

Abstract
Purpose
DNA strand breaks appear to be important in mediating radiosensitization during thymidine deprivation. This work examines the role of DNA repair, and altered thymidine analogs in altering the response to radiation during thymidine deprivation.
Methods
Mismatch repair deficient and proficient cell lines HEC59 and HC-2.4 were treated with FUdR, AZT and irradiation either alone or in combination and outcomes of clonogenic survival and cell cycle distributions were determined.
Results
Survival outcomes for all treatments were similar for both cell lines, suggesting hMSH2 does not significantly influence thymidine deprivation toxicity or radiosensitiation. The chain terminating thymidine analog azidothymidine (AZT) increased the toxicity of FUdR and increased DNA fragmentation. The combination of FUDR and AZT afforded greater radiosensitization than either drug alone. Drug enhancement ratios, the degree of excess radiation induced cell death in drug treated cultures compared to radiation alone for HEC59 was 1.2, 1.4 and 1.8 for AZT, FUdR and the combination. Enhancement ratios for HC-2.4 were 1.3, 1.5 and 1.8 for AZT, FUdR and the combination.
Conclusion
AZT, a chain terminating thymidine analog, can enhance the radiosensitizing affects of thymidine deprivation. DNA strand breaks may play an important role in the mechanism of thymidine deprivation induced radiosensitization.
Keywords: Radiosensitization, fluorodeoxyuridine, azidothymidine, mismatch repair, thymidine deprivation
Introduction
Thymidine deprivation is cytotoxic to all actively growing cells and enhances the toxicity of ionizing radiation. Thymidine deprivation is the basis of chemoradiotherapy treatments for several cancer types. Chemotherapeutic agents which are thought to act at least in part by depleting thymidine include the fluoropyrimidines fluorouracil and fluorodeoxyuridine (FUdR), capecitabine, gemcitabine, as well as methotrexate and pemetrexed. The exact mechanism responsible for cytotoxicity and radiosensitization during thymidine deprivation remains elusive.
One event thought to occur during thymidine deprivation is incorporation of dUTP into DNA, essentially using the uracil nucleotide as a thymidine analog (1). Indeed, most DNA polymerases have poor ability to distinguish between dUTP and dTTP. dTMP is synthesized from dUMP, therefore agents that block the conversion of dUMP to dTMP simultaneously deplete thymidylate and increase dUMP and ultimately dUTP. This favors dUTP incorporation into DNA. Once integrated into DNA, uracil becomes a target for base excision repair (2). This repair proceeds as uracil glycosylase cleaves the glycosidic bond between uracil and the deoxyribose, leaving the phosphodiester backbone of the DNA helix intact, but with a highly reactive abasic site in place of the uracil. Apurinic/apyrimidinic (AP) endonuclease cleaves the DNA strand containing the abasic site to create a single strand nick. The strand containing the nick is displaced by the action of DNA polymerase. The displaced strand is cleaved by a FLAP endonuclease such as Rad27 in yeast. DNA polymerase and DNA ligase can then act to synthesize and seal the affected strand. Several of these repair steps include cleaving bonds in DNA that could potentially lead to greater and more threatening damage than that posed by the original errant dUTP.
The toxicity of ionizing radiation is thought to be due, at least in part, to DNA nicks and breaks. The DNA breaks occurring during repair of uracil and/or other bases from DNA during thymidine deprivation may combine with the damage imparted from ionizing radiation to create synergistic toxicity. Inhibition of uracil glycosylase in glioma cells decreased the degree of radiosensitization imparted by thymidine deprivation (3), suggesting the activity of attempted repair contributes to radiosensitization toxicity. In addition, S. cerevisiae cells lacking DNA strand incision activities for repair of uracil and oxidatively damaged bases from DNA showed little radiosensitization during thymidine deprivation (4). If repair mediated DNA breaks are responsible for some of the combined toxicity of thymidine deprivation and radiation, then maneuvers to further increase strand breaks during thymidine deprivation should also increase cytotoxicity and radiosensitization.
Depletion of thymidine pools favors incorporation of thymdine analogs other than dUTP if they are available. An example is iododeoxyuridine, which can be incorporated into DNA of replicating cells. If cells are treated with FUdR first, to deplete thymidine pools, the relative incorporation of iododeoxyuridine increases (5). One possible means to increase the radiosensitizing effects of thymidine deprivation may be to increase the number of strand breaks using a chain terminating thymidine analog. This hypothesis was tested by using azidothymidine (AZT), a thymidine analog that lacks the 3′ hydroxyl group necessary for chain elongation, in essence creating a strand break at each site of AZT incorporation (6). Combining thymidine deprivation with AZT indeed enhanced radiosensitization. The role of hMsh2 mediated DNA mismatch repair in contributing to the toxicity of thymidine deprivation and radiosensitization was also examined.
