pα values were summed for X3, X4, and X5 amino acids in each compound using literature data for each amino acid (14, 15). Helicity (%), ΔΔGα (kcal/mol), Pα (relative units) values are respectively 91, -2.31, 4.80 (10, A3A4A5); 100, -2.16, 4.70 (11, A3L4A5); 80, -2.04, 4.64 (12, A3M4A5); 84, -1.87, 4.42 (13, A3Q4A5); 76, -1.95, 4.65 (14, A3F4A5); 32, -1.54, 3.20 (15, A3G4A5); 32, -1.12, 2.04 (16, G3S4A5); 30, -1.05, 1.32 (17, S3S4S5); 14, 0.0, 1.41 (18, G3G4G5). Data for C and D were collected in 10 mM phosphate buffer (pH 7.4) at 25 °C (11).
Lactam bridges have been used before within longer peptides to enhance helicity, but there has been no consensus as to which amino acids should be linked for maximum helicity, where and in what order they should be positioned in a sequence, and which sidechains favor α-helicity (10). Instead, helical content and optimal bridge location was thought to be context dependent. However, we have previously shown in a model peptide (11) that alternative lactam linkages that might conceivably aid α-helix induction are inferior to the (K1 → D5) linkage in 1 (Fig. 1C). Furthermore, variations to the three central alanine amino acids within the pentapeptide cyclic unit are well tolerated (Fig. S6). Most importantly, we demonstrate here that amino acid substitutions known to favor α-helicity in proteins also produce the highest α-helicity in the cyclic pentapeptide module (Fig. 1D and Fig. S2). There was a linear correlation between free energies of protein α-helix stability with cyclic peptide α-helix stability (Fig. 1D), indicating that 1 is a generic α-helical scaffold capable of mimicking different α-helices in different proteins.
Adapting Cyclic Pentapeptides to Mimic Biological Sequences.
This cyclic pentapeptide module can be used in a number of ways to create 1–4 turn α-helices (Fig. 2), these lengths being most common for helical protein sequences recognized by other proteins or nucleic acids. Compound 1 presents three central variable amino acids for recognition (Fig. 2A). Two cycles joined “back-to-back” (Fig. 2B) present six variable amino acids in a 10 residue helix (17) but, if separated by two amino acids, there are eight variable amino acids in a 12-residue helix (Fig. 2C). These different arrangements allow versatility in designing α-helix mimics to expose different side chains on different helix faces, whereas positioning lactam bridges at alternative sites to minimise steric clashes with receptors. An advantage of this approach is that no unnatural components are used to fix helicity, the constraining linkers being composed entirely of amino acid side chains.
Fig. 2.

α-helices composed of cyclic pentapeptides. Different distributions of variable side chains (gray spheres) and linking bridges (green) allow design of different helix mimics for binding to different receptors. Models built in Insight (16) using lowest energy NMR structures of 1 (Fig. 1).
To assess the general utility of this approach to preorganizing water-stable α-helical fragments of proteins, we chose four α-helices from viral, bacterial, and human proteins with diverse functions and quite different receptors (Figs. 3–6). Our objective was to compare short peptide sequences corresponding to different protein α-helices (or putative α-helices) in both unconstrained (19, 21, 23, 25) and α-helix-constrained (20, 22, 24, 26) forms. By comparatively assessing the extent of induction of α-helical structure, serum stability, and biological activity, we could determine if this approach had general utility to mimicking protein α-helices in small molecules.
Fig. 3.

RSV fusion peptides. (A) RSV F glycoprotein HR-C (residues F483–V495) mimicked by 20 vs.unconstrained 19. (B and C) The unconstrained sequence 19 had no structure whereas 20 showed an α-helical CD spectrum and NMR structure. The CD spectra of the linear and helix-constrained RSV peptide suggested that the constrained analogue had significantly more helicity. The structure was confirmed through 1H NMR. (D) Helix-constrained 20 inhibited recombinant RSV F mediated fusion in a cell-to-cell assay at picomolar concentrations (190 pM, pIC50 ≤ 9.72 ± 0.03; blue) whereas unconstrained 19 showed no inhibition < 1 μM (black). Error bars represented ± S.E.M with n≥3.
