

Abstract
Background
A key issue for cardiovascular genetic medicine is ascertaining if a putative mutation indeed causes dilated cardiomyopathy (DCM). This is critically important as genetic DCM, usually presenting with advanced, life-threatening disease, may be preventable with early intervention in relatives known to carry the mutation.
Methods and Results
We recently undertook bidirectional resequencing of TNNT2, the cardiac troponin T gene, in 313 probands with DCM. We identified six TNNT2 protein-altering variants in nine probands, all who had early onset, aggressive disease. Additional family members of mutation carriers were then studied when available. Four of the nine probands had DCM without a family history, and five had familial DCM. Only one mutation (Lys210del) could be attributed as definitively causative from prior reports. Four of the five missense mutations were novel (Arg134Gly, Arg151Cys, Arg159Gln, Arg205Trp), and one was previously reported with hypertrophic cardiomyopathy (Glu244Asp). Based on the clinical, pedigree and molecular genetic data these five mutations were considered possibly or likely disease causing. To further clarify their potential pathophysiologic impact, we undertook functional studies of these mutations in cardiac myocytes reconstituted with mutant troponin T proteins. We observed decreased Ca2+ sensitivity of force development, a hallmark of DCM, in support of the conclusion that these mutations are disease-causing.
Conclusions
We conclude that the combination of clinical, pedigree, molecular genetic and functional data strengthen the interpretation of TNNT2 mutations in DCM.
Keywords: dilated cardiomyopathy, genetics
Introduction
A key issue in cardiovascular genetic medicine is determining if a putative mutation in a gene known to be associated with dilated cardiomyopathy (DCM) is indeed causative of disease. While a molecular genetic diagnosis may not necessarily guide treatment or impact outcome of a proband with advanced disease, pre-symptomatic at-risk relatives who carry the disease-causing mutation can undergo heightened clinical surveillance, and with evidence of early disease, medical and/or anti-arrhythmic device therapy may prevent disease progression or life-threatening complications. This issue is critically important because DCM usually presents with advanced, life-threatening disease. Recent guidelines suggest that a genetic etiology should be considered even in the absence familial DCM,1 further favoring the benefits of genetic testing in this population. That treatment may prevent or delay DCM progression in pre-symptomatic at-risk relatives provides a powerful rationale for molecular genetic testing.
Despite these clear benefits, molecular diagnosis of DCM in a proband is hampered by extensive allelic heterogeneity (see2-4 for review). Several dozen mutations, almost all of them private missense, have been reported in the genes more commonly implicated in genetic DCM such as LMNA,5-8 MHY7,9-12 or TNNT2,9, 11-15 encoding the lamin A/C, beta myosin heavy chain, and cardiac troponin T proteins, respectively. Further, locus heterogeneity is a prominent feature of genetic DCM, as private mutations in more than 20 other genes have also been implicated.2-4 Such complicating factors make assessing pathogenicity difficult. While assigning pathogenicity of a mutation is usually based on its rarity in the population, the alteration of a conserved amino acid, and family studies showing segregation with disease among multiple affected relatives or negative results in the unaffected parents of a sporadic case, the approach we used in our original resequencing report,12 this approach heavily relies on prediction models, significant clinical follow-up resources and the cooperation of family members. A large DCM mutation database provides a practical alternative, but years of data accumulation will be necessary before its full implementation. Therefore, providers currently making use of genetic testing in their practice must decide in favor of or against increased frequency of cardiovascular screening despite limited data.
With the advent of genetic cardiomyopathy guidelines,1 and with genetic testing rapidly becoming available for greater numbers of DCM genes16 at lower cost, these issues will only become more problematic. Because of these issues, we reasoned that relevant functional studies, when combined with pedigree and molecular genetic data, may be valuable in interpreting molecular results in DCM, particularly when affected family members are not available to assess segregation.
Troponin T cardiomyopathy is an aggressive, early onset disease accompanied by considerable morbidity and mortality. Elegant functional studies have previously demonstrated characteristic shifts in calcium sensitivity and force of contraction with DCM and hypertrophic cardiomyopathy (HCM) resulting from TNNT2 mutations.17-23 We therefore selected this gene for study, and now present the combined clinical, family, molecular genetics and functional data for troponin T DCM.
Materials and Methods
Patient Population
Written, informed consent was obtained from all subjects, and the OHSU Institutional Review Board approved the project. The study included 313 probands (291 Caucasians, of whom seven were of Hispanic descent; 16 African-Americans, three Asians and three Native Americans/Alaskan Natives), and used methods of clinical categorization of FDC versus IDC as previously described.12, 24 A composite clinical description of 304 probands and their families and our FDC/IDC categories have previously been described24 in detail; 302 of those 304 and an additional 11 IDC and FDC probands were included in the present study.
Genetic analysis
Genomic DNA was extracted from whole blood and was sequenced in both directions to detect nucleotide variants in TNNT2, cardiac troponin T, as previously described.12 All exons and intron/exon boundaries were PCR amplified by standard methods at SeattleSNPs under contract to the NHLBI resequencing service. Amino acid numbering was after Townsend et al 25 and gene structure was after Gomes et al 26 (Figure 1.A).
Figure 1. The TNNT2, cardiac troponin T gene structure, cardiac troponin T amino acid conservation, and pedigrees of cardiac troponin T associated cardiomyopathy.