Methods
Cell lines
HEC59 is a human endometrial cancer cell line kindly provided by Dr. Thomas Kunkel. HEC59 lacks a functional allele of hMSH2. HC-2.4 is derived from HEC59 by transfer of an entire human chromosome 2 (containing a wild-type hMSH2 allele) into HEC59 (7). HC-2.4 has lower mutational frequency, consistent with correction of the DNA repair deficiency in HEC59. The transferred chromosome 2 also contains a gene for neomycin resistance, allowing for selection. HC-2.4 was maintained under neomycin selection, but selection was removed for drug exposure and clonogenic survival studies. Cells were grown in DMEM/F-12 supplemented with 10% defined FBS, penicillin and streptomycin. Cells were incubated in 95% air and 5% CO2 in a humidified incubator at 37 °C.
Drug Treatment
Logarithmically growing cultures of HEC59 and/or HC-2.4 were used for these studies. Cells were harvested from plates, counted and replated into fresh plates and fresh media at 106 cells per ml. Cells were allowed to attach for 24 hours. For FUdR exposure, freshly prepared FUdR in PBS was added to cells 24 hours after plating. For combination treatment with azidothymidine (AZT), freshly prepared AZT was added following 24 hours of FUdR exposure. Control cultures with AZT only were treated in parallel and received AZT 48 hours after plating.
Clonogenic survival
Colony formation assay was performed by harvesting exponentially growing cells, plating them to fresh medium and allowing them to attach for 24 hours. Drug treatment with FUdR was then initiated at a concentration of 30 micromolar unless otherwise stated. AZT treatment, with or without FUdR, was initiated 48 hours after plating, at a concentration of 1 millimolar unless otherwise stated. After the defined exposure time, all cells (attached and non-attached) were harvested, counted using a Coulter cell counter (Beckman Coulter, Inc., Fullerton, CA, USA) and replated to fresh drug-free medium and allowed to grow for 21 days. Plates were then stained with Coomassie stain and colonies consisting of greater than 50 cells were counted.
Irradiation
For the radiation experiments, cells were exposed to ionizing radiation (0–10 Gy) using a Pantak high-frequency 22-kV and 10-mA X-ray generator.
Flow Cytometry
Approximately one million cells were collected. All treated cells were collected by centrifugation, media was aspirated and the pellet was resuspended in 100 microliters PBS. Three mls of −20°C 70% EtOH (ethanol) was added and incubated for 1 hour at 4°C. Cells were then washed twice with 2mls PBS and resuspended in 100 microliters PBS. The cells were then treated with RNase A at a concentration of 0.5 mg/ml. An equal volume of a 100μg/ml propidium iodide (PI) solution was added to the cell suspension and incubated 4°C for 30 minutes in the dark. The cells were then analyzed on a Becton Dickson FACscan flow cytometer. To determine the relative distribution of cells in various phases of the cell cycle, the relative fluorescence intensities corresponding to cells in G1, S and G2 were determined using the untreated control. The number of cells within the respective range of PI intensities was determined and divided by the total number of cells counted.
Analysis
We used Poisson regression to model the observed number of colonies as a multivariate function of the predictor variables: hec59, dose and treatment, given the number of plated cells. A separate intercept was included in the model for each radiation experiment to account for the variability from experiment to experiment. Statistically significant global tests of equality across treatments were followed up with pairwise comparisons to identify specific treatment differences. Linear-quadratic approach was used to characterize survival following radiation with and without sensitizers (8) Coefficients alpha and beta were determined corresponding to linear and quadratic portions of the survival curves respectively.
Results
The mismatch repair deficient endometrial cancer cell line HEC59 lacks hMSH2 function and a derivative cell line, HC-2.4, contains an extra chromosome 2 bearing a wild-type allele of hMSH2 able to restore repair function. These cell lines were treated with FUdR to deplete thymidine and subsequent survival was determined using colony formation assays as shown in figure 2A. Survival was similar for both cell lines. In order to examine the additive toxicity of additional treatments, we limited further experiments to an FUdR concentration of 30 micromolar. At this concentration, HEC59 and HC-2.4 had similar FUdR sensitivities. These data suggest hMsh2 mediated repair does not contribute to the toxicity of FUdR treatment.
Figure 2.

