

Abstract
Hormones integrate the activities of their target cells through receptor-modulated cascades of protein interactions that ultimately lead to changes in cellular function. Understanding how the cell assembles these signaling protein complexes is critically important to unraveling disease processes, and to the design of therapeutic strategies. Recent advances in live-cell imaging technologies, combined with the use of genetically encoded fluorescent proteins, now allow the assembly of these signaling protein complexes to be tracked within the organized microenvironment of the living cell. Here, we review some of the recent developments in the application of imaging techniques to measure the dynamic behavior, colocalization, and spatial relationships between proteins in living cells. Where possible, we discuss the application of these different approaches in the context of hormone regulation of nuclear receptor localization, mobility, and interactions in different subcellular compartments. We discuss measurements that define the spatial relationships and dynamics between proteins in living cells including fluorescence colocalization, fluorescence recovery after photobleaching, fluorescence correlation spectroscopy, fluorescence resonance energy transfer microscopy, and fluorescence lifetime imaging microscopy. These live-cell imaging tools provide an important complement to biochemical and structural biology studies, extending the analysis of protein-protein interactions, protein conformational changes, and the behavior of signaling molecules to their natural environment within the intact cell.
CELLS RESPOND TO hormones through signaling pathways that orchestrate the recruitment and assembly of regulatory proteins into biomolecular “machines.” These signaling pathways function in part to influence the dynamic equilibrium between assembly and disassembly of the protein machines. This may require the repositioning of physically separated machine components into common subcellular compartments to allow their assembly and function within the proper cellular environment.
Recent initiatives outlined in the National Institutes of Health (NIH) Roadmap (http://nihroadmap.nih.gov) focus on ways to solve the structures of protein machines. To achieve these goals, it will be critical to understand how the cell architecture influences the formation of these protein complexes, but the contributions of the intracellular environment typically are lost during traditional biochemical analysis. Rapid advances in live-cell imaging technologies, combined with the use of genetically encoded fluorescent proteins (FPs) now allow the assembly of protein complexes to be tracked within the organized microenvironment of the living cell. These tools provide an important complement to the biochemical studies, extending the analysis of protein-protein interactions, protein conformational changes and the behavior of signaling molecules to their natural environment within the intact cell.
The FPs have become widely used as noninvasive markers in living cells, and their successful integration into variety of living systems illustrates that the expression of these proteins in cells is well tolerated (reviewed in Ref. 1). Through the modification of existing FPs and the cloning of new color variants, the genetically encoded FPs provide protein markers that emit light from the blue to the red range of the visible spectrum (2–6). These noninvasive markers can be used in truly remarkable ways to identify and follow specific cells in living animals by repeated microscopic imaging over periods of days to weeks (7–9). In addition, these FP tags can be used in combination with a variety of different imaging methods to track the functional recruitment, colocalization, and interactions of specific protein partners within subcellular compartments in living cells. These cellular imaging methods complement the biochemical techniques that are traditionally used to define protein interactions, but importantly, extend the observations to the interactions of proteins at specific subcellular sites within the living cell (reviewed in Refs. 6 and 10–12). This review aims to provide an overview of recent developments in live-cell imaging techniques as they are applied to the analysis of the subcellular dynamics and interactions of proteins in living cells. We will use examples from studies of nuclear protein interactions in living cells to describe how these techniques are advancing the understanding of hormone regulation.
MIAGING PROTEIN BEHAVIOR IN LIVING CELLS
The study of FP-tagged proteins in living cells can be accomplished readily with currently available laser scanning confocal microscope (LSCM) or wide-field microscope (WFM) digital imaging systems. For live-cell imaging where both temporal and spatial information is gathered, the detection of the fluorescence signals must be balanced against the potential for photodamage to the sample. In this regard, the excitation light path used by both LSCM and WFM illuminates the entire specimen under the objective, so it is important to minimize the exposure of living cells and tissues to potentially damaging light. The major difference between the two systems is that the WFM collects all the emitted fluorescence from the sample, including the out-of-focus light from above and below the focal plane, whereas the LSCM uses a pinhole in the light path to prevent most of the out-of-focus signal from reaching the detector. The out-of-focus signal, which increases in thicker and more complex tissue samples, degrades the contrast in the acquired image. This limits the effectiveness of WFM to samples less than about 30 μm thick, and either LSCM or multiphoton excitation microscopy will be the better choice for thicker samples (12–15). For thin samples, however, such as cells growing in monolayer, WFM is among the most sensitive methods available (12–15). Furthermore, the image contrast can be improved by mathematically reassigning out-of-focus signal to other three-dimensional planes using image deconvolution computer algorithms (reviewed in Refs. 12 and 16–18).
Both the LCSM and WFM approaches have been used in the analysis of the subcellular location of FP-labeled nuclear receptors (NRs) and coregulatory proteins. As with all experiments using fusion proteins, it is important to verify that the addition of the 27-kDa FP does not perturb the parent protein function. Although expressed FP-fusion proteins often retain the subcellular targeting of the parent protein, the activities of the fusion protein may be affected in subtle ways, and it can be challenging to evaluate protein function. For the nuclear receptors, fusion protein function is most often assessed using response element-containing promoters linked to reporter genes, allowing the comparison of ligand-dependent activity to the parent protein. No single assay, however, can define all aspects of fusion protein activity, so experimental results obtained with the FP-labeled NRs must be interpreted carefully. When expressed in cells and imaged by fluorescence microscopy, the FP-NRs adopted the same subcellular locations identified by antibody-staining of the endogenous receptors in fixed cells (reviewed in Ref. 19). Furthermore, the live-cell imaging studies confirmed many prior biochemical studies showing that ligand binding increased the transport of some classes of NRs from the cytoplasm to the nucleus, providing a clear example of how cell signaling controls the compartmentalization of critical components of molecular machines. Other classes of NRs, including the estrogen receptor and most nonsteroid NRs, were observed to be constitutively nuclear. Again, live-cell imaging provided a new perspective on the behavior of these NRs, by showing that in most cases, ligand binding resulted in repositioning of the receptors to discrete sites within the cell nucleus (reviewed in Refs. 19–21). The functional importance of this repositioning is currently a very active area of research (22, 23).
These microscopic observations showed the apparently random overlap of the discrete sites occupied by the FP-NRs with the sites of active transcription typically observed in a cell nucleus (22). Here, however, the optical resolution of the light microscope becomes a limiting factor. The wavelength of light determines the minimal unit of optical resolution, called the point spread function (PSF). For the ideal conventional light microscope, the PSF is elliptical, about 200 nm in the x-y plane and about 600 nm in the z-axis. The best image that can be achieved is an ensemble of the individual PSFs, and increasing magnification will not change this resolution limit (18). Several strategies have been developed that overcome the limits of optical resolution, and these are discussed below. One straightforward approach is to increase the size of the target. Cell lines have been engineered that carry a multicopy transgene array inserted into a single site in the genome. The arrays often form blocks of heterochromatin that can be identified cytologically, and the association of green FP (GFP)-fusion proteins with the array can be monitored in real time (24–26). For example, an array consisting of 256 copies of the lactose operator repeat was introduced into mammalian cells and the single copy of the array was visualized by expression of a GFP-lactose repressor fusion protein (27). Building on this approach, Janicki and colleagues (28) developed a system to visualize the morphology of the integrated array, the kinetics of mRNA synthesis by the transcription unit, and the accumulation of the protein product to be visualized simultaneously. This artificial system provided information about the operation of a simple transcription unit in real time. Importantly, the use of integrated arrays containing more complex natural promoters offers the opportunity to define the temporal and spatial recruitment of a wider range of activities (26).
QUANTIFYING THE DYNAMIC BEHAVIOR OF PROTEINS
The transcriptional activities of the ligand-regulated NRs involve their interactions with chaperones, DNA, chromatin-remodeling factors, and the general transcription machinery. These interactions change the mobility of the NRs within the nuclear compartment, and optical methods, described below, have been developed that allow the dynamics of proteins to be monitored inside the living cell (10, 12, 29). The application of these methods to the study of the NRs showed that these proteins have very dynamic behavior in the living cell. These live-cell imaging studies demonstrated that ligand binding to the NRs affects their dynamic interactions, which typically are measured over time frames of seconds or less. This has led to the realization that NR action is best thought of as a series of transient interactions with chromatin and coregulatory protein complexes (reviewed in Refs. 30–32). These transient interactions were confirmed in an in vitro assay using extremely rapid (<1 μsec) cross-linking of the glucocorticoid receptor (GR) to chromatin templates with high-intensity UV laser light (33). This rapid association with DNA contrasts superficially with results obtained by the chromosomal immunoprecipitation (ChIP) approach, in which ligand-initiated cycles of association of the NRs and their coregulatory proteins with specific promoters are typically on the order of 20–40 min (31, 34–36). The ChIP method requires several minutes for the cross-linking step, and therefore represents a summation of the population of transient binding events that occur during that period (31). Thus, the ChIP technique provides a global average of the dynamic exchanges between interacting factors, and measurement of the transient binding events are best obtained by the fluorescence techniques discussed below.