Fig. 4.

S. pneumoniae competence stimulating peptide-1. (A) S. pneumoniae CSP-1 (21) is a quorum sensing peptide that is proposed to form an α-helix in lipid-like environments (pdb 2a1c). (B) Peptide 21 had no structure in water by CD, whereas cyclic analogue (22) formed a well-defined α-helix, shown by CD spectra and (C) NMR structures. (D) Growth curves for S.pneumoniae treated with 10 μM CSP-1 mimetic (22, green), unconstrained analogue (21, red) and PBS control (black) showing at least 10-fold more potent inhibition by 22. Error bars represented ± S.E.M with n≥3.
Fig. 5.

HIV-1 Rev (A) HIV-1 Rev residues 34–50 bind as an α-helix to stem loop IIB of RRE (pdb 1etg). (B and C) The native sequence (23) had little structure in water but analogue (24) had significant α-helicity that extended beyond the cyclic regions. (D) Affinities of 23 and 24 were assessed using biotin-labeled RRE and 10 nM Ac-TRQARRNRRRWRERQR-AAAC(Fluorescein)-NH2. Constrained mimetic (24) had submicromolar affinity (pIC50 ≤ 6.65 ± 0.04; red) whereas unconstrained analogue (23) had submicromolar affinity (pIC50 ≤ 6.05 ± 0.07; black), an advantage in free energy of 3.4 kJ.mol- 1.K-1. Error bars represented ± S.E.M with n≥3.
RSV Fusion Protein.
The RSV F glycoprotein is critical for fusion and entry of RSV to host cells (18). The native, prefusion configuration of F on the virion surface is trimeric. Host cell receptor engagement induces conformational reorganization of the trimer so that three HR-C and three HR-N domains associate in a postfusion 6 α-helix bundle. Each helical HR-C domain sits in a groove created by two of the three HR-N helices. This structural rearrangement drives virus-cell membrane fusion, which is inhibited by formation of the six α-helix bundle (18). The unconstrained peptide 19 (Fig. 3A) corresponding to HR-C residues F483-V495 was synthesized with amides at either end. The corresponding back-to-back double lactam bridged 12-residue peptide, Ac-cyclo-(2,6; 7,11)-Nal[KDEFD][KSIRD]V-NH2 (20, Fig. 3A) with 1-naphthylalanine (Nal) at the C-terminus, was synthesized as a putative helix-constrained analogue. Circular dichroism spectra (Fig. 3B) show unconstrained sequence 19 with no well defined structure in water, whereas constrained peptide 20 displayed well defined α-helical CD spectra (Fig. 3B).
A three dimensional solution structure was determined for compound 20 in water (Fig. 3C) using 2D NMR spectroscopy. Compound 20 showed substantial interresidue (68) and medium range (42) NOEs and eight intramolecular hydrogen bonds (Table S6, Table S7, Table S8, Fig. S7, Fig. S8, Fig. S9, Fig. S10, and Fig. S11), whereas the unconstrained peptide 19 showed few NMR parameters, consistent with negligible structure. The helix-constrained peptide 20 was a potent inhibitor of RSV fusion (IC50 190 pM, Fig. 3D) compared with unconstrained 19 that had no inhibition below 1 μM. This approximately 10,000-fold enhanced inhibition of viral cell-to-cell fusion makes 20 the most potent inhibitor of RSV fusion known (19).
Competence Stimulating Protein from Streptococcus pneumoniae.
Competence stimulating peptide (CSP-1) is a 17-residue quorum sensing pheromone (21, Fig. 4A) from Streptococcus pneumoniae (20). This secreted peptide acts on a cell-surface histidine kinase known as ComD, and at low concentrations promotes bacterial growth. At higher concentrations (≥10 μM) than required for competency, it has antibacterial activity (21). The specific binding of CSP-1 to surface protein ComD is unusual for antibacterial peptides and is a unique mechanism for antibactericidal activity. Peptide 21 has a number of residues expected to be helical in proteins (E, Nle, R, L, K, Q), and shows some helical propensity in a simulated lipid environment (pdb 2a1c), but little α-helical structure in water (Fig. 4B).