Drug cytotoxicity dose response. A) Cells were plated and allowed to adhere for 24 hours then treated for 48 hours with varying doses of FUdR or control as described in Methods. After treatment, all cells were collected, washed, counted and replated to determine survival. B) Similar treatments were conducted with varying doses of AZT.
Azidothymidine, AZT, is a thymidine analog which is imported and phosphorylated by thymidine kinase and can subsequently be used as a thymidine analog during DNA synthesis. Since AZT does not contain a 3′ hydroxyl group, it cannot serve as a substrate for chain elongation. Therefore, further DNA synthesis on that chain is interrupted, resulting in a single strand break. Cellular sensitivity to AZT as determined by clonogenic survival (Figure 2B) was similar for HEC59 and HC-2.4. A modest sensitivity was seen at 1 mM in both strains. This is a concentration that can be achieved in human serum in clinically relevant oral doses. Since both cell lines showed similar sensitivities, hMsh2 does not apparently contribute to the toxicity of AZT.
When endogenous levels of thymidine are low, thymidine analogs may be used for DNA synthesis in its place. For example, dUTP is thought to be incorporated into DNA during thymidine deprivation. AZT acting as a thymidine analog for DNA synthesis during thymidine deprivation could also act to introduce strand breaks after being inserted into the nascent DNA strand. To examine this possibility, HEC59 and HC-2.4 lines were treated with FUdR for 24 hours at 30 micromolar FUdR. AZT was then added to a concentration of 1 mM and cells were incubated in both drugs for an additional 24 hours. Therefore, treatment consisted of FUdR for 48 hours with AZT also present for the final 24 hours. When the two treatments are given concurrently, the toxicity is additive (Figure 3).
Figure 3.

Combined treatment increases toxicity. Cells were treated with FUdR (30 micromolar), AZT (1 mM) or the combination as outlined in Figure 1 and in Methods. Clonogenic survival was determined.
Flow cytometry for DNA content was used to further characterize the response of these cell lines to drug treatment. As expected, FUdR treatment resulted in cell cycle delay with an increase in accumulation of cells in G1 and S. AZT treatment alone increased the proportion of cells in S phase, again consistent with an S-phase replication block. Combining FUdR and AZT resulted in a significant decrease in cells in G2, an increase in cells in S and a notable increase in the proportion of cells with subG1 content of DNA. This is consistent with AZT acting as a block to DNA synthesis and increasing the proportion of cells with fragmented DNA (Figure 4 and Table 1).
Figure 4.

Cell cycle changes resulting from drug treatment and recovery. HEC59 cells were exposed to 30 micromolar FUdR, 1 mM AZT or a combination. Samples of cells were collected after 48 hours of FUdR treatment alone, 24 hours of AZT treatment alone and concurrent treatment as shown in figure 1. Control cultures not treated with drug were also analyzed. Cells were collected, fixed and stained with propidium iodide as described in methods. HEC59+2 cultures were analyzed also with similar results. The scale for x- and y-axes are constant for all conditions shown. Parallel experiments in HEC59+2 gave similar results.
Table 1.
Drug treatment and subsequent outgrowth results in DNA fragmentation and changes in cell cycle distribution. Cells were treated with drugs as shown in Figure 1 (30 micromolar FUdR for 48 hours, 1 mM AZT for 24 hours or combined treatment with 30 micromolar FUdR for 48 hours and 1 mM AZT delivered during the last 24 hours of FUdR), collected, fixed in ethanol, stained with propidium iodide as described in methods and analyzed by flow cytometry. Untreated control was used as a reference to determine proportion of cells in various stages of the cell cycle.
Changes in cell cycle distribution at different times following various drug treatments. Thirty micromolar FUdR (F) and 1 mM AZT (A) were used as outlined in Figure 1. Cells scored with greater than G2 content comprise the remainder to give 100% for each condition.