Fluorescence Recovery after Photobleaching
The use of photobleaching techniques to visualize and quantify dynamic processes in cells was introduced in the 1970s (37). The strategy is straightforward: fluorescence recovery after photobleaching (FRAP) exploits the ability of LSCM to rapidly and irreversibly photobleach a small region of interest (ROI) within the cell, and then follow the recovery of fluorescence in the ROI that occurs as nonbleached proteins migrate in from adjacent regions (Fig. 1A and Table 1). Provided that the system under study is in equilibrium, the rate of influx of the nonbleached proteins allows an estimation of the mobility of the labeled protein population.
Fig. 1.
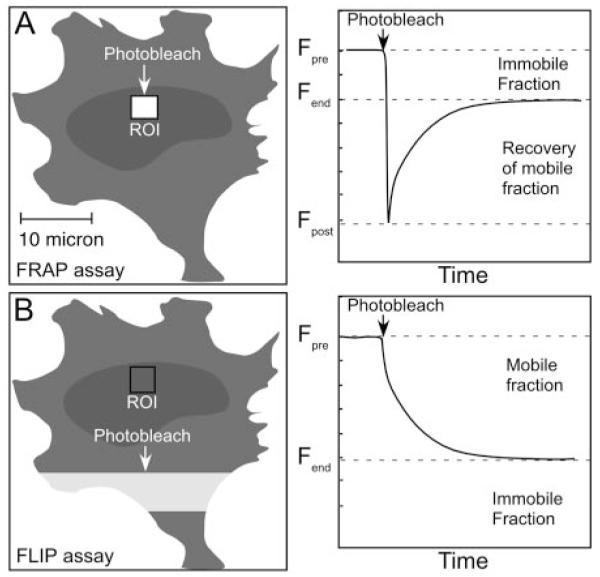
Photobleaching Analysis Reveals the Mobility of FP-Labeled Proteins
A, FRAP measures the recovery of fluorescence within a ROI after rapid photobleaching of the FP-labeled protein within that ROI. This monitors the movement of nonbleached FP-labeled protein (mobile fraction) from adjacent regions into the photobleached ROI over time. Tight binding of the bleached protein to cellular structures impedes complete recovery of fluorescence (immobile fraction). B, The FLIP assay measures the loss of fluorescence from a ROI after the continuous photobleaching of an adjacent region. The loss of fluorescence from the ROI over time defines kinetics of the mobile fraction of the FP-labeled protein. Incomplete loss of fluorescence defines the immobile fraction of FP-labeled protein that does not move into the continuously photobleached ROI. For both FRAP and FLIP, mathematical modeling of the kinetics of fluorescence recovery/loss should consider the possibility of one or more mobile and immobile fractions.
Table 1.
Methods for Imaging Molecular Dynamics and Interactions in Living Cells
| Technique | What It Measures | Advantages | Disadvantages | Refs. |
|---|---|---|---|---|
| FRAP FLIP, iFRAP | Protein mobility and compartmentalization |
Standard method on most LSCM systems |
Complex decay kinetics, photodamage artifacts |
12, 38, 39, 10, 46, 47 |
| Photoactivation | Allows user to switch on FP, molecular highlighters with high contrast |
Limited fluorophores available, complex kinetics |
29, 46 | |
| FCS | Protein mobility and complex formation |
Requires low FP concentrations, high sensitivity, spatial, temporal resolution |
Difficult to implement, complex decay kinetics, sensitive to photobleaching |
55, 56, 57 |
| FRET SBT correction |
FRET quantifies spatial relationships between fluorophores (<80Å) arising from protein interactions and protein conformation. |
Computer algorithms available, can follow specific interactions with time |
Requires careful controls to establish SBT correction. Values vary with algorithm and detection equipment. |
71–74, 78 |
| pbFRET | Easiest and most accurate way to quantify FRET |
Endpoint assay, cell movement artifacts |
81, 82 | |
| FLIM-FRET | Sensitive to different local microenvironments |
Independent of intensity; can provide most rigorous information about interactions. |
Complex, multicomponent lifetimes difficult to interpret and may be additionally affected by cell environment |
86 – 90 |
Although the methodology is well established and easily performed, the interpretation of photobleaching data can be challenging. For instance, sometimes the recovery of fluorescence is incomplete, indicating the presence of one or more immobile populations of proteins that are bound stably to structures within the photobleached region (Fig. 1A, immobile fraction). This immobile fraction prevents newly arriving unbleached proteins from binding to those structures, leading to the incomplete recovery of fluorescence. Immobile fractions also may arise from physical limits to protein diffusion imposed by subcellular compartments within the ROI. Thus, it may be necessary to account for the presence of one or more immobile, or lower mobility, fractions in the mathematical modeling of the kinetics of fluorescence recovery. In addition, it may not be possible to completely photobleach the ROI in a short time period, so there can be exchange with the unbleached compartment during the photobleach period. The FRAP analysis can be further complicated by changes in the confocal image plane during the observation period because of cell movement. Moreover, there may be additional photobleaching while monitoring the recovery phase that must be corrected for during data analysis, and there is always the potential for photodamage to light-sensitive cellular processes. The protein mobilities determined by FRAP experiments, as with other measurements discussed in this review, are best described as the average of many interactions and influences. Therefore, FRAP and other analyses should be carefully interpreted keeping in mind the complex behavior of proteins in the intact systems (10, 12, 38, 39).
Despite these limitations, FRAP has been very useful for characterizing the mobility of proteins in the cell (30, 40). FRAP analysis has revealed that many NRs, including the GR, estrogen, thyroid hormone, and retinoic acid receptors, are highly mobile within the cell nucleus (22, 26, 41–43). FRAP studies of the GR demonstrated that ligand binding reduced its mobility in the nucleus by increasing its association with other nuclear structures (26, 30, 41). Furthermore, by visualizing GR associated with multimerized promoter elements in a chromatin array stably integrated into the genome, it was possible to use FRAP to determine the mobility of other coregulatory proteins that were interacting with the GR (43–45). This approach has helped establish the timing and order of the association and dissociation of protein complexes involved in glucocorticoid and estrogen activation of gene transcription. Together, these studies illustrate how the FRAP technique can provide important insights into the functional consequences of protein dynamics in living cells.
Fluorescence Loss after Photobleaching
Other photobleaching techniques can provide information that complements the findings obtained using FRAP. The related techniques of fluorescence loss in photobleaching (FLIP) and inverse FRAP (iFRAP) are modifications of the FRAP technique that measure the loss of fluorescence from a ROI after photobleaching of an adjacent region (Fig. 1B and Table 1). The FLIP technique involves the repeated bleaching of an area within the cell to deplete fluorescent proteins that move through that area (reviewed in Refs. 10, 12, 46). The fluorescence in the ROI is monitored during the bleaching of the adjacent area, and if the regions are linked, fluorescence will decrease in the ROI as the FP moves out and into the region of continuous bleaching (Fig. 1B). Proteins that are stably associated with a cellular structure will bleach more slowly than those that are more freely mobile. Both the FRAP and FLIP methods were used to follow the exchange of GFP-labeled GR with an integrated chromatin array in the living cell nucleus, providing complementary measurements of the transient interactions of GR with the chromatin template (26).
In contrast, the iFRAP approach involves the photobleaching of almost the entire cell, except a small ROI. The loss of fluorescence from the unbleached ROI is monitored, providing a direct measurement of diffusion out of that specific region (47). The FLIP and iFRAP approaches reduce concerns about photodamage artifacts possible in FRAP experiments because the measurements are acquired from regions of cells that are not photobleached (38). Because many nuclear proteins are functionally compartmentalized within well-defined subnuclear domains, the iFRAP approach provides an excellent method to determine the kinetics of protein flux through these domains. For example, iFRAP was used to determine the dynamic properties of many different protein components that form a subnuclear domain, the Cajal body (CB) (48). Those studies demonstrated three distinct kinetic classes describing the association of 14 different protein components of the CBs, with residence times that ranged from seconds to minutes.