By contrast, the back-to-back constrained peptide 22 is significantly α-helical (61% based on CD molar ellipticity at 222 nm) in water (Fig. 4B), confirmed by determination of its solution structure in water by 2D NMR spectroscopy (Fig. 4C, Table S9, Table S10, Table S11, Fig. S12, Fig. S13, Fig. S14, and Fig. S15). The structure shows a high degree of α-helicity for the ten residues within the lactam-bridged region, with fraying of the seven residues at the ends outside of the constrained sequence. This fraction of residues in the helix is similar to the fraction of helicity calculated from the CD spectrum. Furthermore, whereas the bacterial pheromone 21 inhibits growth of S. pneumoniae at μM concentrations (IC50 ∼ 10 μM, Fig. 4D), the helix-constrained analogue 22 has more potent antibacterial activity and abolishes S. pneumoniae growth at 10 μM (IC50 ≤ 1 μM), representing > 10-fold enhanced antibacterial activity over unconstrained 21.
RNA Binding Protein HIV-1 Rev.
A 17-residue peptide (22) HIV Rev (34–50) (23, Fig. 5A) is the component of the 116 amino acid protein HIV-1 Rev that binds to RNA (to stem IIB region of Rev Responsive Element, RRE of RNA) (23) and transports mRNA from the nucleus to the cytoplasm of an HIV-infected host cell. Blocking export into the cytosol prevents translation to the viral enzymes and structural proteins that make up new virus particles. With 13 helix-favoring residues (10 Arg, 1 Glu, 1 Ala, 1 Gln), 23 is partially α-helical in water containing 20% TFE (24) but has negligible α-helicity (< 6%) in water alone (Fig. 5B).
By contrast, when constrained by two cycle-forming lactam bridges spaced two residues apart (Fig. 5A), analogue 24 is highly α-helical (92%) in water (Fig. 5B), and our NMR-derived structure shows that α-helicity extends beyond the cyclic regions (Fig. 5C, Table S12, Table S13, Table S14, Fig. S16, Fig. S17, Fig. S18, Fig. S19, and Fig. S20). In 23, Arg5 is known to be very important for RNA binding and in 24 it had to be replaced by Lys5 to allow formation of the K → D lactam bridge. Despite this replacement, helix-constrained compound 24 retained nM affinity for RRE (Fig. 5D), almost 4- to 5-fold higher than for 23 (Fig. 5D) and 1,000-fold higher than its corresponding linear analogue Ac-TRQAKRNRDRRKRERD-NH2 (no binding at high μM concentrations). Thus, the helix-constraining strategy using different spacing between the two lactam-bridged cycles was also effective in vitro in inducing α-helicity and RNA-binding affinity for an HIV viral epitope.
ORL-1 Binding Nociceptin Protein.
We also chose to mimic a “superagonist” of the G protein-coupled receptor known as ORL-1 based on the native ligand nociceptin(1–17), a peptide hormone important in sensory perception and pain. This unstructured peptide has a putative α-helical address domain (ARKSARKLANQ) and an unstructured N-terminal message domain (FGGF), both of which may become structured as a helix and turn respectively, upon binding in or near the membrane (25, 26). In water native nociceptin(1–17) has negligible helicity (< 6%), as has a more potent agonist analogue containing a parafluoro-phenylalanine at position 4 and lysine at position 15 (25, Fig. 6). By contrast, a constrained analogue of 25, compound 26 (Fig. 6A), incorporating two lactam bridges with two residues between them, shows 45% α-helicity in water (NMR structure data in Table S15, Table S16, Table S17, Fig. S21, Fig. S22, Fig. S23, Fig. S24, and Fig. S25). This relatively low helicity for mimetic 26 versus mimetics 20, 22, and 24 is attributed to the unstructured “triggering” message domain at the N-terminus, as well as accumulation of positive charges on one helix face. Nevertheless, the induction of at least some helix structure in this peptide suggested that it should be more active than nociceptin(1–17). Function was investigated by nociceptin(1–17) induced intracellular ERK phosphorylation in mouse Neuro-2a neuroblastoma cells (Fig. 6D). There was a 9-fold improvement inagonist potency for helix-constrained 26 over unconstrained 25 (EC50 40 pM vs. 360 pM). To our knowledge compound 26 is the most potent nociceptin agonist identified to date, and could be a useful biological probe or therapeutic lead in disease indications where ORL-1 and nociceptin are involved.
