

Abstract
Myeloid-derived suppressor cells (MDSC) producing arginase I are increased in the peripheral blood of patients with renal cell carcinoma (RCC). MDSC inhibit T-cell function by reducing the availability of l-arginine and are therefore considered an important tumor escape mechanism. We aimed to determine the origin of arginase I–producing MDSC in RCC patients and to identify the mechanisms used to deplete extracellular l-arginine. The results show that human MDSC are a subpopulation of activated polymorphonuclear (PMN) cells expressing high levels of CD66b, CD11b, and VEGFR1 and low levels of CD62L and CD16. In contrast to murine MDSC, human MDSC do not deplete l-arginine by increasing its uptake but instead release arginase I into the circulation. Activation of normal PMN induces phenotypic and functional changes similar to MDSC and also promotes the release of arginase I from intracellular granules. Interestingly, although activation of normal PMN usually ends with apoptosis, MDSC showed no increase in apoptosis compared with autologous PMN or PMN obtained from normal controls. High levels of VEGF have been shown to increase suppressor immature myeloid dendritic cells in cancer patients. Treatment of RCC patients with anti-VEGF antibody bevacizumab, however, did not reduce the accumulation of MDSC in peripheral blood. In contrast, the addition of interleukin-2 to the treatment increased the number of MDSC in peripheral blood and the plasma levels of arginase I. These results may provide new insights on the mechanisms of tumor-induced anergy/tolerance and may help explain why some immunotherapies fail to induce an antitumor response.
Introduction
Immunotherapy with interleukin-2 (IL-2) is a standard of treatment for patients with metastatic renal cell carcinoma (RCC). However, response is limited to 15% to 23% of patients (1). This in part may be explained by a state of immune anergy/tolerance, a well-described phenomenon in cancer patients. We and others have reported that patients with RCC and pancreatic cancer have increased numbers of polymorphonuclear (PMN) cells in the peripheral blood, which copurify with peripheral blood mononuclear cells (PBMC) on a Ficoll-Hypaque density gradient (2, 3). These cells have been named myeloid-derived suppressor cells (MDSC; ref. 4). MDSC express high levels of arginase I and induce T-cell anergy by depleting l-arginine, which impairs T-cell proliferation and cytokine production and reduces the expression of T-cell receptor (TCR) CD3ζ chain (2, 3). Increased numbers of MDSC in the peripheral blood of RCC patients correlated with low l-arginine and high ornithine levels in plasma, and a profound T-cell dysfunction (3).
Arginase hydrolyzes the amino acid l-arginine to ornithine and urea. There are two isoforms of arginase, cytoplasmic arginase I and mitochondrial arginase II, encoded by two different genes. MDSC expressing arginase I deplete l-arginine from the microenvironment and profoundly inhibit T-cell functions (5, 6). Inhibition of arginase I restores T-cell function in vitro and induces an antitumor response in vivo (6). l-arginine depletion by murine MDSC is the result of an increased uptake through the cationic amino acid transporter 2B (CAT-2B; refs. 5, 6). However, the mechanisms of reduction of l-arginine by human MDSC remain unclear.
Extensive work by Gabrilovich and colleagues has shown a strong association between increased levels of vascular endothelial growth factor (VEGF) and high numbers of immature dendritic cells in peripheral blood of patients with gastric, lung, and head and neck cancer (7, 8). Blocking of VEGF in various murine tumor models decreased the counts of suppressive dendritic cells and induced antitumor activity (9). Clinical trials have shown the clinical efficacy of anti-VEGF antibody, bevacizumab (10), in patients with RCC (11). However, it is unclear whether VEGF regulates MDSC accumulation in RCC patients.