Photoactivation
Another approach to study the mobility of proteins within a ROI inside the living cell involves the use of photoactivatable FPs (reviewed in Refs. 29 and 46). Some of the naturally occurring FPs display a characteristic called kindling, where they are first nonfluorescent, but become brightly fluorescent in response to intense illumination (49, 50). Recently, a strongly photoactivatable mutant of GFP (PA-GFP), based on the weak kindling behavior of the wild-type jellyfish GFP (51) was developed that is activated 100-fold by a brief intense pulse of 400 nm light (52). This allows the user to “switch on” the FP in a ROI, and then follow the photoactivated protein as it diffuses away from the ROI (Fig. 2 and Table 1). Photoactivation and photobleaching techniques were recently used in combination to examine the dynamics of the p80 coilin protein, a molecular marker for CBs. Both techniques demonstrated that GFP-coilin in the nucleoplasm was constantly cycling in and out of the CB and revealed three different kinetic components, ranging from highly mobile to more stable GFP-coilin, presumably assembled into structures (48, 53, 54). The ability to use either photobleaching or photoactivation to change the fluorescent state of the marker offers great opportunity to quantify protein behavior in live cells.
Fig. 2.

Photoactivatable FP as Molecular Highlighters
PA-GFP is activated 100-fold by a brief intense pulse of 400 nm light to a restricted ROI. This allows the FP-labeled protein to be selectively activated in the ROI. The measurement of fluorescence intensity in and away from the ROI with time after photoactivation defines, respectively, the immobile fraction of the FP-labeled protein and the kinetics of the mobile protein.
Fluorescence Correlation Spectroscopy (FCS)
FCS is another optical approach that directly measures the dynamics of fluorescent-labeled molecules in living cells, but in this case without the need to photobleach the sample. FCS is essentially a photon-counting technique that monitors the fluorescence signal emanating from a very small (~1 fl) optically defined volume (Fig. 3 and Table 1). The FCS approach uses the microscope objective lens to focus the laser beam into the specimen, creating a diffraction limited excitation volume. The emitted fluorescence signal from the observation volume fluctuates as the labeled molecules diffuse in and out, and the duration of the fluctuations are related to the average time individual molecules reside within volume. This residence time can be used to determine the diffusion coefficient for the fluorescent-labeled proteins in the volume (55, 56). Importantly, the interactions of proteins change their diffusion coefficients, and the residence time of the FP-tagged protein will also reflect the size of the protein complex it is associated with. Thus, FCS monitors the average association and dissociation of proteins, which vary with interaction kinetics and with protein complex composition and size, in an optically defined volume within the living cell (Fig. 3). Here, photobleaching of the molecules must be avoided because this mimics the exit of proteins from the focal volume. The analysis of slow-moving protein complexes requires acquisition times sufficiently long to measure the process, which increases the potential for photobleaching artifacts. FCS uses lower laser powers than other techniques and requires much lower concentrations of the FPs to acquire data (55, 56). The characteristics of sensitivity and spatial and temporal resolution make FCS a valuable approach for live cell studies. However, as with all sophisticated techniques, it is important to learn the limits of the method, and understand the complexities of the data analysis (57).
Fig. 3.

FCS Measures Fluorescence Signals within a Very Small Optically Defined Observation Volume
The amount of fluorescence detected from the observation volume over time will fluctuate with the concentration, retention, and rate of diffusion of the FP-labeled protein within the observation volume (X in the diagram). Correlation functions calculated from the fluctuation data are analyzed to determine the overall diffusion coefficients and concentrations of the proteins within the specified observation volume.
Innovations on the FCS principle allow fluorescence fluctuations to be measured as a function of space instead of time (58, 59). This spatial autocorrelation yields information about the aggregation state of proteins within the cell at the time the image was acquired. Another recent innovation, in which the brightness of the fluctuating unit was measured, has been used to establish the stoichiometry of GFP-NRs in living cells (60). Such fluorescence fluctuation spectroscopy showed that the retinoid X receptor ligand binding domain (LBD) could interact as a dimer in living cells but that ligand addition shifts the equilibrium toward homodimerization, particularly at lower retinoid X receptor concentrations. Conversely, the retinoic acid receptor LBD is monomeric even in the presence of ligand, whereas the LBD of testicular factor 4, an orphan nuclear receptor, exists as a mixture of monomer and dimers. Thus, the fluorescence fluctuation spectroscopy innovation permits not only the identification of altered mobility upon ligand binding, but also of a change in the oligomeric status of the FP-labeled protein that may be associated with that change in mobility.
The techniques discussed above typically follow a single protein, which is potentially interacting with many different protein partners within the cell. Thus, it is desirable to characterize the relative mobilities of several different proteins, each labeled with a different-colored fluorescent protein. In this regard, it is possible to monitor the signals from two or even three differently labeled proteins (61). The repeated appearance of coincident signals from the different FP-labeled proteins within the optically defined observation volume provides evidence for the interactions of the proteins. Establishing whether specific proteins physically interact, not just whether a protein diffuses more slowly as a result of interaction with unknown factors, can also be inferred or measured by other live cell imaging techniques discussed below. This ability is central to tracking the formation and dissociation of specific components of molecular machines within living cells.
VISUALIZING PROTEINS THAT ARE COLOCALIZED
Multicolor imaging of two or more proteins, each labeled with a different FP, provides a powerful method to determine how cell signaling regulates the positioning of proteins in common subcellular compartments. For example, several recent studies have used multicolor imaging to characterize the organization NRs and their coregulatory proteins in distinct subnuclear compartments. Many studies have shown that the binding of agonist or antagonist ligands leads to a rapid accumulation of the NRs in specific subnuclear sites (reviewed in Ref. 20), which may be common to different members of the NR family (62). Furthermore, it was shown that agonist binding to the NRs resulted in rapid redistribution of some transcription coactivators to the same subnuclear sites (22, 42, 45, 63), possibly through direct protein-protein interactions (64–66). Studies of corepressor protein colocalization with the estrogen receptor α also showed that the agonist-bound receptor recruited corepressor proteins from matrix-associated deacetylase compartments into a microspeckled compartment via a mechanism that was dependent on receptor DNA binding activity (23). The functional importance of any of these colocalization events remains unknown, although combined imaging and extraction studies suggested that liganded NRs, and their associated cofactors, bind to some form of nuclear matrix (22). Similar mechanisms may apply to other transcription factors, where the expression of one protein shifts the steady-state location of coregulatory factors, changing their concentrations relative to target genes (67–69).
DEFINING THE SPATIAL RELATIONSHIPS BETWEEN PROTEINS IN LIVING CELLS
Detecting the colocalization of different proteins, either by multicolor fluorescence microscopy or FCS, can only demonstrate that the proteins occupy the same PSF. Because the minimum dimension of the PSF in conventional microscopy is about 200 nm, considerable distances may actually separate proteins that appear to be colocalized, so inference about protein interactions must come from other techniques. Newer optical techniques have been developed that promise severalfold improvement in the optical resolution (70), but much higher resolution is necessary to detect protein-protein interactions in living cells.
Fluorescence Resonance Energy Transfer (FRET)
FRET microscopy is one method of gaining the angstrom-scale resolution that is necessary to detect protein-protein interactions. FRET microscopy measures the direct transfer of excitation energy from a donor fluorophore attached to one protein to an acceptor fluorophore attached to an interacting protein. FRET results from the electromagnetic interactions between the donor and acceptor fluorophores, and this limits the distance over which energy transfer can occur to less than about 8 nm. Thus, the detection of FRET provides measurements of the spatial relationship of the fluorophores on the scale of angstroms (Fig. 4A and Table 1). The essential requirement for efficient transfer of energy from a donor to an acceptor FP is a substantial overlap of the donor emission and acceptor absorption spectra (reviewed in Refs. 71–74). This is also the Achilles heel of FRET measurements because the required spectral overlap leads to significant background in the FRET signal, resulting from spectral bleedthrough (SBT) noise that is contributed by both the donor and acceptor fluorophores.
Fig. 4.