Fig. 6.

GPCR-ligand nociceptin. (A) Nociceptin residues 1–17 (25) is a superagonist of ORL-1. (B) The native nociceptin(1-17) sequence had no structure in water but a recent structure in 10 mM SDS suggests R8-Q17 has α-helical propensity. (C) Cyclic mimetic 26 showed α-helicity between R8-Q17 whereas N-terminal heptapeptide had some turn-like structure. (D) Helix-constrained nociceptin analogue 26 was a 9-fold more potent ORL-1 agonist (EC50 40 pM, pEC50 ≤ 10.39 ± 0.14; orange), than unconstrained 25 (360 pM, pEC50 ≤ 9.43 ± 0.17; black) as assessed by ERK phosphorylation in mouse neuroblastoma cells Neuro-2a. Error bars represented ± S.E.M with n≥3. F(4-F) = parafluorophenylalanine.
Discussion
A solution structure is reported for the smallest known α-helix (1) in water, the first three dimensional structure for a single isolated α-helical turn with just five amino acids. The peptide backbone structure was virtually identical to an idealized α-helix. Cyclic pentapeptide 1 is stable in water and added acid, base, or trypsin. It did not unwind in strong protein denaturants (e.g., 8 M guanidine) and was not degraded in serum over 24 h. It was maximally helical with no CD spectral change caused by the helix-promoting solvent TFE. Importantly, this five-residue helical scaffold tolerated different amino acids at the three central positions and there was a linear correlation between amino acids that favored helicity in the cycle and amino acids that favor helicity in proteins. The cycle was most helical with lysine at the N-terminus, aspartate at the C- terminus, and amides at both ends.
When helical cyclic pentapeptides were coupled together, they mimic α-helical epitopes from viral, bacterial, and human proteins. Strategically fixing these epitopes in water-stable, preorganized, α-helical structures resulted in similar functions, with as high or even higher potencies/affinities, as proteins bearing the same helical sequences. Generic application of this approach to protein mimicry was shown for helical epitopes of (I) the F fusion protein of respiratory syncytial virus, with pM inhibition of viral fusion; (ii) a quorum sensing pheromone (CSP-1) of the bacterium S. pneumoniae, with inhibition of bacterial growth at sub-μM concentrations; (iii) the RNA-binding viral protein HIV-1 Rev, with nM affinity for the RNA segment Rev Responsive Element; and (iv) the human hormone nociceptin, with intracellular ERK phosphorylation in neuroblastoma cells at pM concentrations.
Based on CD spectra, none of the unconstrained peptides (19, 21,23, 25) were α-helical in water (Figs. 3B, 4B, 5B, and 6B), but under the same conditions all of the constrained peptides (20, 22, 24, 26) showed α-helicity (45–92%, Figs. 3B, 4B, 5B, and 6B). Helicity was induced by either two back-to-back cyclic pentapeptides (20, 22) or two adjacent cyclic pentapeptides separated by two amino acids (24, 26). Helix preorganization resulted in significant enhancements (9-, 10-, 1,000-, and 10,000-fold) in affinity/activity over unconstrained analogues, these functional responses being similar or greater than for the native proteins from which they were derived. Full-length HR-C peptide from RSV F protein inhibits RSV fusion with IC50 > 2.9 μM in a cell fusion assay (27), consistent with little helicity outside of the HR-N trimer. Full-length CSP-1 protein inhibits S. pneumoniae growth at only ≥10 μM (21). The 116 residue HIV-Rev protein binds to RNA with Kd approximately 1 nM (28). Nociceptin(1–17) is an agonist at only nM concentrations (25). We also found the helix-constrained compounds to be more stable in human serum than their linear analogues, the latter being typically degraded within 1 h whereas the constrained peptides were stable for > 24 h, except for the exocyclic residues, which were cleaved by proteases over several hours.