A. The TNNT2, cardiac troponin T gene structure. The TNNT2 gene is shown, and the adult isoform of cardiac troponin T, encoded by exons 2-17, is a total of 288 amino acids in length. The locations of the 6 mutations presented are labeled with their respective letters (A through I); other published DCM mutations are labeled J through N. Mutations reported to cause hypertrophic cardiomyopathy are labeled 1 through 32. Glu244Asp, previously reported in association HCM and reported here in association with DCM is identified as both (I, 24). Exon numbering25 and amino acid numbering26 is per prior reports. Binding regions for troponin C, troponin I, tropomyosin and cardiac actin are shown. HCM mutations are shown in Supplemental Table A.
B. Amino acid conservation. Altered amino acids are shown for each missense mutation on the first line. The wild-type human sequence is shown, followed by rat, mouse, chicken, zebrafish and drosophila. All variants were conserved to zebrafish or drosophila except Arg159Gln (rat, mouse).
C. Pedigrees of cardiac troponin T associated cardiomyopathy. Pedigrees have been labeled by letter, which correspond to their respective mutation as shown in panel A and given in Tables 1 and 2. Squares represent males, circles females. An arrowhead denotes the proband. A diagonal line marks deceased individuals. Solid symbols indicate idiopathic dilated cardiomyopathy with or without heart failure; shaded symbols represent any cardiovascular abnormality. Open symbols represent unaffected individuals. The presence or absence of the pedigree's TNNT2 mutation is indicated by a + or - symbol, respectively. Obligate carriers are noted in parenthesis, (+). Pedigree G, previously published by others,5 is not shown.
Samples from probands identified by the resequencing service as carriers of protein-altering variants, as well as any available samples from their relatives were resequenced in our laboratory for confirmation and segregation analysis. Nucleotide changes were only evaluated if they were absent from all 253 control samples analyzed at the resequencing center (188 Caucasian, 24 African-American, 22 Asian and 19 Hispanic). Putative disease-causing nucleotide alterations identified in African-American samples were further evaluated in an additional 169 control African-American DNA samples in our laboratory, for a total of 193 African-American controls (386 chromosomes).
Functional studies in porcine skinned cardiac fiber preparations
Human cardiac wild type troponin T (HCTnT-WT) was previously cloned in our laboratory,27 and used as a template for overlapping PCR with primers designed to replace those amino acids present in the DCM troponin T mutant proteins. Recombinant human cardiac troponin T (HCTnT), troponin I and troponin C proteins were prepared as previously reported.26, 28 Formation of human troponin I and troponin C complexes was performed as previously described.28 Skinned papillary muscles were obtained from the left ventricles of fresh pig hearts obtained at a local slaughterhouse. Small bundles of fibers were isolated and treated overnight in a pCa 8.0 relaxing solution containing 1% Triton X-100 as previously described.26, 28 After the overnight treatment, fibers were then transferred to a pCa 8.0 solution containing 50% glycerol, without Triton X-100 and stored at - 20°C. This preparation was used to dissect out porcine muscle fiber bundles that were mounted onto a force transducer (Supplemental Figure 1A). Fiber force development was evaluated with increasing Ca2+ concentration from pCa 8.0 - 4.0 (human wild-type and mutant troponin T proteins were incorporated into the fibers as described in the supplemental figure A2). Data were analyzed using the following equation: % Change in force = 100 X [Ca2+]n / ([Ca2+]n + [Ca2+50]n) where “[Ca2+50]” is the free [Ca2+] which produces 50% force and “n” is the Hill coefficient, as previously described.26, 28 The experimental results were reported as X ± S.E.M., and analyzed for significance using unpaired Student's t test at p < 0.05.
Results
Molecular genetic data
Bidirectional sequencing of TNNT2 was carried out on DNA specimens from 313 unrelated probands with IDC or FDC, as described in our preliminary report.12 Six protein-altering variants, none of which were present in 253 control specimens, were identified in nine of 313 probands (2.9%) (Table 1),9, 13, 15, 18, 29 of whom eight were Caucasian (one of Hispanic descent, Pedigree B), and one was African-American (Pedigree I). The variant in Pedigree I was not identified in an additional 193 African-American control DNAs. Of the six variants, five were missense mutations, all of which altered highly conserved amino acids (Figure 1.B). The sixth was the Lys210del mutation, previously reported in multiple families associated with DCM.9, 13, 18 Three of the six variants have previously been reported in association with DCM9, 13, 15, 18 or HCM29 (Table 1). TNNT2 mutations identified here and those previously reported in DCM and HCM show marked allelic heterogeneity (Figure 1.A).
Table 1. Nonsynonymous mutations in TNNT2, cardiac troponin T.