FRET Microscopy Detects the Direct Transfer of Excitation Energy from a Donor Fluorophore to Acceptor Fluorophores that Is Limited to Distances of Less than about 80 Å
A, When the donor is excited (blue arrow) and energy transfer occurs (red arrow), the donor fluorescence signal is quenched and there is sensitized emission from the acceptor (green arrow). FRET signals can be quantifying by measuring sensitized emission using SBT correction methods, or by measuring donor dequenching using pbFRET, as described in the text. B, FRET also can be detected by measuring the fluorescence lifetime of the donor fluorophore population. Comparing the fluorescence lifetime of a donor alone (red curve) with the donor in the presence of an acceptor (blue curve) indicates the quenching of the donor-protein population by the interactions with the acceptor-tagged proteins. C, For populations of cells that express varying levels of donor- and acceptor-labeled proteins, the FRET signals from individual cells will be proportional to the amount of acceptor-labeled protein available, reaching a maximum when all donor-labeled protein interacts with acceptor-labeled protein (closed circles). This relationship will not be true for coexpressed, but noninteracting labeled proteins (closed squares). This relationship aids the interpretation of the biochemical and structural contributors to FRET signals measured within a subcellular compartment.
SBT Correction
The accurate measurement of FRET signals requires methods to remove the SBT background signals, and several different computer algorithms have been designed for this purpose (75–77). A comprehensive comparison of these and other correction methods was recently published (78). The common approach is to acquire reference images of control cells expressing either the donor- or the acceptor-labeled proteins alone. This information is then used to define and remove the contributions of the donor and acceptor SBT background from the FRET signal obtained from experimental cells that express both the donor and acceptor-labeled proteins. This SBT correction is specific to the collection conditions used on a particular instrument, and control measurements must be made for each experiment. Cell movement, focal plane drift, and a lack of precisely registered images used for FRET determinations are potential sources of artifacts that will appear as regions of either very high or negative FRET in energy transfer images, and must be critically evaluated (79).
Because subtraction of large SBT contributions can introduce artifacts into the estimates of the corrected FRET signals, these correction approaches will work best when the fluorescence intensities of the donor- and acceptor-labeled proteins are similar, and accurately measured above the background noise (78). In addition, energy transfer from the donor to the acceptor fluorophore is accompanied by a lowering of the amount of donor fluorescence. Therefore, accurate removal of the donor contribution to the FRET signal requires knowledge of the donor signal that is lost to energy transfer. As indicated below, methods for calculating this efficiency of energy transfer present problems for the collection of sequential images, and the different algorithms mentioned above vary in their methods for accounting for this correction.
One method amenable to live cell imaging is to subtract only the acceptor contributions to the FRET and donor fluorescence channels (80). This leaves only the donor contributions to the FRET and donor fluorescence channels and these remaining fluorescence amounts can be readily compared with that detected in cells expressing only the donor fluorophore. When energy is transferred from the donor to the acceptor, the resultant increase in fluorescence in the FRET channel is at the expense of donor channel fluorescence. This is detected as a FRET/donor intensity ratio higher than that observed from the donor control cells. These ratios are readily measured but will vary with collection and instrument parameters and are therefore impossible to compare when collected on different equipment.
Acceptor Photobleaching
Comparison of FRET results that are obtained using different imaging systems is best done by determining the efficiency of energy transfer with the technique of acceptor photobleaching FRET (pbFRET; Table 1). If the acceptor fluorophore is destroyed by photobleaching, the donor signal will be increased because its energy is no longer transferred to the acceptor. Therefore, comparing donor fluorescence intensity before and after photobleaching precisely measures the proportion of donor energy lost to FRET (81, 82). Measuring this efficiency of FRET by the photobleaching approach requires the selective bleaching of the acceptor because any bleaching of the donor fluorophore will lead to an underestimation of the donor dequenching (79, 83, 84).
Because the acceptor is irreversibly bleached, the pbFRET method cannot be repeated on the same cell, and thus represents an end-point experiment. Therefore, for live cell measurements, pbFRET cannot be used to track interactions with time. The pbFRET method, however, is valuable for verifying FRET results obtained by other methods. A recent study used the combination of pbFRET and FRAP techniques to characterize the stability of protein complexes in plant protoplast membranes (85). This technique, called FRET-FRAP, uses selective bleaching of the acceptor within a defined ROI inside the cell, and monitors the dequenching of the donor signal within that ROI. The kinetics of requenching of the donor is then measured as the bleached acceptor is exchanged for the nonbleached acceptor. This approach has the advantage of measuring FRET in two ways (donor dequenching and requenching), and has the added benefit of providing information about the stability of the protein complex being investigated.
Fluorescence Decay Measurements
Conventional fluorescence microscopy uses differences in intensity of the probes to reveal microscopic morphology and report the location of particular molecular components. Measurements of the fluorescence lifetime of a probe, the time that a probe spends in the excited state before returning to the ground state, can also provide detailed information about cellular environments and physical processes that influence the fluorophore (86, 87). Microscopic techniques measuring fluorescence lifetime, collectively referred to as fluorescence lifetime imaging microscopy (FLIM), can separate the probe excited-state lifetimes into different decay components (Fig. 4B and Table 1). Thus, where conventional microscopy detects FPs with similar fluorescence intensity distribution throughout a cell, FLIM may detect regional differences in the fluorescence lifetimes, which would indicate different local microenvironments. There are several earlier review articles that provide a comprehensive guide to methods for measuring fluorescence lifetimes (86–90).
Because the fluorescence lifetime of a fluorophore is sensitive to environmental and physical processes that influence the excited-state, the FLIM approach provides a method to detect FRET (12, 89, 90). For FLIM-FRET measurements, the presence of an acceptor will shift the mean lifetime for the donor population to shorter lifetimes because energy transfer dissipates the donor excited-state energy (Fig. 4B). These measurements can potentially provide very detailed information about molecular interactions in living cells. For instance, measuring fluorescence lifetimes of the FP-labeled protein populations within the living cell may expose several different molecular species, each with characteristic lifetimes. Repeated measurements could reveal dynamic changes in fluorescence lifetimes within the protein populations and can be used to evaluate changes in protein-protein interactions over time.
In practice, however, most of the FPs that have been characterized in living cells exhibit multiexponential fluorescence decays, and this greatly complicates the interpretation of fluorescence lifetime measurements (91–93). For example, the cyan FP (CFP) was found to exhibit different fluorescent states, which limit its utility as a donor in FLIM-FRET experiments (94, 95). For some biological applications, it may be adequate to assume that FRET measurements obtained by lifetime decay kinetics conform to a simple two-component model, which describes donor lifetimes as populations of proteins that are either quenched or unquenched by FRET (96). Here again, the effect of FRET to shorten the average lifetime of the donor population can be verified using the acceptor photobleaching method described above. The selective photobleaching of the acceptor should lead to a shift of the donor lifetime distribution to the unquenched donor population (97). One strategy to reduce the complexity of lifetime data analysis is to alter FP structure to simplify their fluorescence decays. For instance, mutations to change the structure of CFP were recently reported to stabilize the protein in a single excited-state conformation (98). This mutant CFP, as well as other similarly generated FPs may be more suitable as probes for future FLIM-FRET studies. As with the other FRET techniques described above, it is important that the user understands the inherent limits of the method and calculations used to quantify FRET signals.
FRET MEASURES PROTEIN STRUCTURE AND INTERACTION
The application of FRET-based approaches will be necessary to solve the structures of protein machines within the intracellular environment. The use of these approaches has already contributed to understanding the effects of ligand on interactions between NR dimers or between the NR and their coactivator proteins (64–66). To date, these studies have confirmed in living cells the presence of interactions surmised from prior biochemical studies. Now that these techniques have become established using known interactions, we can expect a variety of new insights that can be uniquely obtained by FRET analysis. This will require the precise understanding of the biochemical parameters that are being measured by FRET microscopy.
It is important to recognize that the detection of FRET provides information about the spatial relationship of the fluorophores labeling the target proteins. Therefore, the FRET measurements to detect the interactions of the parent proteins will be affected by where the FPs are attached to those proteins. Furthermore, there will be substantial cell-to-cell heterogeneity in the FRET measurements, which reflects the variation in the levels of the independently expressed donor- and acceptor-labeled proteins. Here, however, the variability in donor and acceptor levels can provide useful information about the nature of the interaction. For example, the detailed analysis of FRET from cells expressing varying levels of acceptor-tagged protein relative to donor-tagged protein showed that the FRET signal increased with increasing amounts of acceptor protein up to a maximum level, where all the donor-tagged proteins were paired with the acceptor-tagged proteins (Fig. 4C). The data fit to a curve defined by a mathematical model that assumes a bimolecular interaction (99). Events that affect the protein interactions shift the position of the curve, whereas events affecting the structure of the interaction complex shift the maximal amount of FRET at saturation. These types of experiments will provide critical biochemical and structural information for specific protein complexes measured with time and at specific locations within living cells. This modeling requires measurements from hundreds of cells expressing variable amounts of acceptor-labeled protein. It also assumes that the interactions occur similarly in all cells, which would not be true for events that might affect a subpopulation of the cells, such as those undergoing mitosis.