This work is a blueprint for design and utility of constrained α-helices that can substitute for α-helical protein sequences as short as five amino acids. Combinations of specific helical cyclic peptide modules led to highly helical compounds with up to 104 fold enhancements in biological activity over unconstrained peptides. Variations to 60% of the sequences within constrained regions are permitted, enabling this mimicking strategy to be applicable to a wide range of α-helical segments of proteins. The promising conformational and chemical stability suggests many diverse applications in biology as molecular probes, drugs, diagnostics, and possibly even vaccines. The constrained peptides herein offer similar binding affinity and/or function as the proteins from which they were derived, with the same amino acid sequences that confer specificity, while retaining stability and solubility akin to small molecule therapeutics. This approach of rationally linking cyclic helix modules together looks to be a promising technology suitable for rational structure-based design using native protein structures and combinatorial helix libraries.
Materials and Methods
Peptide Synthesis.
Performed as described (11, 17) by Fmoc chemistry. The phenyl isopropyl ester of aspartic acid and methyl trityl group of lysine were removed from the peptide-resin with 3% TFA in dichloromethane (DCM) (5 × 2 min). Cyclization was effected on-resin using Benzotriazole-1-yl-oxy-tris-(dimethylamino)-phosphonium hexafluorophosphate (BOP) or 1-hydroxy-7-aza-benzotriazole (HOAt), base N,N-Diisopropylethyamine (DIPEA), and DMSO/NMP (1∶1). The procedure was repeated for multiple cyclizations. Crude peptides were purified by rp-HPLC (Rt1: Vydac C18 column, 300 Å. 22 × 250 mm, 214 nm, Solvent A = 0.1% TFA in H2O, Solvent B = 0.1% TFA, 10% H2O in acetonitrile. Gradient: 0% B to 70% B over 35 min). Compounds were > 95% purity by analytical HPLC. Retention times and mass spectral data are in Table S1.
Circular Dichroism.
Measurements were made for compounds (50–400 μM, 500 μL) dissolved in 10 mM phosphate buffer (pH 7.4) in a 0.1 cm Jasco quartz cell with a Jasco J-710 spectropolarimeter (λ 185–260 nm) calibrated with (1S) - (+) - 10-camphorsulfonic acid. Peptide concentrations were determined by NMR [pulse length based concentration determination (PULCON)] (29). Fractional helicity was calculated from molar ellipticities at λ 222 nm (17).
Structure Calculations.
Peptides (2–3 mg) were dissolved in 600 μL H2O/D2O (9∶1) and the pH adjusted to 4.0–5.0. 1D and 2D (TOCSY, NOESY, dqf-COSY)  -NMR spectra were recorded on a Bruker Avance 600 spectrometer with water suppression (WATERGATE) (30) at 288–298 K. Spectra were processed using Topspin (Bruker) and NOE intensities were collected manually. 1H chemical shifts were referenced to 4,4-dimethyl-4-silapentane-1-sulfonic acid (DSS) (δ 0.00 ppm) in water.
-NMR spectra were recorded on a Bruker Avance 600 spectrometer with water suppression (WATERGATE) (30) at 288–298 K. Spectra were processed using Topspin (Bruker) and NOE intensities were collected manually. 1H chemical shifts were referenced to 4,4-dimethyl-4-silapentane-1-sulfonic acid (DSS) (δ 0.00 ppm) in water.  coupling constants were measured from 1D 1H NMR and dqf-COSY spectra. Distance restraints used in calculating solution structures were derived from NOESY spectra (mixing time 250–300 ms). Calculations were performed using XPLOR (31) with modifications to generate lactam bridges. Final structures had no distance (> 0.2 Å) or angle (> 5°) violations and were deposited with all NMR data in Biological Magnetic Resonance Bank. Accession numbers: 3z0q0z1 (compound 1), 3z0q0z2 (20), 3z0q0z3 (22), 3z0q0z4 (24), and 3z0q0z0 (26).
coupling constants were measured from 1D 1H NMR and dqf-COSY spectra. Distance restraints used in calculating solution structures were derived from NOESY spectra (mixing time 250–300 ms). Calculations were performed using XPLOR (31) with modifications to generate lactam bridges. Final structures had no distance (> 0.2 Å) or angle (> 5°) violations and were deposited with all NMR data in Biological Magnetic Resonance Bank. Accession numbers: 3z0q0z1 (compound 1), 3z0q0z2 (20), 3z0q0z3 (22), 3z0q0z4 (24), and 3z0q0z0 (26).