| Pedigree | Molecular Genetic Data | Clinical and Pedigree Data | Functional Data | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Nucleotide change* | Amino Acid Change† | Diagnosis, FDC or IDC | Race | Segregation‡ | Disease-Associated Status in Prior Report12 | Conservation§ | Previously reported cardiovascular disease | Reference | Calcium sensitivity | |
| A | 13712C>G | Arg134Gly | FDC | Non-Hispanic White | Yes | Likely | Yes | decreased | ||
| B | 13763C>T‖ | Arg151Cys | IDC | Hispanic White | Likely | Yes | ||||
| C | 14679G>A | Arg159Gln | IDC | Non-Hispanic White | Possibly | Yes | decreased | |||
| D | 16080C>T | Arg205Trp | IDC | Non-Hispanic White | Likely | Yes | DCM¶ | 15 | decreased | |
| E | 16096_16098delAGA | Lys210del | FDC | Non-Hispanic White | Likely | N/A | DCM | 9, 13, 15, 18 | decreased | |
| F | 16096_16098delAGA | Lys210del | FDC | Non-Hispanic White | Yes | Likely | N/A | DCM | 9, 13, 15, 18 | decreased |
| G | 16096_16098delAGA | Lys210del | FDC | Non-Hispanic White | Yes | Likely | N/A | DCM | 9, 13, 15, 18 | decreased |
| H | 16096_16098delAGA | Lys210del | FDC | Non-Hispanic White | Yes | Likely | N/A | DCM | 9, 13, 15, 18 | decreased |
| I | 16742G>T | Glu244Asp | IDC | Non-Hispanic African American | Possibly | Yes | HCM | 29 | decreased | |
Nucleotide numbering is per the SeattleSNPs resequencing service.
Amino acid numbering is per previous publications.
Segregation means multiple affected carrying mutation and/or multiple nonaffected not carrying mutation; entry left blank because of insufficient clinical data and/or DNA specimens to assess segregation.
see Figure 1.B.
Homozygous mutation.
The Arg205Trp reported here is a novel mutation; the earlier study reported Arg205Leu.
Clinical data
Preliminary clinical assignments of were made for each mutation12 and are provided (Table 1). Clinical characteristics (Table 2) and pedigree structures (Figure 1.C) are presented.
Table 2. Clinical Characteristics.
| Subject | Age of diagnosis, years | DCM | ECG / Arrhythmia | LVEDD, mm (Z-score) | LV septum, posterior wall thickness, mm | Ejection Fraction | Mutation present (yes, no, unknown) | Comment |
|---|---|---|---|---|---|---|---|---|
| Pedigree A: ARG-134-GLY | ||||||||
| A.1 | 63 | yes | NA | 57 mm (1.90) | NA | 25% | yes | |
| A.4 | 34 | yes | AF, VT, bigem, PM, ICD | 58 mm (1.69) | 10/10 | 30% | yes | |
| A.5 | 34 | NA | NA | NA | NA | NA | unknown | Heart transplant at 34 yrs; no medical records |
| A.6 | 34 | yes | NSSTT | 63mm (3.14) | 8/9 | 35% | yes (obligate carrier) | Died of SCD at age 36 |
| A.8 | 12 | yes | 1AVB | 51mm (1.78) | 7/7 | 23% | yes | Asymptomatic |
| A.10 | 6 | yes | NA | 58 mm (6.05) | 5/6 | 19% | yes | Heart transplant at 8 yrs |
| Pedigree B: ARG-151-CYS | ||||||||
| B.6 | 56 | no | LAD, LVH | NA | 7/7 | 60% | yes | |
| B.7 | 52 | no | NA | 51 mm (2.11) | 9/9 | 57% | yes | Asymptomatic at 53 yrs |
| B.8 | 16/20 | no | NL | 50 mm (0.23) | 7/7 | 70% | yes | Asymptomatic |
| B.9 | 19 | yes | NSSTT, LVD, low voltage | 65 mm (6.00) | NA | 24% | yes, homozygous | Heart transplant at 19 years |
| B.10 | 16/20 | no | NA | 47 mm (0.77) | 6/5 | 66% | yes, homozygous | Asymptomatic |
| Pedigree C: ARG-159-GLN | ||||||||
| C.3 | 20 | yes | NA | 65 mm (5.60) | 9/8 | 25% | yes | HF onset associated with pregnancy |
| Pedigree D: ARG-205-TRP | ||||||||
| D.3 | 0.5 | yes | 1AVB, LVH | 78 mm (7.01) | 4/4 | 18% | yes | Congenital DCM at 4 mo; heart transplant at 12 yrs |
| Pedigree E: Lys 210 deletion | ||||||||
| E.1 | ? | yes | NA | NA | NA | NA | unknown | Died awaiting heart transplant at 52 yrs |
| E.2 | 31 | yes | LAFB, LVH, NSSTT | 83 mm (7.10) | 11/10 | 14% | yes | Heart transplant at 31 yrs |
| E.3 | 5 | NA | NA | NA | NA | NA | unknown | No medical records; died 3 mo after diagnosis |
| Pedigree F: Lys 210 deletion | ||||||||
| F.1 | 50 | yes | NA | NA | NA | NA | unknown | DCM at autopsy |
| F.3 | 34 | NA | NA | NA | NA | NA | unknown | Died after heart transplant at age 34; no medical records |
| F.4 | 37 | yes | RAD | 66 mm (3.95) | 9/11 | 30% | yes | Asymptomatic, age 50 |
| F.6 | 26 | yes | 1AVB, LBBB | 81 mm (6.47) | 11/11 | 15% | yes | Heart failure |
| Pedigree H: Lys 210 deletion | ||||||||
| H.1 | 35 | |||||||
| H.6 | 74 | yes | LAD, LAFB, NSSTT | NA | NA | 28% | yes (obligate carrier) | Diagnosed at 74 years; no coronary angiogram |
| H.8 | N/A | NA | NA | NA | NA | NA | yes (obligate carrier) | Died at 34 years of cancer |
| H.11 | 59 | yes | 1AVB, NSSTT | 58 mm (4.05) | 14/11 | 20% | yes | Heart Failure |
| H.12 | 16 | yes | 1AVB, RAE, LAE, LVH | NA | NA | NA | unknown | Died at 16 years of advanced HF; DCM by autopsy |
| H.15 | 52 | yes | LBBB, NSSTT | 75 mm (5.00) | 9/9 | 16% | yes | Heart failure |
| H.16 | 47 | yes | NL | 59 mm (2.54) | 7/7 | 50% | yes | Asymptomatic |
| H.17 | 1 | yes | NA | NA | NA | NA | unknown | Died at 1 yr, diagnosis by autopsy |
| H.18 | 12 | yes | 1AVB, LAE, LVH, NSSTT | NA | NA | NA | unknown | Died of DCM at 12 yrs |
| H.19 | 21 | yes | LAE, RAD | NA | NA | 20% | unknown | Died of DCM at 21 yrs |
| Pedigree I: GLU-244-ASP | ||||||||
| I.3 | 13 | yes | LVH, RVH, NSSTT | 70 mm (5.66) | NA | 7% | yes | Severely reduced systolic function; heart failure, VAD, heart transplant 13 at yrs |