Another important FRET-based approach for characterizing changes in protein structure in living cells involves monitoring energy transfer between donor and acceptor FPs attached to different sites in the same biosensor protein. In this case, because the relative levels of donor and acceptor are fixed, FRET can be detected as the ratio of acceptor to donor fluorescence. The ratio imaging automatically corrects for the SBT background, greatly simplifying FRET measurements with the biosensor proteins. This intramolecular approach has been used with tremendous utility in the development of FRET-based biosensors designed to detect conformation changes in the biosensor in specific compartments within living cells resulting from binding of a ligand or modification by a signaling pathway (reviewed in Refs. 6, 10, 12, and 84). This intramolecular approach was recently used with dual-labeled estrogen receptors to show that ligand binding induces a very rapid intramolecular folding of that NR (100). One caveat to this approach, however, concerns the potential formation of oligomers of the dual-labeled proteins, where the contributions from both intramolecular and intermolecular FRET will be measured. Another concern is the potential for pro-teolytic cleavage of the dual-tagged protein, which will lead to separation of the FPs and potential loss of FRET signals. The intramolecular FRET approach provides the ability to coordinately measure structure and interaction with time and space in living cells and will greatly contribute to our understanding of the events accompanying sequential steps in the functioning of these molecular machines.
Despite the power of these FRET-based approaches, it is important to point out that, by themselves, they cannot prove the direct interaction of the labeled proteins. Fluorophores labeling noninteracting, but closely associated, proteins may fall within the range of FRET (<80Å). In contrast, fluorophores on interacting proteins may be separated by distances of more than 80Å and may be beyond the detection by FRET. In addition, there is the potential for the FPs themselves to form dimers, especially when fusion proteins are expressed at high concentrations in a restricted volume, such as inside a cellular organelle, or in diffusion-limited compartments, such as in the two-dimensional space of biological membranes (101, 102). The substitution of the alanine residue at position 206 of the jellyfish-based FPs with lysine blocked the potential dimer formation without changing any other characteristic of the FPs (101). Given these considerations, it is important that FRET results indicating the direct protein-protein interactions between specific protein partners are supported by those from other techniques, such as coimmunoprecipitation, epitopetagged protein pull-down, and two-hybrid assays. However, these traditional approaches are subject to potential artifacts due the nonphysiological conditions of protein extraction and analysis. Despite its limits, live-cell FRET-based imaging provides the most physiological relevant method for studying protein interactions currently available.
NUCLEAR RECEPTOR DYNAMICS AND INTERACTION
The initiatives highlighted by the NIH Roadmap illustrate the importance of combining traditional biochemistry and structural biology with fluorescence measurements of protein interaction and structure in the microenvironments within living cells. It is through the combination of these techniques that investigators are beginning to define at the molecular level how NR structure influences its protein interactions, and the dynamics of these interactions over time at specific subcellular locations in the living cell. To date, fluorescence microscopy has shown that the NRs and their associated coregulatory proteins undergo a ligand-regulated redistribution to specific subcellular compartments. FRAP and related techniques have shown the ligand to affect the dynamic association of the NRs with those compartments, and with their DNA targets, chaperones, cofactors, and components of the basal transcription machinery. FRET has shown that ligand alters NR structure and interaction and is beginning to order those events in relationship to the recruitment of factors to promoters, and these measurements will ultimately complement those obtained by in vitro methods such as ChIP. Overall, the emerging ability to measure the dynamics of protein interactions and the structures of the interacting complexes directly in living cells will enable a much better understanding of the molecular machines governing NR activity and of life itself.
Acknowledgments
We thank Cindy Booker, Ignacio Demarco, and Dr. Ty Voss for critical comments. We also thank Dr. Ammasi Periasamy in the W. M. Keck Center for Cellular Imaging at the University of Virginia for his help.
The work performed in the authors’ laboratories is funded by National Institutes of Health Grants DK 47301 (to R.N.D.) and DK 54345 (to F.S.).
Abbreviations
- CB
Cajal body
- CFP
cyan FP
- FCS
fluorescence correlation spectroscopy
- FLIM
fluorescence lifetime imaging microscopy
- FP
fluorescent protein
- FRAP
fluorescence recovery after photobleaching
- FRET
fluorescence resonance energy transfer
- GFP
green FP
- GR
glucocorticoid receptor
- iFRAP
inverse FRAP
- LBD
ligand binding domain
- LSCM
laser scanning confocal microscope
- NR
nuclear receptor
- PA-GFP
photoactivatable GFP
- PSF
point spread function
- ROI
region of interest
- pbFRET
photobleaching FRET
- SBT
spectral bleedthrough
- WFM
wide-field microscope
REFERENCES
- 1.Hadjantonakis AK, Nagy A. The color of mice: in the light of GFP-variant reporters. Histochem Cell Biol. 2001;115:49–58. doi: 10.1007/s004180000233. [DOI] [PubMed] [Google Scholar]
- 2.Karasawa S, Araki T, Nagai T, Mizuno H, Miyawaki A. Cyan-emitting and orange-emitting fluorescent proteins as a donor/acceptor pair for fluorescence resonance energy transfer. Biochem J. 2004;381:307–312. doi: 10.1042/BJ20040321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Matz MV, Lukyanov KA, Lukyanov SA. Family of the green fluorescent protein: journey to the end of the **rainbow. Bioessays. 2002;24:953–959. doi: 10.1002/bies.10154. [DOI] [PubMed] [Google Scholar]
- 4.Patterson G, Day RN, Piston D. Fluorescent protein spectra. J Cell Sci. 2001;114:837–838. doi: 10.1242/jcs.114.5.837. [DOI] [PubMed] [Google Scholar]
- 5.Shaner NC, Campbell RE, Steinbach PA, Giepmans BN, Palmer AE, Tsien RY. Improved monomeric red, orange and yellow fluorescent proteins derived from Disco-soma sp. red fluorescent protein. Nat Biotechnol. 2004;22:1567–1572. doi: 10.1038/nbt1037. [DOI] [PubMed] [Google Scholar]
- 6.Zhang J, Campbell RE, Ting AY, Tsien RY. Creating new fluorescent probes for cell biology. Nat Rev Mol Cell Biol. 2002;3:906–918. doi: 10.1038/nrm976. [DOI] [PubMed] [Google Scholar]
- 7.Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, Sanes JR. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28:41–51. doi: 10.1016/s0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- 8.Trachtenberg JT, Chen BE, Knott GW, Feng G, Sanes JR, Welker E, Svoboda K. Long-term in vivo imaging of experience-dependent synaptic plasticity in adult cortex. Nature. 2002;420:788–794. doi: 10.1038/nature01273. [DOI] [PubMed] [Google Scholar]