| HEC59 flow cytometry averages | ||||
|---|---|---|---|---|
| HEC59 | ||||
| %subG1 | %G1 | %S | %G2 | |
| Con | 6 | 30 | 13 | 21 |
| F 24 | 8 | 29 | 21 | 16 |
| F 48 | 11 | 34 | 22 | 12 |
| F 72 | 32 | 21 | 19 | 15 |
| A 48 | 10 | 25 | 24 | 12 |
| A 72 | 10 | 29 | 16 | 21 |
| F+ A 48 | 36 | 26 | 17 | 9 |
| F+A 72 | 45 | 17 | 17 | 14 |
| HC-2.4 flow averages | ||||
|---|---|---|---|---|
| HC-2.4 | ||||
| %subG1 | G1 | S | G2 | |
| Con | 3 | 35 | 13 | 28 |
| f 24 | 3 | 32 | 21 | 20 |
| F48 | 4 | 38 | 23 | 15 |
| F72 | 23 | 26 | 20 | 16 |
| A 48 | 9 | 29 | 18 | 17 |
| A 72 | 16 | 28 | 16 | 19 |
| F+A 48 | 13 | 31 | 22 | 17 |
| F+A 72 | 18 | 29 | 17 | 17 |
Previous studies in our laboratory examining the toxicity of thymidine deprivation in a variety of DNA repair mutants of S. cerevisiae suggested that a significant amount of cell killing occurs after release from thymidine deprivation rather than during the time of thymidine depletion itself (2). To examine this possibility in mammalian cancer cells, HEC59 and HC-2.4 were treated with drug(s) as described above. Media was removed after drug exposure and replaced with drug free media. Cells were then incubated for an additional 24 hours, then collected and analyzed by flow cytometry for DNA content. Consistent with our findings in S. cerevisiae, a significant increase in cells containing subG1 content of DNA is seen in cells treated with FUdR. The proportion of cells with subG1 content of DNA is even higher after a 24 outgrowth in cells that had been treated with both FUdR and AZT. A representative experiment examining DNA content in treated HEC59 cells is shown in figure 4. Similar results were found for HC-2.4 cells. These findings suggest that a significant amount of DNA damage from thymidine deprivation occurs during attempted recovery from thymidine depletion. The flow cytometry data confirm this combination of thymidine analogs produces greater DNA damage, suggesting the potential for greater radiosensitization.
Both cell lines were examined for their sensitivity to radiation using the clonogenic survival assay. As has been reported previously (9), HEC59 and HC-2.4 have similar sensitivities to ionizing radiation (Figure 5).
Figure 5.

Radiation sensitivity of HEC59 and HEC59+2 cells is comparable. Cells were plated and allowed to attach for 24 hours. Plates were then exposed to varying doses of radiation as described in Methods. Plates were stained and colonies counted after 14 days.
Treatment with either FUdR or AZT prior to exposure to ionizing radiation increases the sensitivity of both HEC59 and HC-2.4 to radiation (Figure 6). Pretreatment with drug was similar to conditions used to examine the sensitivity of these lines to drug only (figure 1). Cells were treated with FUdR for 48 hours prior to irradiation or 24 hours with AZT prior to irradiation or a combination treatment consisting of FUdR only for 24 hours followed by combined AZT + FUdR for an additional 24 hours. Following drug exposure, cells were irradiated to varying doses and subjected to a clonogenic survival assay. Pretreatment with either drug sensitizes cells to ionizing radiation, whereas pretreatment with both drugs significantly increases sensitivity to radiation and enhances killing (Table 2). Survival curves were analyzed to determine alpha and beta coefficients corresponding to linear and quadratic portions of the survival curves (8). The changes engendered by drug treatment to alpha and beta values are also shown (Table 3). These findings are consistent with the hypothesis that DNA strand breaks contribute to the toxicity of ionizing radiation exposure during thymidine deprivation.
Figure 6.

Radiosensitization by thymidine analogs. A) Cells were treated with 30 micromolar FUdR for 48hours, irradiated. Treated cells were then collected, counted and replated. Relative survival was determined compared to no drug and plotted as shown. B) Cells were treated with 1 mM AZT for 24 hours then irradiated and treated as described for A. C) Cells were treated with 30 micromolar FUdR for 48 hours and 1 mM AZT concurrently during the second 24 hour period. Cells were treated as described above.
Figure 1.
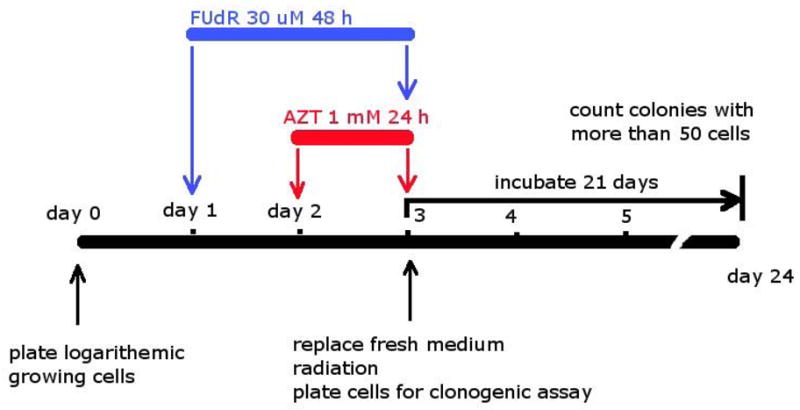
Diagram of study design and treatment delivery.