RSV Fusion Assay.
Performed as reported (32). Subconfluent monolayers of HEK293 cells were cotransfected with pCICO.Fopt.FL (a codon optimized full-length F gene expression plasmid driven by the CMV promoter) (33) and pGL4.80 (an expression plasmid encoding the luciferase gene under the control of the T7 promoter) using the transfection reagent Fugene-6 (Roche) with a total DNA:transfection reagent ratio of 1∶3. At 4 h posttransfection, HEK293 and BsrT7 (stable cell line constitutively expressing T7 polymerase) cells were mixed and replated into 96 well microtitre plates in the presence of mimetic. At 24 h posttransfection luminescence was measured in the presence of the Enduren substrate using the TriLux MicroBeta workstation (PerkinElmer).
S. pneumoniae Proliferation.
Cultures of S.pneumoniae were grown overnight from frozen aliquots to produce a stock solution. This was used to seed fresh cultures that were incubated at 37 °C and allowed to grow until OD590 = 0.05, at which point the compounds (or controls) were added. Growth was monitored at OD590 over 6 h.
ORL-1 Activation.
Neuro-2a cells were cultured in DMEM, 10% FBS supplemented with nonessential amino acids. Cells were plated (80,000 cells/well, 2 mL volume), in 12-well plates and allowed to adhere for 24–36 h. Prior to assay, the medium was removed and replaced with serum-free media for 12–15 h. Cells were challenged with compounds for 35 min at 37 °C. pERK and total ERK concentrations were determined using the AlphaScreen Surefire assay and the ratio of pERK to total ERK was normalized to PBS control (0%) and 1 μM Nociceptin1-17-OH (100%).
Rev RRE Competitive Binding Assay.
Adapted from a protocol previously reported (34). Briefly, 10 nM labeled Rev (suc-TRQARRNRRRRWRERQRAAAAC-(fluorescein)-NH2) and 7.5 nM RRE-biotin 5′-GGU AUG GGC GCA GCG CAA GCU GAC GGU ACA GGC C-3′-biotin (Ambion) in assay buffer (30 mM Hepes pH 7.5, 100 mM KCl, 40 mM NaCl, 10 mM ammonium acetate, 10 mM guanidine HCl, 2 mM MgCl2, 0.5 mM EDTA, 50 μg/mL Escherichia coli tRNA and 0.01% Igepal) were treated with test compounds at different concentrations. AlphaScreen® FITC Acceptor beads (10 μg, PerkinElmer) were added and the plate was incubated for 30 min. AlphaScreen® FITC Donor beads (10 μg, PerkinElmer) were added to each well and the plate incubated for 30 min and read using Envision® (PerkinElmer).
Supplementary Material
Acknowledgments.
We thank Renée Beyer for fluorescent Rev peptide and Stephen Kidd for S. pneumoniae. We thank the Australian Research Council (ARC) for Grants FF0668733, DP0210330, and DP0770936; and the National Health Medical Research Council for Grant 511194. D.P.F. is supported by an ARC Federation Fellowship, and G.R.-G. by a postdoctoral fellowship from Ministerio de Educación y Ciencia (MEC) and Fundación Española para la Ciencia y la Tecnología (FECYT) (Spain).
Footnotes
Conflict of interest statement: N.E.S. and D.P.F. are inventors on a 2005 patent application WO2005090388 owned by The University of Queensland.
This article is a PNAS Direct Submission.
Data deposition: NMR data for all 5 NMR structures have been deposited in the Biological Magnetic Resonance Bank [accession numbers 3z0q0z1 (compound 1), 3z0q0z2 (20), 3z0q0z3 (22), 3z0q0z4 (24) and 3z0q0z0 (26)].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1002498107/-/DCSupplemental.