- 9.Walsh MK, Lichtman JW. In vivo time-lapse imaging of synaptic takeover associated with naturally occurring synapse elimination. Neuron. 2003;37:67–73. doi: 10.1016/s0896-6273(02)01142-x. [DOI] [PubMed] [Google Scholar]
- 10.Lippincott-Schwartz J, Snapp E, Kenworthy A. Studying protein dynamics in living cells. Nat Rev Mol Cell Biol. 2001;2:444–456. doi: 10.1038/35073068. [DOI] [PubMed] [Google Scholar]
- 11.van Roessel P, Brand AH. Imaging into the future: visualizing gene expression and protein interactions with fluorescent proteins. Nat Cell Biol. 2002;4:E15–E20. doi: 10.1038/ncb0102-e15. [DOI] [PubMed] [Google Scholar]
- 12.Goldman RD, Spector DL, editors. Live cell imaging: a laboratory manual. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 2005. [Google Scholar]
- 13.Andrews PD, Harper IS, Swedlow JR. To 5D and beyond: quantitative fluorescence microscopy in the post-genomic era. Traffic. 2002;3:29–36. doi: 10.1034/j.1600-0854.2002.30105.x. [DOI] [PubMed] [Google Scholar]
- 14.Swedlow JR, Platani M. Live cell imaging using wide-field microscopy and deconvolution. Cell Struct Funct. 2002;27:335–342. doi: 10.1247/csf.27.335. [DOI] [PubMed] [Google Scholar]
- 15.Gerlich D, Ellenberg J. 4D imaging to assay complex dynamics in live specimens. Nat Cell Biol. 2003:S14–S19. [PubMed] [Google Scholar]
- 16.Swedlow JR. Quantitative fluorescence microscopy and image deconvolution. Methods Cell Biol. 2003;72:349–367. doi: 10.1016/s0091-679x(03)72017-2. [DOI] [PubMed] [Google Scholar]
- 17.Swedlow JR, Hu K, Andrews PD, Roos DS, Murray JM. Measuring tubulin content in Toxoplasma gondii:a comparison of laser-scanning confocal and wide-field fluorescence microscopy. Proc Natl Acad Sci USA. 2002;99:2014–2019. doi: 10.1073/pnas.022554999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wallace W, Schaefer LH, Swedlow JR. A working-person’s guide to deconvolution in light microscopy. Bio-techniques. 2001;31:1076–1082. doi: 10.2144/01315bi01. [DOI] [PubMed] [Google Scholar]
- 19.DeFranco DB. Navigating steroid hormone receptors through the nuclear compartment. Mol Endocrinol. 2002;16:1449–1455. doi: 10.1210/mend.16.7.0880. [DOI] [PubMed] [Google Scholar]
- 20.Hager GL, Lim CS, Elbi C, Bedmann CT. Trafficking of nuclear receptors in living cells. J Steroid Biochem Mol Biol. 2000;74:249–254. doi: 10.1016/s0960-0760(00)00100-x. [DOI] [PubMed] [Google Scholar]
- 21.Hager GL, Smith CL, Fragoso G, Wolford R, Walker D, Barsony J, Htun H. Intranuclear trafficking and gene targeting by members of the steroid/nuclear receptor superfamily. J Steroid Biochem Mol Biol. 1998;65:125–132. doi: 10.1016/s0960-0760(97)00178-7. [DOI] [PubMed] [Google Scholar]
- 22.Stenoien DL, Mancini MG, Patel K, Allegretto EA, Smith CL, Mancini MA. Subnuclear trafficking of estrogen receptor-α and steroid receptor coactivator-1. Mol Endocrinol. 2000;14:518–534. doi: 10.1210/mend.14.4.0436. [DOI] [PubMed] [Google Scholar]
- 23.Voss TC, Demarco IA, Booker CF, Day RN. Corepressor subnuclear organization is regulated by estrogen receptor via a mechanism that requires the DNA-binding domain. Mol Cell Endocrinol. 2005;231:33–47. doi: 10.1016/j.mce.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 24.Belmont AS. Visualizing chromosome dynamics with GFP. Trends Cell Biol. 2001;11:250–257. doi: 10.1016/s0962-8924(01)02000-1. [DOI] [PubMed] [Google Scholar]
- 25.Gasser SM. Visualizing chromatin dynamics in inter-phase nuclei. Science. 2002;296:1412–1416. doi: 10.1126/science.1067703. [DOI] [PubMed] [Google Scholar]
- 26.McNally JG, Müller WG, Walker D, Wolford R, Hager GL. The glucocorticoid receptor: rapid exchange with regulatory sites in living cells. Science. 2000;287:1262–1265. doi: 10.1126/science.287.5456.1262. [DOI] [PubMed] [Google Scholar]
- 27.Robinett CC, Straight A, Li G, Willhelm C, Sudlow G, Murray A, Belmont AS. In vivo localization of DNA sequences and visualization of large-scale chromatin organization using lac operator/repressor recognition. J Cell Biol. 1996;135:1685–1700. doi: 10.1083/jcb.135.6.1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Janicki SM, Tsukamoto T, Salghetti SE, Tansey WP, Sachidanandam R, Prasanth KV, Ried T, Shav-Tal Y, Bertrand E, Singer RH, Spector DL. From silencing to gene expression: real-time analysis in single cells. Cell. 2004;116:683–698. doi: 10.1016/s0092-8674(04)00171-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lippincott-Schwartz J, Patterson GH. Development and use of fluorescent protein markers in living cells. Science. 2003;300:87–91. doi: 10.1126/science.1082520. [DOI] [PubMed] [Google Scholar]
- 30.Hager GL, Elbi C, Becker M. Protein dynamics in the nuclear compartment. Curr Opin Genet Dev. 2002;12:137–141. doi: 10.1016/s0959-437x(02)00278-2. [DOI] [PubMed] [Google Scholar]
- 31.Hager GL, Nagaich AK, Johnson TA, Walker DA, John S. Dynamics of nuclear receptor movement and transcription. Biochim Biophys Acta. 2004;1677:46–51. doi: 10.1016/j.bbaexp.2003.09.016. [DOI] [PubMed] [Google Scholar]
- 32.Hinojos CA, Sharp ZD, Mancini MA. Molecular dynamics and nuclear receptor function. Trends Endocrinol Metab. 2005;16:12–18. doi: 10.1016/j.tem.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 33.Nagaich AK, Walker DA, Wolford R, Hager GL. Rapid periodic binding and displacement of the glucocorticoid receptor during chromatin remodeling. Mol Cell. 2004;14:163–174. doi: 10.1016/s1097-2765(04)00178-9. [DOI] [PubMed] [Google Scholar]
- 34.Metivier R, Penot G, Hubner MR, Reid G, Brand H, Kos M, Gannon F. Estrogen receptor-α directs ordered, cyclical, and combinatorial recruitment of cofactors on a natural target promoter. Cell. 2003;115:751–763. doi: 10.1016/s0092-8674(03)00934-6. [DOI] [PubMed] [Google Scholar]
- 35.Reid G, Hubner MR, Metivier R, Brand H, Denger S, Manu D, Beeddouin J, Ellenberg J, Gannon F. Cyclic, proteasome-mediated turnover of unliganded and liganded ERα on responsive promoters is an integral feature of estrogen signaling. Mol Cell. 2003;11:695–707. doi: 10.1016/s1097-2765(03)00090-x. [DOI] [PubMed] [Google Scholar]
- 36.Shang Y, Hu X, DiRenzo J, Lazar MA, Brown M. Cofactor dynamics and sufficiency in estrogen receptorregulated transcription. Cell. 2000;103:843–852. doi: 10.1016/s0092-8674(00)00188-4. [DOI] [PubMed] [Google Scholar]
- 37.Axelrod D, Koppel DE, Schlessinger J, Elson E, Webb WW. Mobility measurement by analysis of fluorescence photobleaching recovery kinetics. Biophys J. 1976;16:1055–1069. doi: 10.1016/S0006-3495(76)85755-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Phair RD, Misteli T. Kinetic modelling approaches to in vivo imaging. Nat Rev Mol Cell Biol. 2001;2:898–907. doi: 10.1038/35103000. [DOI] [PubMed] [Google Scholar]
- 39.Weiss M. Challenges and artifacts in quantitative photobleaching experiments. Traffic. 2004;5:662–671. doi: 10.1111/j.1600-0854.2004.00215.x. [DOI] [PubMed] [Google Scholar]
- 40.Misteli T. Protein dynamics: implications for nuclear architecture and gene expression. Science. 2001;291:843–847. doi: 10.1126/science.291.5505.843. [DOI] [PubMed] [Google Scholar]
- 41.Schaaf MJ, Cidlowski JA. Molecular determinants of glucocorticoid receptor mobility in living cells: the importance of ligand affinity. Mol Cell Biol. 2003;23:1922–1934. doi: 10.1128/MCB.23.6.1922-1934.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maruvada P, Bedmann CT, Hager GL, Yen PM. Dynamic shuttling and intranuclear mobility of nuclear hormone receptors. J Biol Chem. 2003;278:12425–12432. doi: 10.1074/jbc.M202752200. [DOI] [PubMed] [Google Scholar]