Table 2.
Thymidine analog treatment sensitizes cells to irradiation. Drug exposures are as described in Figure 1. The enhancement ratio was calculated as described in methods.
Drug Enhancement Ratios for radiation cytotoxicity, calculated from curve-fit data at 10% survival comparing drug treated irradiated cells to irradiated cells only. Conditions for drug treatment are shown in Figure 1. Cells were treated with 30 micromolar FUdR for 48 hours or 1mM AZT for 24 hours or combined FUdR with AZT during the last 24 hours prior to radiation exposure.
| HEC59 | HC-2.4 | |
|---|---|---|
| AZT | 1.2 | 1.3 |
| FUdR | 1.4 | 1.5 |
| FUdR + AZT | 1.8 | 1.8 |
Table 3.
Values for alpha and beta derived by examining survival curves for various drug + radiation combinations. Values were determined as described in methods and (8).
Alpha and Beta values derived from radiation survival curves. Drug and radiation conditions used are shown in Figure 1.
| HEC59 | ||
|---|---|---|
| Group | Alpha | Beta |
| Control | 0.062 | 0.124 |
| AZT | 0.52 | 0.048 |
| FUdR | 0.55 | 0.069 |
| AZT + FUdR | 0.40 | 0.25 |
| HC-2.4 | ||
|---|---|---|
| Group | Alpha | Beta |
| Control | 0.12 | 0.14 |
| AZT | 0.58 | 0.065 |
| FUdR | 0.61 | 0.086 |
| AZT + FUdR | 0.46 | 0.27 |
Discussion
This study examines the possibility of increasing the toxicity and radiosensitization of thymidine deprivation through combining FUdR and azidothymidine (AZT). Concurrent AZT and FUdR treatment provides at least an additive increase in cytotoxicity and radiosensitization. The increase in toxicity implicates DNA strand breaks as an important component of the mechanism of toxicity and radiosensitization during thymidine deprivation. Furthermore, characterization of DNA content during drug treatment suggests that AZT indeed contributes to greater DNA fragmentation during and immediately following thymidine deprivation.
A myriad of cellular events occur during thymidine deprivation. Understanding which of these events contribute to the increase in radiosensitivity seen in thymidine deficient cells is important to further improve the efficacy and selective advantage of this treatment. DNA strand incision activities in base excision repair involved with the removal of uracil from DNA partially contribute to thymidine deprivation mediated radiosensitization in the yeast S. cerevisiae (4). In addition, base excision repair enzymes acting to remove oxidatively damaged bases also contribute to radiosensitization. S. cerevisiae cells lacking the major glycosylase enzymes responsible for removing oxidatively damaged bases (Ntg1 and Ntg2) showed a reduced level of radiosensitization (4). Cells lacking the enzyme responsible for strand incision during uracil base excision repair (Apn1) also showed reduced radiosensitization. Cells lacking Ntg1, Ntg2 and Apn1 showed no increase in radiation sensitivity during thymidine deprivation, suggesting a major component of radiosensitization during thymidine deprivation occurs due to repair- mediated DNA strand breaks. Similar findings were seen in a pair human glioma cell lines differing only in expression of a protein inhibitor of uracil glycosylase (3). The cell line expressing the inhibitor, and therefore producing less repair mediated breaks, showed reduced radiosensitization during thymidine deprivation. The finding reported here showing AZT increases radiosensitization during thymidine deprivation is consistent with our findings in yeast and supports the model that DNA strand breaks are an important mediator of radiosensitization. In both the yeast and glioma models described above, the toxicity of thymidine deprivation alone and the toxicity of thymidine deprivation combined with radiation respond differently to the alterations in DNA repair activity, suggesting thymidine deprivation and radiosensitization create toxicity by distinct pathways.
When cells die from thymidine deprivation may provide additional clues regarding the nature of thymidine deprivation. Previous work in S. cerevisiae using a vital stain suggests that cells depleted of thymidine undergo cell cycle arrest but remain metabolically active during drug exposure. However, once drug is removed and the cells are returned to nutrient replete growth medium, cells undergo increased DNA fragmentation and lose the ability to metabolize vital dye (2). The findings in yeast suggest cytotoxicity occurs as cells attempt recovery from thymidine depletion. Furthermore, mutants deficient in uracil base excision at the apyrimidinic/apurinic endonuclease (apn1) step are extraordinarily sensitive to thymidine deprivation and show an essentially complete inability to recover from the cell cycle arrest induced by thymidine deprivation. HEC59 and HC-2.4 cells also show cell cycle arrest during thymidine deprivation. The largest increase in cells containing fragmented DNA, as evidenced by sub-G1 content of DNA, occurs after removal of FUdR. This agrees well with previous findings in yeast and again suggests that it is the return to growth and division that poses the greatest threat to thymidine deprived cells. Adding radiotherapy to cells treated with FUdR and AZT may act to increase the burden of DNA damage, further aggravating the problem of completing DNA repair and cell cycle recovery.