1AVB, first degree atrioventricular block; AF, atrial fibrillation; bigem, bigemeny; DCM, dilated cardiomyopathy; ICD, implantable cardiac defibrillator; LAD, left axis deviation; LAE, left atrial enlargement; LAFB, left anterior fasciular block; LBBB, left bundle branch block; LVEDD, left ventricular end-diastolic dimension; LVH, left ventricular hypertrophy; NSSTT, non-specific ST-T changes; PM, pacemaker; RAD, right axis deviation; RAE, right atrial enlargement; RVH, right ventricular hypertrophy; VAD, ventricular assist device; VT, ventricular tachycardia.
The variant in pedigree A (Arg134Gly) segregated with disease in other affected family members and was therefore considered likely disease causing.
The proband in Pedigree B whose parents were second cousins, was homozygous for an Arg151Cys alteration, and was the only clinically affected member of her family. DNA analysis indicated that each parent was heterozygous for the alteration. We previously reported this mutation as likely disease-causing12 in part because of the homozygous state and in part because of the characteristic clinical behavior of TNNT2 mutations to cause early onset, aggressive disease as observed in the proband. However, the pathogenicity of this mutation was not certain, as one sibling also carried the homozygous mutation and had no evidence of disease. Further, the proband's asymptomatic parents were carrying the mutation in the heterozygous state. All previous reports of TNNT2 mutations, including all others herein, were heterozygous and had been sufficient to cause the DCM phenotype, and the older age of the proband's parents should have increased their probabilities of manifesting disease.
The Arg159Gln TNNT2 nucleotide alteration in the proband of Pedigree C was considered possibly disease-causing12 (Table 1) as it predicted the replacement of a conserved amino acid; however, no additional pedigree information was available to assess segregation with disease.
The proband in pedigree D carried an Arg205Trp alteration. Because a previous report had identified an Arg205Leu mutation in association with DCM, we therefore assigned Arg205Trp as likely disease causing. However, neither functional nor segregation data to support this assignment were available for our proband or in the prior report.15
The Lys210del mutation had been shown to be disease-causing in several pedigrees with excellent segregation data,9, 13, 15 and was observed in unrelated probands from four families (E, F, H; G has been described by others9 and is not shown).
The TNNT2 Glu244Asp variant in the proband of Pedigree I was previously reported in a patient with HCM,29 however, the proband had a very dilated left ventricle and marked reduction in systolic function consistent with DCM (Table 2). This mutation was previously assigned by us as likely disease-causing12 (Table 1) because of the prior HCM report. However, segregation data were lacking in the HCM report and in our pedigree.
The median age of onset of the 30 affected individuals with DCM from the nine pedigrees was 32.5 years, with a mean age of 31.1 years (range 0.5 to 72 years) (Table 2). Twelve of 30 (40%) had disease onset by age 20 years, 14 of 30 (47%) had disease onset by 30 years, and 21 of 30 (70%), had disease onset by 40 years.
Functional Data
The Ca2+ sensitivity of force development obtained in porcine cardiac skinned fibers, when comparing the pCa-force relationship of fibers exchanged with human wild-type or DCM mutant troponin T proteins reconstituted with the human cardiac troponin I and troponin C complex, demonstrated reduced sensitivity to calcium consistent with DCM in our18, 20, 21 and other17-19, 22, 23 prior reports. The Arg151Cys, Arg159Gln and Arg205Trp decreased Ca2+ sensitivity of force development of 0.09, 0.07 and 0.07 log units of [Ca2+], respectively (Table 3). The Arg134Gly mutation did not display changes in Ca2+ sensitivity compared to the wild-type, but increased the maximal force development (Table 3). The same functional phenotype (no changes in Ca2+ sensitivity and increased force) was previously observed with the Glu244Asp mutation.20 This could be associated with a distinct functional phenotype of some DCM mutants or a specific functional phenotype for mutations that start as hypertrophic and progress to dilated cardiomyopathy.30 The pCa50 ([Ca2+] needed to reach 50% of maximal force), Hill coefficient (a cooperativity index of thin filament activation) and maximal force values are shown in Figure 2 and Table 3. To address whether the troponin DCM mutants had altered affinity for the thin filament and subsequent poor incorporation into the fibers, we measured the unregulated tension at low Ca2+ concentration (pCa 8.0) after displacement and before reconstitution with the troponin I and troponin C binary complex. As more exogenous troponin T incorporates into the thin filament and more endogenous troponin is displaced, there is a greater amount of tension generated at low Ca2+ levels since there is less troponin available to inhibit force (see supplemental figure A for additional detail). Fibers incubated with the Arg134Gly mutant showed a reduced ability (38% Ca2+ unregulated force) to displace the endogenous porcine troponin complex compared to the human wild-type protein (87% Ca2+ unregulated force) (Table 3). The other troponin T mutants did not show changes in their incorporation into the thin filament. This finding indicates that the Arg134Gly mutant may exert its effects through changes in protein-protein interaction that alter thin filament and contractile function via mechanisms independent of calcium sensitivity to give the human DCM phenotype.