- 43.Stenoien DL, Nye AC, Mancini MG, Patel K, Dutertre M, O’Malley BW, Smith CL, Belmont AS, Mancini MA. Ligand-mediated assembly and real-time cellular dynamics of estrogen receptor α-coactivator complexes in living cells. Mol Cell Biol. 2001;21:4404–4412. doi: 10.1128/MCB.21.13.4404-4412.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Becker M, Bedmann C, John S, Walker DA, Vigneron M, McNally JG, Hager GL. Dynamic behavior of transcription factors on a natural promoter in living cells. EMBO Rep. 2002;3:1188–1194. doi: 10.1093/embo-reports/kvf244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Elbi C, Walker DA, Romero G, Sullivan WP, Toft DO, Hager GL, DeFranco DB. Molecular chaperones function as steroid receptor nuclear mobility factors. Proc Natl Acad Sci USA. 2004;101:2876–2881. doi: 10.1073/pnas.0400116101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lippincott-Schwartz J, Altan-Bonnet N, Patterson GH. Photobleaching and photoactivation: following protein dynamics in living cells. Nat Cell Biol Suppl. 2003:S7–S14. [PubMed] [Google Scholar]
- 47.Dundr M, Hoffmann-Rohrer U, Hu Q, Grummt I, Rothblum LI, Phair RD, Misteli T. A kinetic framework for a mammalian RNA polymerase in vivo. Science. 2002;298:1623–1626. doi: 10.1126/science.1076164. [DOI] [PubMed] [Google Scholar]
- 48.Dundr M, Hebert MD, Karpova TS, Stanek D, Xu H, Shpargel KB, Meier UT, Neugebeder KM, Matera AG, Misteli T. In vivo kinetics of Cajal body components. J Cell Biol. 2004;164:831–842. doi: 10.1083/jcb.200311121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chudakov DM, Belousov VV, Zaraisky AG, Novoselov VV, Staroverov DB, Zorov DB, Lukyanov S, Lukyanov KA. Kindling fluorescent proteins for precise in vivo photolabeling. Nat Biotechnol. 2003;21:191–194. doi: 10.1038/nbt778. [DOI] [PubMed] [Google Scholar]
- 50.Chudakov DM, Feofanov AV, Mudrik NN, Lukyanov S, Lukyanov KA. Chromophore environment provides clue to “kindling fluorescent protein” riddle. J Biol Chem. 2003;278:7215–7219. doi: 10.1074/jbc.M211988200. [DOI] [PubMed] [Google Scholar]
- 51.Yokoe H, Meyer T. Spatial dynamics of GFP-tagged proteins investigated by local fluorescence enhancement. Nat Biotechnol. 1996;14:1252–1256. doi: 10.1038/nbt1096-1252. [DOI] [PubMed] [Google Scholar]
- 52.Patterson GH, Lippincott-Schwartz J. A photoactivatable GFP for selective photolabeling of proteins and cells. Science. 2002;297:1873–1877. doi: 10.1126/science.1074952. [DOI] [PubMed] [Google Scholar]
- 53.Handwerger KE, Murphy C, Gall JG. Steady-state dynamics of Cajal body components in the Xenopus germinal vesicle. J Cell Biol. 2003;160:495–504. doi: 10.1083/jcb.200212024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Deryusheva S, Gall JG. Dynamics of coilin in Cajal bodies of the Xenopus germinal vesicle. Proc Natl Acad Sci USA. 2004;101:4810–4814. doi: 10.1073/pnas.0401106101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Berland KM. Fluorescence correlation spectroscopy: a new tool for quantification of molecular interactions. Methods Mol Biol. 2004;261:383–398. doi: 10.1385/1-59259-762-9:383. [DOI] [PubMed] [Google Scholar]
- 56.Bacia K, Schwille P. A dynamic view of cellular processes by in vivo fluorescence edto- and cross-correlation spectroscopy. Methods. 2003;29:74–85. doi: 10.1016/s1046-2023(02)00291-8. [DOI] [PubMed] [Google Scholar]
- 57.Hess ST, Webb WW. Focal volume optics and experimental artifacts in confocal fluorescence correlation spectroscopy. Biophys J. 2002;83:2300–2317. doi: 10.1016/S0006-3495(02)73990-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wiseman PW, Brown CM, Webb DJ, Hebert B, Johnson NL, Squier JA, Ellisman MH, Horwitz AF. Spatial mapping of integrin interactions and dynamics during cell migration by image correlation microscopy. J Cell Sci. 2004;117:5521–5534. doi: 10.1242/jcs.01416. [DOI] [PubMed] [Google Scholar]
- 59.Wiseman PW, Squier JA, Ellisman MH, Wilson KR. Two-photon image correlation spectroscopy and image cross-correlation spectroscopy. J Microsc. 2000;200:14–25. doi: 10.1046/j.1365-2818.2000.00736.x. [DOI] [PubMed] [Google Scholar]
- 60.Chen Y, Wei L-N, Müller JD. Probing protein oligomerization in living cells with fluorescence fluctuation spectroscopy. Proc Natl Acad Sci USA. 2003;100:15492–15497. doi: 10.1073/pnas.2533045100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Heinze KG, Jahnz M, Schwille P. Triple-color coincidence analysis: one step further in following higher order molecular complex formation. Biophys J. 2004;86:506–516. doi: 10.1016/S0006-3495(04)74129-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ochiai I, Matsuda K, Nishi M, Ozawa H, Kawata M. Imaging analysis of subcellular correlation of androgen receptor and estrogen receptor α in single living cells using green fluorescent protein color variants. Mol Endocrinol. 2004;18:26–42. doi: 10.1210/me.2002-0262. [DOI] [PubMed] [Google Scholar]
- 63.Schedfele F, Chang C-Y, Liu W, Baxter JD, Wan Y, Nordeen S, Day RN, McDonnell DP. Temporally distinct and ligand-specific recruitment of nuclear receptor interacting peptides and co-factors to subnuclear domains containing the estrogen receptor. Mol Endocrinol. 2000;14:2024–2039. doi: 10.1210/mend.14.12.0572. [DOI] [PubMed] [Google Scholar]
- 64.Bai Y, Giguere V. Isoform-selective interactions between estrogen receptors and steroid receptor coactivators promoted by estradiol and ErbB-2 signaling in living cells. Mol Endocrinol. 2003;17:589–599. doi: 10.1210/me.2002-0351. [DOI] [PubMed] [Google Scholar]
- 65.Llopis J, Westin S, Ricote M, Wang Z, Cho CY, Kurokawa R, Mullen TM, Rose DW, Rosenfeld MG, Tsien RY, Glass CK. Ligand-dependent interactions of coactivators steroid receptor coactivator-1 and peroxisome proliferator-activated receptor binding protein with nuclear hormone receptors can be imaged in live cells and are required for transcription. Proc Natl Acad Sci USA. 2000;97:4363–4368. doi: 10.1073/pnas.97.8.4363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Weatherman RV, Chang C-Y, Clegg NJ, Carroll DC, Day RN, Baxter JD, McDonnell DP, Scanlan TS, Schedfele F. Ligand-selective interactions of estrogen receptor detected in living cells by fluorescence resonance energy transfer. Mol Endocrinol. 2002;16:487–496. doi: 10.1210/mend.16.3.0813. [DOI] [PubMed] [Google Scholar]
- 67.Francastel C, Magis W, Groudine M. Nuclear relocation of a transactivator subunit precedes target gene activation. Proc Natl Acad Sci USA. 2001;98:12120–12125. doi: 10.1073/pnas.211444898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Isogai Y, Tjian R. Targeting genes and transcription factors to segregated nuclear compartments. Curr Opin Cell Biol. 2003;15:1–8. doi: 10.1016/s0955-0674(03)00052-8. [DOI] [PubMed] [Google Scholar]
- 69.Schedfele F, Enwright JF, III, Wang X, Teoh C, Srihari R, Erickson RL, MacDougald OA, Day RN. CCAAT/enhancer binding protein α assembles essential cooperating factors in common subnuclear domains. Mol Endocrinol. 2001;15:1665–1676. doi: 10.1210/mend.15.10.0716. [DOI] [PubMed] [Google Scholar]
- 70.Martin S, Failla AV, Spori U, Cremer C, Pombo A. Measuring the size of biological nanostructures with spatially modulated illumination microscopy. Mol Biol Cell. 2004;15:2449–2455. doi: 10.1091/mbc.E04-01-0045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jares-Erijman EA, Jovin TM. FRET imaging. Nat Biotechnol. 2003;21:1387–1395. doi: 10.1038/nbt896. [DOI] [PubMed] [Google Scholar]