Data presented here suggest AZT increases DNA fragmentation during thymidine deprivation. The most straightforward interpretation is that AZT is incorporated into DNA as a thymidine analog when cellular thymidine pools are low. Incorporation of other thymidine analogs in addition to dUTP has been described by others. For example, the incorporation of iodouracil into DNA is significantly increased during thymidine deprivation (5). Other mechanisms may contribute to the combination of AZT to FUdR. AZT has recently been shown to impart mitochondrial damage (12), with resultant mitochondrial dysfunction and oxidative stress contributing to long-term AZT toxicity. Both mitochondrial DNA and other targets appear to be important for the mitochondrial toxicity of AZT. It is possible that mitochondrial events are also contributing to the toxicity of AZT + FUdR. Indeed, the toxicity of thymidine deprivation induced by 5-fluorouracil alone can be abrogated by a mitochondrially directed anti-oxidant (13), supporting the potential role of mitochondrial oxidative stress induced by AZT as a possible mechanism for combined toxicity.
Base excision repair of uracil from DNA clearly plays a major role in the cytotoxicity and radiosensitization of thymidine deprivation. Similar strand cleavage events occur during mismatch repair pathways, including msh2. HNPCC tumors arising in individuals with defects in mismatch repair may respond differently to thymidine deprivation based chemotherapy (10). Meyers, et al. (11) examined the sensitivity of MSH2 deficient and matched repair proficient cell lines to FUdR. They found HEC59 was more resistant to thymidine deprivation induced by protracted exposure than HC-2.4 using a growth inhibition assay. Shorter exposure times similar to those used in our study resulted in virtually no difference in FUdR sensitivity between repair proficient and repair deficient cells. Prolonged exposure to tomudex, a thymidylate synthase inhibitor, was more toxic to HEC59 (msh2 mutant) cells, consistent with our findings. Meyers, et al. also showed the Msh2-Msh6 enzyme complex was active with fluorouracil:guanine as a substrate. The increased sensitivity of HC-2.4 seen in their system with protracted exposure may therefore be due to incorporation of FUdR and subsequent Msh2 mediated repair occurring during prolonged exposure but not during brief FUdR exposures. Although we did not test protracted exposure in our system, their observations with repair mediated breaks increasing toxicity are consistent with our observation of AZT induced breaks increasing toxicity.
AZT and 5FU has been shown by others to increase cytotoxicity and DNA damage. Andreuccetti et. al. (6) described a synergistic interaction with this combination in cultured colon cancer cell lines. 5FU and AZT given concurrently also create greater DNA damage in nucleated blood cells of colon cancer patients (14). AZT also increases the toxicity of methotrexate in a methotrexate resistant cell line (15). AZT also enhances the toxicity of 5 fluoro 5 deoxyuridine (DFUR) presumably through changes in thymidine phosphorylase activitiy (16). AZT is a radiation sensitizer. It has been shown to increase the radiosensitivity of a head and neck cancer line HEp-2 (17). These authors have suggested the radiosensitization of AZT may result from an inhibition of telomerase after radiation exposure. Others have also seen an increase in the effects of radiation with AZT (18), although the extent of radiosensitization is less than that from FUdR. AZT and methotrexate has also been combined clinically in HIV related non-Hodgkins lymphoma. Tosi et. al. (19) found the combination of AZT and methotrexate to be tolerable and had activity against lymphoma with a 77% response rate. No clinical study to our knowledge has examined the safety or efficacy of the combination of thymidine deprivation with AZT and radiotherapy. The experiments reported here were limited to cancer cell lines, but similar processes may occur in normal cells. Greater normal tissue toxicity during combined fluoropyrimidine and radiotherapy in patients also receiving AZT may occur. Similarly, combinations of other dideoxythymidine analogs with agents that produce thymidine deficiency could potentially lead to greater normal tissue toxicity. 5-FU based chemotherapy and radiotherapy in HIV positive patients treated for anal cancer did not appear to create excess toxicity (20). More recently, Seo, et al. (21), examined the clinical outcome in 36 immunocompetent and immumocomprimised patients treated for anal cancer. Ten patients in their cohort received highly active antiretroviral therapy (HAART). Although the agents comprising HAART for these subjects were not specified, thymidine based reverse transcriptase are common components. No additional toxicity was found in subjects treated with HAART. (21). Additional clinical data is needed. The data presented in this report offer insight into the mechanism of thymidine deprivation mediated radiosensitization and also provide a rationale for clinical studies involving all three agents.