Table 3. Summary of calcium-force relationship curves in porcine ventricular myocyte fibers displaced with human cardiac troponin T wild type or troponin T mutations and reconstituted with human cardiac troponin I/troponin C complex.
The pCa50, nH, % maximal force and % Ca2+ unregulated force values are the average of a number of independent fiber experiments, and the errors are reported as S.E. values. The Ca2+ unregulated force was calculated after the human cardiac troponin T (HCTnT) treatment by the following equation: (FpCa8/FpCa4) X 100, where the FpCa8 (x in Figure 5) [and FpCa4 (y in Figure 6)] are the force at pCa 8.0 and pCa 4.0 solutions, respectively. The Ca2+ unregulated force reflects the efficiency of the human cardiac troponin T to displace the whole native troponin complex and its ability to incorporate into the fiber.
| Human cardiac troponin T proteins | pCa50 | Hill coefficient, nH | Δ pCa50a | % Maximal Force Recovery | % Ca2+ Unregulated Force | Number of Experiments |
|---|---|---|---|---|---|---|
| Native (porcine) | 5.83 ± 0.01 | 3.44 ± 0.09 | + 0.06 | 100.0 | - | 40 |
| Wild-type | 5.77 ± 0.02 | 2.50 ± 0.07 | - | 67.6 ± 3.7 | 87.2 ± 3.2 | 9 |
| Arg134Gly | 5.72 ± 0.03 | 3.09 ± 0.07 | - 0.05 | 79.7 ± 2.5* | 38.1 ± 3.0* | 9 |
| Arg 151Cys | 5.68 ± 0.02* | 2.64 ± 0.16 | - 0.09 | 65.7 ± 3.4 | 79.8 ± 6.4 | 7 |
| Arg159Gln | 5.70 ± 0.02* | 2.53 ± 0.06 | - 0.07 | 65.0 ± 3.4 | 91.1 ± 4.3 | 7 |
| Arg205Trp | 5.70 ± 0.02* | 2.33 ± 0.07 | - 0.07 | 63.2 ± 3.2 | 92.3 ± 3.2 | 8 |
| Lys210delb | - | - | -0.12b | 46.8b | 89.6b | - |
| Glu244Aspc | - | - | 0c | 83.4c | 100.0c | - |
Figure 2. Normalized pCa-Force relationship in skinned cardiac muscle fibers.
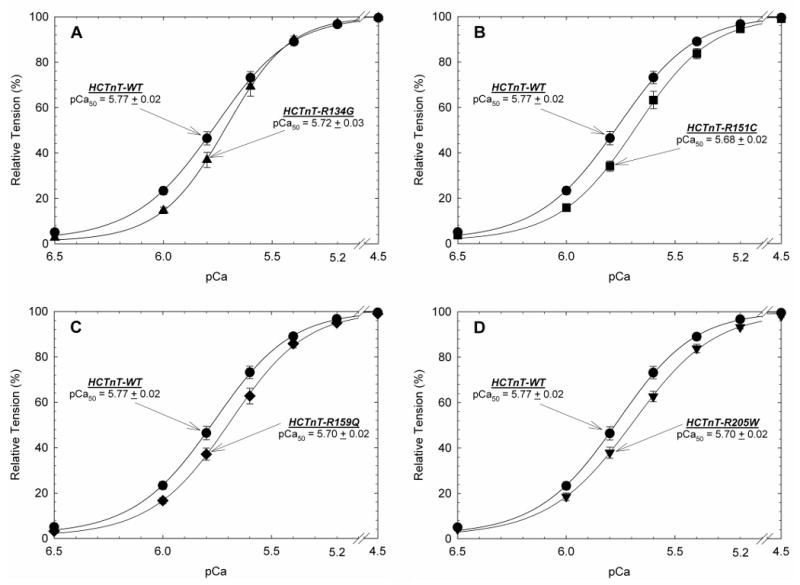
The wildtype (WT) and DCM-HCTnTs replaced native CTnT in the procedure shown in Figure 2.A) HCTnT-WT containing fibers were compared to DCM-mutant HCTnT-R134G B) HCTnT-WT containing fibers compared to DCM-mutant HCTnT-R151C C) HCTnT-WT containing fibers compared to DCM-mutant HCTnT-R159Q D) HCTnT-WT containing fibers compared to DCM-mutant HCTnT-R205W. The Ca2+ dependence of force development remained unchanged in native porcine fibers and after HCTnT-WT substitution (See Table 3). Data in each panel is an average of 7-9 experiments and the mean is shown as mean ± S.E.