- 72.Periasamy A, Day RN. Visualizing protein interactions in living cells using digitized GFP imaging and FRET microscopy. Methods Cell Biol. 1999;58:293–314. doi: 10.1016/s0091-679x(08)61962-7. [DOI] [PubMed] [Google Scholar]
- 73.Pollok BA, Heim R. Using GFP in FRET-based applications. Trends Cell Biol. 1999;9:57–60. doi: 10.1016/s0962-8924(98)01434-2. [DOI] [PubMed] [Google Scholar]
- 74.Truong K, Ikura M. The use of FRET imaging microscopy to detect protein-protein interactions and protein conformational changes in vivo. Curr Opin Struct Biol. 2001;11:573–578. doi: 10.1016/s0959-440x(00)00249-9. [DOI] [PubMed] [Google Scholar]
- 75.Elangovan M, Wallrabe H, Chen Y, Day RN, Barroso M, Periasamy A. Characterization of one- and two-photon excitation fluorescence resonance energy transfer microscopy. Methods. 2003;29:58–73. doi: 10.1016/s1046-2023(02)00283-9. [DOI] [PubMed] [Google Scholar]
- 76.Gordon GW, Berry G, Liang XH, Levine B, Herman B. Quantitative fluorescence resonance energy transfer measurements using fluorescence microscopy. Biophys J. 1998;74:2702–2713. doi: 10.1016/S0006-3495(98)77976-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Xia Z, Liu Y. Reliable and global measurement of fluorescence resonance energy transfer using fluorescence microscopes. Biophys J. 2001;81:2395–2402. doi: 10.1016/S0006-3495(01)75886-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Berney C, Danuser G. FRET or no FRET: a quantitative comparison. Biophys J. 2003;84:3992–4010. doi: 10.1016/S0006-3495(03)75126-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kenworthy AK. Imaging protein-protein interactions using fluorescence resonance energy transfer microscopy. Methods. 2001;24:289–296. doi: 10.1006/meth.2001.1189. [DOI] [PubMed] [Google Scholar]
- 80.Youvan DC, Silva CM, Bylina EJ, Coleman WJ, Dilworth MR, Yang MM. Calibration of fluorescence resonance energy transfer in microscopy using genetically engineered GFP derivatives on nickel chelating beads. Biotechnology et alia. 1997;3:1–18. www.et-al.com. [Google Scholar]
- 81.Bastiaens PI, Jovin TM. Microspectroscopic imaging tracks the intracellular processing of a signal transduction protein: fluorescent-labeled protein kinase C β I. Proc Natl Acad Sci USA. 1996;93:8407–8412. doi: 10.1073/pnas.93.16.8407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bastiaens PI, Majoul IV, Verveer PJ, Soling HD, Jovin TM. Imaging the intracellular trafficking and state of the AB5 quaternary structure of cholera toxin. EMBO J. 1996;15:4246–4253. [PMC free article] [PubMed] [Google Scholar]
- 83.Day RN, Periasamy A, Schedfele F. Fluorescence resonance energy transfer microscopy of localized protein interactions in the living cell nucleus. Methods. 2001;25:4–18. doi: 10.1006/meth.2001.1211. [DOI] [PubMed] [Google Scholar]
- 84.Miyawaki A, Tsien RY. Monitoring protein conformations and interactions by fluorescence resonance energy transfer between mutants of green fluorescent protein. Methods Enzymol. 2000;327:472–500. doi: 10.1016/s0076-6879(00)27297-2. [DOI] [PubMed] [Google Scholar]
- 85.Vermeer JE, van Munster EB, Vischer NO, Gadella TWJ. Probing plasma membrane microdomains in cowpea protoplasts using lipidated GFP-fusion proteins and multimode FRET microscopy. J Microsc. 2004;214:190–200. doi: 10.1111/j.0022-2720.2004.01318.x. [DOI] [PubMed] [Google Scholar]
- 86.Clegg RM, Holub O, Gohlke C. Fluorescence **lifetime-resolved imaging: measuring lifetimes in an image. Methods Enzymol. 2003;360:509–542. doi: 10.1016/s0076-6879(03)60126-6. [DOI] [PubMed] [Google Scholar]
- 87.Dong CY, French T, So PT, Buehler C, Berland KM, Gratton E. Fluorescence-lifetime imaging techniques for microscopy. Methods Cell Biol. 2003;72:431–464. doi: 10.1016/s0091-679x(03)72021-4. [DOI] [PubMed] [Google Scholar]
- 88.Bastiaens PI, Squire A. Fluorescence lifetime imaging microscopy: spatial resolution of biochemical processes in the cell. Trends Cell Biol. 1999;9:48–52. doi: 10.1016/s0962-8924(98)01410-x. [DOI] [PubMed] [Google Scholar]
- 89.Centonze VE, Sun M, Masuda A, Gerritsen H, Herman B. Fluorescence resonance energy transfer imaging microscopy. Methods Enzymol. 2003;360:542–560. doi: 10.1016/s0076-6879(03)60127-8. [DOI] [PubMed] [Google Scholar]
- 90.Wouters FS, Bastiaens PI. Fluorescence lifetime imaging of receptor tyrosine kinase activity in cells. Current Biol. 1999;9:1127–1130. doi: 10.1016/s0960-9822(99)80484-9. [DOI] [PubMed] [Google Scholar]
- 91.Heikal AA, Hess ST, Webb WW. Multiphoton spectroscopy and excited state dynamics of enhanced green fluorescent protein (EGFP): acid-base specificity. Chem Phys. 2001;274:37–55. [Google Scholar]
- 92.Striker G, Subramaniam V, Seidel CAM, Volkmer A. Photochromicity and fluorescence lifetimes of green fluorescent protein. J Phys Chem B. 1999;103:8612–8617. [Google Scholar]
- 93.Suhling K, Siegel J, Phillips D, French PM, Leveque-Fort S, Webb SE, Davis DM. Imaging the environment of green fluorescent protein. Biophys J. 2002;83:3589–3595. doi: 10.1016/S0006-3495(02)75359-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hyun Bae J, Rubini M, Jung G, Wiegand G, Seifert MH, Azim MK, Kim JS, Zumbusch A, Holak TA, Moroder L, Huber R, Budisa N. Expansion of the genetic code enables design of a novel “gold” class of green fluorescent proteins. J Mol Biol. 2003;328:1071–1081. doi: 10.1016/s0022-2836(03)00364-4. [DOI] [PubMed] [Google Scholar]
- 95.Tramier M, Kemnitz K, Durieux C, Coppey-Moisan M. Picosecond time-resolved microspectrofluorometry in live cells exemplified by complex fluorescence dynamics of popular probes ethidium and cyan fluorescent protein. J Microsc. 2004;213:110–118. doi: 10.1111/j.1365-2818.2004.01271.x. [DOI] [PubMed] [Google Scholar]
- 96.Clayton AH, Hanley QS, Verveer PJ. Graphical representation and multicomponent analysis of single-frequency fluorescence lifetime imaging microscopy data. J Microsc. 2004;213:1–5. doi: 10.1111/j.1365-2818.2004.01265.x. [DOI] [PubMed] [Google Scholar]
- 97.Chen Y, Periasamy A. Characterization of two-photon excitation fluorescence lifetime imaging microscopy for protein localization. Microsc Res Tech. 2004;63:72–80. doi: 10.1002/jemt.10430. [DOI] [PubMed] [Google Scholar]
- 98.Rizzo MA, Springer GH, Granada B, Piston DW. An improved cyan fluorescent protein variant useful for FRET. Nat Biotechnol. 2004;22:445–449. doi: 10.1038/nbt945. [DOI] [PubMed] [Google Scholar]
- 99.Schedfele F, Wang X, Liu X, Day RN. Conformation of CCAAT/enhancer binding protein α dimers varies with intranuclear location in living cells. J Biol Chem. 2003;278:10578–10587. doi: 10.1074/jbc.M207466200. [DOI] [PubMed] [Google Scholar]
- 100.Michalides R, Griekspoor A, Balkenende A, Verwoerd D, Janssen L, Jalink K, Floore A, Velds A, van’t Veer L, Neefjes J. Tamoxifen resistance by a conformational arrest of the estrogen receptor α after PKA activation in breast cancer. Cancer Cell. 2004;5:597–605. doi: 10.1016/j.ccr.2004.05.016. [DOI] [PubMed] [Google Scholar]
- 101.Zacharias DA, Violin JD, Newton AC, Tsien RY. Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells. Science. 2002;296:913–916. doi: 10.1126/science.1068539. [DOI] [PubMed] [Google Scholar]
- 102.Kenworthy A. Peering inside lipid rafts and caveolae. Trends Biochem Sci. 2002;27:435–437. doi: 10.1016/s0968-0004(02)02178-3. [DOI] [PubMed] [Google Scholar]