Acknowledgments
The authors deeply appreciate the skillful and kind assistance from the University of Iowa Flow Cytometry Core. The Free Radical and Radiation Biology faculty and staff were instrumental in the conduct and interpretation of this work. This research was supported by the University of Iowa Holden Comprehensive Cancer Center and the National Cancer Institute (7K08CA111404-02). A portion of this work was presented at the 2005 Annual ASRO meeting in Denver, Colorado
Footnotes
Conflict of Interest Notification
The authors have no conflicts to report.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ingraham HA, Dickey L, Goulian M. DNA fragmentation and cytotoxicity from increased cellular deoxyuridylate. Biochemistry. 1986;25:3225–3230. doi: 10.1021/bi00359a022. [DOI] [PubMed] [Google Scholar]
- 2.Dornfeld KJ, Johnson M. AP endonuclease deficiency results in extreme sensitivity to thymidine deprivation. Nucleic Acids Research. 2005;33:6644–6653. doi: 10.1093/nar/gki975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Radany ER, Dornfeld KJ. Distinct Roles of Uracil-DNA Base Excision Repair in Fluoropyrimidine-Mediated Radiosensitization and Direct Cytotoxicity in Human Tumor Cells. International Journal of Radiation Oncology, Biology and Physics. 2005 October 1;63:S482–S483. [Google Scholar]
- 4.Allen B, Johnson M, Marsh A, Dornfeld KJ. Base excision repair of both uracil and oxidatively damaged bases contribute to thymidine deprivation induced radiosensitization. International Journal of Radiation Oncology, Biology, Physics. 2006;65:1544–1552. doi: 10.1016/j.ijrobp.2006.03.051. [DOI] [PubMed] [Google Scholar]
- 5.Perillo-Adamer F, Delaloye AB, Genton CS, Schaffland AO, Dupertuis YM, Buchegger F. Short fluorodeoxyuridine exposure of different human glioblastoma lines induces high-level accumulation of S-phase cells that avidly incorporate 125I-iododeoxyuridine. Eur J Nucl Med Mol Imaging. 2006 May;33(5):613–20. doi: 10.1007/s00259-005-0009-y. [DOI] [PubMed] [Google Scholar]
- 6.Andreuccetti M, Allegrini G, Antonuzzo A, Malvaldi G, Conte PF, Danesi R, Del Tacca M, Falcone A. Azidothymidine in combination with 5-fluorouracil in human colorectal cell lines: in vitro synergistic cytotoxicity and DNA-induced strand-breaks. Eur J Cancer. 1996 Jun;32A(7):1219–26. doi: 10.1016/0959-8049(96)00018-4. [DOI] [PubMed] [Google Scholar]
- 7.Umar A, Koi M, Risinger JI, Glaab WE, Tindall KR, Kolodner RD, Boland CR, Barrett JC, Kunkel TA. Correction of hypermutability, N-methyl-N′-nitro-N-nitrosoguanidine resistance, and defective DNA mismatch repair by introducing chromosome 2 into human tumor cells with mutations in MSH2 and MSH6. Cancer Res. 1997 Sep 15;57(18):3949–55. [PubMed] [Google Scholar]
- 8.Lindstrom MJ, Kunugi KA, Kinsella TJ. Global comparison of radiation and chemotherapy dose-response curves with a test for interaction. Radiat Res. 1993 Aug;135(2):269–77. [PubMed] [Google Scholar]
- 9.Yan T, Schupp JE, Hwang HS, Wagner MW, Berry SE, Strickfaden S, Veigl ML, Sedwick WD, Boothman DA, Kinsella TJ. Loss of DNA mismatch repair imparts defective cdc2 signaling and G(2) arrest responses without altering survival after ionizing radiation. Cancer Res. 2001 Nov 15;61(22):8290–7. [PubMed] [Google Scholar]