Discussion
A great deal of progress has recently been made in understanding the genetic basis of DCM with mutations identified in more than 20 genes (reviewed in 2-4). This progress has led to the recent release of clinical guidelines for the genetic cardiomyopathies,1 and to a rapid expansion of clinical molecular genetic testing. However, molecular diagnosis of DCM is challenging. For example, genetic DCM, usually presenting as adult-onset disease, varies greatly in its age of onset. Thus, even if first degree relatives are available for molecular genetic testing, family members carrying the putative disease-causing mutation may have little or no evidence of disease, confounding efforts to assess segregation of the mutation with disease and therefore confirmation of the variant as disease-causing. Yet paradoxically, it is these family members who may most benefit from clinical surveillance and early intervention to prevent disease progression. Further, although not yet reported for TNNT2, sequencing the many genes implicated in DCM will likely reveal increasing numbers of variants of unknown significance. Ongoing research integrating molecular, clinical and functional data, as shown here, may facilitate clinical management by adding to the interpretation of gene variations thought to represent disease-causing DCM mutations.
In this study the molecular, clinical and pedigree data for troponin T mutations in these families were supplemented with functional studies with mutant troponin T proteins reconstituted into porcine cardiac myocytes. This approach yielded highly informative calcium sensitivity and maximal force response data that was helpful to augment molecular genetic data for the identified novel mutations.
Aside from conduction and rhythm perturbations observed with LMNA and SCN5A mutations,2-4 few genotype/phenotype correlations exist for genetic DCM. However, one useful genotype/phenotype correlation may be the fully penetrant, early onset, aggressive disease observed in this and prior reports9, 15 of troponin T DCM. In the 30 affected subjects herein, the median age of onset was 32.5 years, with an average age of onset of 31.1 years; almost one-half (47%) of those affected had disease onset by 30 years of age. This compares with an earlier analysis from our research database of 304 probands and 166 family members affected with FDC/IDC, where the average age at diagnosis was 43 years.24
The mechanism of the early onset and aggressive nature of troponin T cardiomyopathy is likely related to the severity of disruption of the force-generating components of the thick and thin filaments. Functional reports of DCM mutations have shown decreased Ca2+ sensitivity of the myofilament in the tropomyosin/troponin regulatory complex.17-19, 22, 23 Cardiac troponin T is the subunit that connects the troponin complex to the thin filament. The additional subunits are troponin C that binds Ca2+, and troponin I that binds to actin and is involved in inhibition of muscle contraction.31-33 This decrease in Ca2+ sensitivity of force development is sufficient to alter the contractility dynamics of the heart leading to systolic dysfunction, as previously observed for the Lys210 deletion and the R141W mutants.18
While skinned porcine myocytes present an ideal model to evaluate functional changes in troponin cardiomyopathy, other systems able to evaluate the functional impact of mutations of other DCM genes (e.g., those encoding contractile, channel, calcium handling and other proteins) could also augment clinical and molecular genetic data to help confirm that a specific sequence variant causes DCM.
In conclusion, the functional data obtained for the six TNNT2 mutations reported in this study suggests that these variants are highly likely to be causative of DCM considering the cumulative data from molecular genetic, clinical, pedigree and functional studies. Adding functional data to existing clinical and genetic data will be key to understanding the molecular etiology of genetic DCM while moving the cardiovascular genetic medicine field ahead.
Supplementary Material
Acknowledgments
We thank the many families and referring physicians for their participation in the Familial Dilated Cardiomyopathy Research Program, without whom these studies would not have been possible, and Michelle Jones and Jingsheng Liang for their expert technical assistance.
Funding Sources: This work was supported by NIH awards RO1-HL58626, 5 M01 RR000334 (Dr. Hershberger) and R01-HL42325 (Dr. Potter) and a postdoctoral fellowship from the American Heart Association AHA-0825368E (Dr. Pinto). Resequencing services were provided by the University of Washington, Department of Genome Sciences, under U.S. Federal Government contract number N01-HV-48194 from the National Heart, Lung, and Blood Institute.
Footnotes
Conflict of Interest Disclosure: None.