References
- 1.Doig AJ, et al. Structure, stability and folding of the alpha-helix. Biochem Soc Symp. 2001;68:95–110. doi: 10.1042/bss0680095. [DOI] [PubMed] [Google Scholar]
- 2.Wang J, Feng JA. Exploring the sequence patterns in the alpha-helices of proteins. Protein Eng. 2003;16:799–807. doi: 10.1093/protein/gzg101. [DOI] [PubMed] [Google Scholar]
- 3.Pal L, Chakrabarti P, Basu G. Sequence and structure patterns in proteins from an analysis of the shortest helices: Implications for helix nucleation. J Mol Biol. 2003;326:273–291. doi: 10.1016/s0022-2836(02)01338-4. [DOI] [PubMed] [Google Scholar]
- 4.Scholtz JM, Baldwin RL. The mechanism of alpha-helix formation by peptides. Annu Rev Biophys Biomol Struct. 1992;21:95–118. doi: 10.1146/annurev.bb.21.060192.000523. [DOI] [PubMed] [Google Scholar]
- 5.Cabezas E, Satterthwait AC. The hydrogen bond mimic approach: Solid-phase synthesis of a peptide stabilized as an α-helix with a hydrazone link. J Am Chem Soc. 1999;121:3862–3875. [Google Scholar]
- 6.Chapman RN, Dimartino G, Arora PS. A highly stable short alpha-helix constrained by a main-chain hydrogen-bond surrogate. J Am Chem Soc. 2004;126:12252–12253. doi: 10.1021/ja0466659. [DOI] [PubMed] [Google Scholar]
- 7.Walensky LD, et al. Activation of apoptosis in vivo by a hydrocarbon-stapled BH3 helix. Science. 2004;305:1466–1470. doi: 10.1126/science.1099191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seebach D, Hook DF, Glattli A. Helices and other secondary structures of beta- and gamma-peptides. Biopolymers. 2006;84:23–37. doi: 10.1002/bip.20391. [DOI] [PubMed] [Google Scholar]
- 9.Davis JM, Tsou LK, Hamilton AD. Synthetic non-peptide mimetics of alpha-helices. Chem Soc Rev. 2007;36:326–334. doi: 10.1039/b608043j. [DOI] [PubMed] [Google Scholar]
- 10.Taylor JW. The synthesis and study of side-chain lactam-bridged peptides. Biopolymers. 2002;66:49–75. doi: 10.1002/bip.10203. [DOI] [PubMed] [Google Scholar]
- 11.Shepherd NE, Hoang HN, Abbenante G, Fairlie DP. Single turn peptide α-helices with exceptional stability in water. J Am Chem Soc. 2005;127:1974–2983. doi: 10.1021/ja0456003. [DOI] [PubMed] [Google Scholar]
- 12.Wallimann P, Kennedy RJ, Miller JS, Shalongo W, Kemp DS. Dual wavelength parametric test of two-state models for circular dichroism spectra of helical polypeptides: Anomalous dichroic properties of alanine-rich peptides. J Am Chem Soc. 2003;125:1203–20. doi: 10.1021/ja0275360. [DOI] [PubMed] [Google Scholar]
- 13.Driver RW, Hoang HN, Abbebante G, Fairlie DP. A cyclic β-strand tripeptide with an α-helix like CD spectrum. Org Lett. 2009;11:3092–3095. doi: 10.1021/ol901181b. [DOI] [PubMed] [Google Scholar]
- 14.O’Neil KT, DeGrado WF. A thermodynamic scale for the helix-forming tendencies of the commonly occurring amino acids. Science. 1990;250:646–651. doi: 10.1126/science.2237415. [DOI] [PubMed] [Google Scholar]
- 15.Chou PY, Fasman GD. Structural and functional role of leucine residues in proteins. J Mol Biol. 1973;74:263–281. doi: 10.1016/0022-2836(73)90372-0. [DOI] [PubMed] [Google Scholar]
- 16.InsightII. Accelrys Inc.; 2002. [Google Scholar]
- 17.Shepherd NE, Abbenante G, Fairlie DP. Consecutive cyclic pentapeptide modules form short α-helices that are very stable to water and denaturants. Angew Chem Int Edit. 2004;43:2687–2690. doi: 10.1002/anie.200352659. [DOI] [PubMed] [Google Scholar]
- 18.Debnath AK. Prospects and strategies for the discovery and development of small-molecule inhibitors of six-helix bundle formation in class 1 viral fusion proteins. Curr Opin Investig Drugs. 2006;7:118–127. [PubMed] [Google Scholar]