- 10.Stigliano V, Assisi D, Cosimelli M, Palmirotta R, Giannarelli D, Mottolese M, Mete LS, Mancini R, Casale V. Survival of hereditary non-polyposis colorectal cancer patients compared with sporadic colorectal cancer patients. J Exp Clin Cancer Res. 2008 Sep 19;27:39. doi: 10.1186/1756-9966-27-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meyers M, Wagner MW, Mazurek A, Schmutte C, Fishel R, Boothman DA. DNA mismatch repair-dependent response to fluoropyrimidine-generated damage. J Biol Chem. 2005 Feb 18;280(7):5516–26. doi: 10.1074/jbc.M412105200. [DOI] [PubMed] [Google Scholar]
- 12.Lund KC, Peterson LL, Wallace KB. Absence of a universal mechanism of mitochondrial toxicity by nucleoside analogs. Antimicrob Agents Chemother. 2007 Jul;51(7):2531–9. doi: 10.1128/AAC.00039-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hwang PM, Bunz F, Yu J, Rago C, Chan TA, Murphy MP, Kelso GF, Smith RA, Kinzler KW, Vogelstein B. Ferredoxin reductase affects p53-dependent, 5-fluorouracil-induced apoptosis in colorectal cancer cells. Nat Med. 2001 Oct;7(10):1111–7. doi: 10.1038/nm1001-1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Falcone A, Lencioni M, Brunetti I, Pfanner E, Allegrini G, Antonuzzo A, Andreuccetti M, Malvaldi G, Danesi R, Del Tacca M, Conte PF. Maximum tolerable doses of intravenous zidovudine in combination with 5-fluorouracil and leucovorin in metastatic colorectal cancer patients. Clinical evidence of significant antitumor activity and enhancement of zidovudine-induced DNA single strand breaks in peripheral nuclear blood cells. Ann Oncol. 1997 Jun;8(6):539–45. doi: 10.1023/a:1008249803523. [DOI] [PubMed] [Google Scholar]
- 15.Miyachi H, Jiao L, Sowers LC, Scanlon KJ. Collateral sensitivity to azidothymidine in methotrexate resistant human leukemia cells. In Vivo. 1992 Jan-Feb;6(1):17–21. [PubMed] [Google Scholar]
- 16.Tsuneyoshi K, Haraguchi M, Hongye Z, Gotanda T, Tachiwada T, Sumizawa T, Furukawa T, Baba M, Akiyama S, Nakagawa M. Induction of thymidine phosphorylase expression by AZT contributes to enhancement of 5′-DFUR cytotoxicity. Cancer Lett. 2006 Dec 8;244(2):239–46. doi: 10.1016/j.canlet.2005.12.020. [DOI] [PubMed] [Google Scholar]
- 17.Liao ZK, Zhou FX, Luo ZG, Zhang WJ, Xiong J, Bao J, Han G, Zhang MS, Xie CH, Zhou YF. Radio-activation of hTERT promoter in larynx squamous carcinoma cells: an ‘indirected-activator’ strategy in radio-gene-therapy. Oncol Rep. 2008 Jan;19(1):281–6. [PubMed] [Google Scholar]
- 18.Zhou FX, Liao ZK, Dai J, Xiong J, Xie CH, Luo ZG, Liu SQ, Zhou YF. Radiosensitization effect of zidovudine on human malignant glioma cells. Biochem Biophys Res Commun. 2007 Mar 9;354(2):351–6. doi: 10.1016/j.bbrc.2006.12.180. [DOI] [PubMed] [Google Scholar]
- 19.Tosi P, Gherlinzoni F, Visani G, Coronado O, Costigliola P, Mazzetti M, Gritti F, Chiodo F. AZT plus methotrexate in HIV-related non-Hodgkin’s lymphomas. Leuk Lymphoma. 1998 Jun;30(1–2):175–9. doi: 10.3109/10428199809050940. [DOI] [PubMed] [Google Scholar]
- 20.Peddada AV, Smith DE, Rao AR, Frost DB, Kagan AR. Chemotherapy and low- dose radiotherapy in the treatment of HIV-infected patients with carcinoma of the anal canal. Int J Radiat Oncol Biol Phys. 1997 Mar 15;37(5):1101–5. doi: 10.1016/s0360-3016(96)00596-2. [DOI] [PubMed] [Google Scholar]
- 21.Seo Y, Kinsella MT, Reynolds HL, Chipman G, Remick SC, Kinsella TJ. Outcomes of chemoradiotherapy with 5-Fluorouracil, mitomycin C for anal cancer in immunocompetent versus immunodeficient patients. Int J Radiat Oncol Biol Phys. 2009 Sep 1;75(1):143–9. doi: 10.1016/j.ijrobp.2008.10.046. [DOI] [PubMed] [Google Scholar]