References
- 1.Hershberger R, Lindenfeld J, Mestroni L, Seidman C, Taylor M, Towbin J. Genetic Evaluation of Cardiomyopathy - A Heart Failure Society of America Practice Guideline. J Cardiac Failure. 2009;15:83–97. doi: 10.1016/j.cardfail.2009.01.006. [DOI] [PubMed] [Google Scholar]
- 2.Burkett EL, Hershberger RE. Clinical and genetic issues in familial dilated cardiomyopathy. J Am Coll Cardiol. 2005;45:969–81. doi: 10.1016/j.jacc.2004.11.066. [DOI] [PubMed] [Google Scholar]
- 3.Morita H, Seidman J, Seidman CE. Genetic causes of human heart failure. J Clin Invest. 2005;115:518–26. doi: 10.1172/JCI200524351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ashrafian H, Watkins H. Reviews of translational medicine and genomics in cardiovascular disease: new disease taxonomy and therapeutic implications cardiomyopathies: therapeutics based on molecular phenotype. J Am Coll Cardiol. 2007;49:1251–64. doi: 10.1016/j.jacc.2006.10.073. [DOI] [PubMed] [Google Scholar]
- 5.Fatkin D, MacRae C, Sasaki T, Wolff M, Porcu M, Frenneaux M, Atherton J, Vidaillet H, Spudich S, Girolami U, Seidman J, Seidman C. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N Engl J Med. 1999;341:1715–24. doi: 10.1056/NEJM199912023412302. [DOI] [PubMed] [Google Scholar]
- 6.Arbustini E, Pilotto A, Repetto A, Grasso M, Negri A, Diegoli M, Campana C, Scelsi L, Baldini E, Gavazzi A, Tavazzi L. Autosomal dominant dilated cardiomyopathy with atrioventricular block: a lamin A/C defect-related disease. J Am Coll Cardiol. 2002;39:981–90. doi: 10.1016/s0735-1097(02)01724-2. [DOI] [PubMed] [Google Scholar]
- 7.Taylor MR, Fain PR, Sinagra G, Robinson ML, Robertson AD, Carniel E, Di Lenarda A, Bohlmeyer TJ, Ferguson DA, Brodsky GL, Boucek MM, Lascor J, Moss AC, Li WL, Stetler GL, Muntoni F, Bristow MR, Mestroni L. Natural history of dilated cardiomyopathy due to lamin A/C gene mutations. J Am Coll Cardiol. 2003;41:771–80. doi: 10.1016/s0735-1097(02)02954-6. [DOI] [PubMed] [Google Scholar]
- 8.Parks SB, Kushner JD, Nauman D, Burgess D, Ludwigsen S, Peterson A, Li D, Jakobs P, Litt M, Porter CB, Rahko PS, Hershberger RE. Lamin A/C mutation analysis in a cohort of 324 unrelated patients with idiopathic or familial dilated cardiomyopathy. Am Heart J. 2008;156:161–9. doi: 10.1016/j.ahj.2008.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kamisago M, Sharma SD, DePalma SR, Solomon S, Sharma P, McDonough B, Smoot L, Mullen MP, Woolf PK, Wigle ED, Seidman JG, Seidman CE. Mutations in sarcomere protein genes as a cause of dilated cardiomyopathy. N Engl J Med. 2000;343:1688–96. doi: 10.1056/NEJM200012073432304. [DOI] [PubMed] [Google Scholar]
- 10.Daehmlow S, Erdmann J, Knueppel T, Gille C, Froemmel C, Hummel M, Hetzer R, Regitz-Zagrosek V. Novel mutations in sarcomeric protein genes in dilated cardiomyopathy. Biochem Biophys Res Commun. 2002;298:116–20. doi: 10.1016/s0006-291x(02)02374-4. [DOI] [PubMed] [Google Scholar]
- 11.Villard E, Duboscq-Bidot L, Charron P, Benaiche A, Conraads V, Sylvius N, Komajda M. Mutation screening in dilated cardiomyopathy: prominent role of the beta myosin heavy chain gene. Eur Heart J. 2005;26:794–803. doi: 10.1093/eurheartj/ehi193. [DOI] [PubMed] [Google Scholar]
- 12.Hershberger R, Parks S, Kushner JD, Li D, Ludwigsen S, Jakobs P, Nauman D, Burgess D, Partain J, Litt M. Coding sequence mutations identified in MYH7, TNNT2, SCN5A, CSRP3, LBD3, and TCAP from 313 patients with familial or idiopathic dilated cardiomyopathy. Clin Translational Science. 2008;1:21–6. doi: 10.1111/j.1752-8062.2008.00017.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hanson E, Jakobs P, Keegan H, Coates K, Bousman S, Dienel N, Litt M, Hershberger R. Cardiac troponin T lysine-210 deletion in a family with dilated cardiomyopathy. J Card Fail. 2002;8:28–32. doi: 10.1054/jcaf.2002.31157. [DOI] [PubMed] [Google Scholar]
- 14.Li D, Czernuszewicz GZ, Gonzalez O, Tapscott T, Karibe A, Durand JB, Brugada R, Hill R, Gregoritch JM, Anderson JL, Quinones M, Bachinski LL, Roberts R. Novel cardiac troponin T mutation as a cause of familial dilated cardiomyopathy. Circulation. 2001;104:2188–93. doi: 10.1161/hc4301.098285. [DOI] [PubMed] [Google Scholar]
- 15.Mogensen J, Murphy RT, Shaw T, Bahl A, Redwood C, Watkins H, Burke M, Elliott PM, McKenna WJ. Severe disease expression of cardiac troponin C and T mutations in patients with idiopathic dilated cardiomyopathy. J Am Coll Cardiol. 2004;44:2033–40. doi: 10.1016/j.jacc.2004.08.027. [DOI] [PubMed] [Google Scholar]
- 16.University of Washington; Seattle: Dilated Cardiomyopathy Overview; p. 1997. Copyright. 1997-2008., Accessed at http://www.genetests.org. [Google Scholar]