- 19.Roymans D, et al. Binding of a potent small molecule inhibitor of six-helix bundle formation requires interactions with both heptad repeats of the RSV fusion protein. Proc Natl Acad Sci USA. 2010;107:308–313. doi: 10.1073/pnas.0910108106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johnsborg O, Kristiansen PE, Blomqvist T, Havarstein LS. A hydrophobic patch in the competence-stimulating peptide, a pneumococcal competence pheromone, is essential for specificity and biological activity. J Bacteriol. 2006;188:1744–1749. doi: 10.1128/JB.188.5.1744-1749.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oggioni MR, et al. Antibacterial activity of a competence-stimulating peptide in experimental sepsis caused by Streptococcus pneumoniae. Antimicrob Agents Chemother. 2004;48:4725–4732. doi: 10.1128/AAC.48.12.4725-4732.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tan R, et al. RNA recognition by an isolated α-helix. Cell. 1993;73:1031–1040. doi: 10.1016/0092-8674(93)90280-4. [DOI] [PubMed] [Google Scholar]
- 23.Battiste JL, et al. α-helix-RNA major groove recognition in an HIV-1 rev peptide-RRE RNA complex. Science. 1996;273:1547–1551. doi: 10.1126/science.273.5281.1547. [DOI] [PubMed] [Google Scholar]
- 24.Scanlon MJ, Fairlie DP, Craik DJ, Englebretsen DR, West ML. NMR solution structure of the RNA-binding peptide from human immunodeficiency virus (type 1) Rev. Biochemistry. 1995;34:8242–8249. doi: 10.1021/bi00026a005. [DOI] [PubMed] [Google Scholar]
- 25.Meunier JC, et al. Isolation and structure of the endogenous agonist of opioid receptor-like ORL1 receptor. Nature. 1995;377:532–535. doi: 10.1038/377532a0. [DOI] [PubMed] [Google Scholar]
- 26.Okada K, et al. Highly potent nociceptin analog containing the Arg-Lys triple repeat. Biochem Biophys Res Commun. 2000;278:493–498. doi: 10.1006/bbrc.2000.3822. [DOI] [PubMed] [Google Scholar]
- 27.Wang E, et al. Both heptad repeats of human respiratory syncytial virus fusion protein are potent inhibitors of viral fusion. Biochem Biophys Res Commun. 2003;302:469–75. doi: 10.1016/s0006-291x(03)00197-9. [DOI] [PubMed] [Google Scholar]
- 28.Luedtke NW, Tor Y. Fluorescence-based methods for evaluating the RNA affinity and specificity of HIV-1 Rev-RRE inhibitors. Biopolymers. 2003;70:103–19. doi: 10.1002/bip.10428. [DOI] [PubMed] [Google Scholar]
- 29.Wider G, Dreier L. Measuring protein concentrations by NMR spectroscopy. J Am Chem Soc. 2006;128:2571–2576. doi: 10.1021/ja055336t. [DOI] [PubMed] [Google Scholar]
- 30.Liu Maili, et al. Improved WATERGATE pulse sequences for solvent suppression in NMR spectroscopy. J Magn Reson. 1998;132:124–129. [Google Scholar]
- 31.Brunger AT. Xplor 3.851. New Haven CT: Yale University; 1992. [Google Scholar]
- 32.Shepherd NE, et al. Modular α-helical mimetics with antiviral activity against respiratory syncitial virus. J Am Chem Soc. 2006;128:13284–13289. doi: 10.1021/ja064058a. [DOI] [PubMed] [Google Scholar]
- 33.Morton CJ, et al. Structural characterisation of respiratory syncytial virus fusion inhibitor escape mutants: homology model of the F protein and a syncytia formation assay. Virology. 2003;311:275–288. doi: 10.1016/s0042-6822(03)00115-6. [DOI] [PubMed] [Google Scholar]
- 34.Mills NL, Shelat AA, Guy RK. Assay optimization and screening of RNA–Protein interactions by AlphaScreen. J Biomol Screen. 2007;12:946–955. doi: 10.1177/1087057107306128. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.