- 17.Morimoto S, Lu QW, Harada K, Takahashi-Yanaga F, Minakami R, Ohta M, Sasaguri T, Ohtsuki I. Ca(2+)-desensitizing effect of a deletion mutation Delta K210 in cardiac troponin T that causes familial dilated cardiomyopathy. Proc Natl Acad Sci U S A. 2002;99:913–8. doi: 10.1073/pnas.022628899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Venkatraman G, Harada K, Gomes AV, Kerrick WG, Potter JD. Different functional properties of troponin T mutants that cause dilated cardiomyopathy. J Biol Chem. 2003;278:41670–6. doi: 10.1074/jbc.M302148200. [DOI] [PubMed] [Google Scholar]
- 19.Lu QW, Morimoto S, Harada K, Du CK, Takahashi-Yanaga F, Miwa Y, Sasaguri T, Ohtsuki I. Cardiac troponin T mutation R141W found in dilated cardiomyopathy stabilizes the troponin T-tropomyosin interaction and causes a Ca2+ desensitization. J Mol Cell Cardiol. 2003;35:1421–7. doi: 10.1016/j.yjmcc.2003.09.003. [DOI] [PubMed] [Google Scholar]
- 20.Harada K, Potter JD. Familial hypertrophic cardiomyopathy mutations from different functional regions of troponin T result in different effects on the pH and Ca2+ sensitivity of cardiac muscle contraction. J Biol Chem. 2004;279:14488–95. doi: 10.1074/jbc.M309355200. [DOI] [PubMed] [Google Scholar]
- 21.Venkatraman G, Gomes AV, Kerrick WG, Potter JD. Characterization of troponin T dilated cardiomyopathy mutations in the fetal troponin isoform. J Biol Chem. 2005;280:17584–92. doi: 10.1074/jbc.M409337200. [DOI] [PubMed] [Google Scholar]
- 22.Mirza M, Marston S, Willott R, Ashley C, Mogensen J, McKenna W, Robinson P, Redwood C, Watkins H. Dilated cardiomyopathy mutations in three thin filament regulatory proteins result in a common functional phenotype. J Biol Chem. 2005;280:28498–506. doi: 10.1074/jbc.M412281200. [DOI] [PubMed] [Google Scholar]
- 23.Robinson P, Griffiths PJ, Watkins H, Redwood CS. Dilated and hypertrophic cardiomyopathy mutations in troponin and alpha-tropomyosin have opposing effects on the calcium affinity of cardiac thin filaments. Circ Res. 2007;101:1266–73. doi: 10.1161/CIRCRESAHA.107.156380. [DOI] [PubMed] [Google Scholar]
- 24.Kushner JD, Nauman D, Burgess D, Ludwigsen S, Parks S, Pantely G, Burkett EL, Hershberger R. Clinical characteristics of 304 kindreds evaluated for familial dilated cardiomyopathy. J Cardiac Failure. 2006;12:422–29. doi: 10.1016/j.cardfail.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 25.Townsend PJ, Farza H, MacGeoch C, Spurr NK, Wade R, Gahlmann R, Yacoub MH, Barton PJ. Human cardiac troponin T: identification of fetal isoforms and assignment of the TNNT2 locus to chromosome 1q. Genomics. 1994;21:311–6. doi: 10.1006/geno.1994.1271. [DOI] [PubMed] [Google Scholar]
- 26.Gomes AV, Venkatraman G, Davis JP, Tikunova SB, Engel P, Solaro RJ, Potter JD. Cardiac troponin T isoforms affect the Ca(2+) sensitivity of force development in the presence of slow skeletal troponin I: insights into the role of troponin T isoforms in the fetal heart. J Biol Chem. 2004;279:49579–87. doi: 10.1074/jbc.M407340200. [DOI] [PubMed] [Google Scholar]
- 27.Gomes AV, Guzman G, Zhao J, Potter JD. Cardiac troponin T isoforms affect the Ca2+ sensitivity and inhibition of force development. Insights into the role of troponin T isoforms in the heart. J Biol Chem. 2002;277:35341–9. doi: 10.1074/jbc.M204118200. [DOI] [PubMed] [Google Scholar]
- 28.Pinto JR, Parvatiyar MS, Jones MA, Liang J, Potter JD. A troponin T mutation that causes infantile restrictive cardiomyopathy increases Ca2+ sensitivity of force development and impairs the inhibitory properties of troponin. J Biol Chem. 2008;283:2156–66. doi: 10.1074/jbc.M707066200. [DOI] [PubMed] [Google Scholar]
- 29.Watkins H, McKenna WJ, Thierfelder L, Suk HJ, Anan R, O'Donoghue A, Spirito P, Matsumori A, Moravec C, Seidman JG, Seidman CE. Mutations in the genes for cardiac troponin T and alpha-tropomyosin in hypertrophic cardiomyopathy. N Engl J Med. 1995;332:1058–64. doi: 10.1056/NEJM199504203321603. [DOI] [PubMed] [Google Scholar]
- 30.Freeman K, Colon-Rivera C, Olsson MC, Moore RL, Weinberger HD, Grupp IL, Vikstrom KL, Iaccarino G, Koch WJ, Leinwand LA. Progression from hypertrophic to dilated cardiomyopathy in mice that express a mutant myosin transgene. Am J Physiol Heart Circ Physiol. 2001;280:H151–9. doi: 10.1152/ajpheart.2001.280.1.H151. [DOI] [PubMed] [Google Scholar]
- 31.Potter JD, Holroyde MJ, Robertson SP, Solaro RJ, Kranias EG, Johnson JD. Thre regulation of cardiac-muscle contraction by troponin. In: Dowben RM, Shay JW, editors. Cell and Muscle Motility. New York: Plenum Publishing; 1982. pp. 245–55. [Google Scholar]
- 32.Gordon AM, Homsher E, Regnier M. Regulation of contraction in striated muscle. Physiol Rev. 2000;80:853–924. doi: 10.1152/physrev.2000.80.2.853. [DOI] [PubMed] [Google Scholar]
- 33.Takeda S, Yamashita A, Maeda K, Maeda Y. Structure of the core domain of human cardiac troponin in the Ca(2+)-saturated form. Nature. 2003;424:35–41. doi: 10.1038/nature01780. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.