Our results show that human MDSC in RCC patients are a subset of activated granulocytes expressing high levels of CD66b, CD11b, and VEGFR1. These cells degranulate and release arginase I, resulting in low levels of l-arginine in plasma. Activation of normal PMN induces phenotypic and functional changes similar to MDSC. Treatment of RCC patients with anti-VEGF antibody decreased the levels of VEGF but did not have an effect on the percentage of MDSC. In contrast, IL-2 treatment markedly increased the percentage of MDSC and the levels of arginase I.
Materials and Methods
Samples and antibodies
Peripheral blood was collected before treatment from 27 patients with advanced metastatic RCC participating in the Cytokine Working Group clinical trial. Control samples were collected from 16 age- and gender-matched normal controls. Carboxy-fluorescein diacetate succinimidyl ester (CFSE)–labeled PBMC or CD66b-depleted PBMC from RCC patients and controls were stimulated with immobilized anti-CD3 (1 μg/mL; OKT-3; Ortho Biotech Products) and anti-CD28 (0.1 μg/mL; BD Biosciences) and proliferation was determined by flow cytometry after 96 h as previously described (12). Supernatants were collected at 72 h to determine IFNγ production. Human RCC cell line 786-O was purchased from the American Type Culture Collection.
Antibodies against CD10, CD11b, CD11c, CD14, CD15, CD16, CD33, CD45Ro, CD45Ra, CD62L, and CD66b were purchased from Becton Dickinson Biosciences. Antibodies against CD13 and CD24 were purchased from eBiosciences. Anti-VEGFR1 (Flt-1) was purchased from R&D Systems. Antibodies against CD4 and CD8 were purchased from Beckman Coulter. Mouse IgG1-FITC/IgG2b-PE, rat IgG1-FITC, and rat-IgG2-PE (Pharmingen-Becton Dickinson) were used as isotype controls. Results were expressed as percent of positive cells and mean fluorescence intensity (MFI). Antibodies against arginase I and arginase II (Santa Cruz Biotechnology) were used for Western blots. Apoptosis was determined by testing the expression of Annexin V by flow cytometry (Becton Dickinson Biosciences), as described by the vendor. Fluorescence acquisition and analysis were done in a Coulter EPICS XL flow cytometer (Beckman Coulter) with a 488-nm argon laser.
Clinical trial
The clinical trial entitled “First Line Treatment with Bevacizumab (B) and High Dose (HD) Bolus Aldesleukin (IL-2) in Metastatic RCC Patients” was conducted by centers affiliated with the cytokine network. Patients enrolled in the trial had been histologically confirmed for metastatic RCC with predominantly clear cell histology with measurable metastatic disease. Treatment consisted of bevacizumab (10 mg/kg) i.v. given every 2 wk followed on week 3 (day 15) and week 5 (day 29) by bevacizumab plus high-dose IL-2 (600,000 IU/kg) i.v. every 8 h as tolerated to a maximum of 14 doses.
Isolation and depletion of human MDSC from PBMC
MDSC were isolated from freshly isolated PBMC by using CD14-negative selection and CD11b-positive selection magnetic beads (Miltenyi Biotech) or by labeling PBMC with anti-CD66b followed by anti-FITC magnetic bead separation, according to the manufacturer’s specifications. Purity of the subsets ranged between 93% and 99% as tested by flow cytometry. Separation of MDSC from frozen samples may cause a significant decrease in MDSC viability; therefore, all experiments were done using fresh peripheral blood samples. Morphology of the cells (MDSC and PMN) was performed by staining of cytospins. Briefly, 5 × 104 cells were centrifuged in a StatSpin CytoFuge centrifuge at 8.5 × g for 4 min and stained using Harleco Hemacolor kit from EMD. Depletion of CD66b+ cells was performed by labeling PBMC with anti-CD66b followed by anti-FITC magnetic bead separation (Miltenyi Biotech). The percentage of CD66b+ cells in the negative selected population ranged between 0.02% and 1.1%.
Isolation and activation of PMN
PMN from healthy donors and patients were isolated by dextran sedimentation. Briefly, peripheral blood was centrifuged over Ficoll-Hypaque (GE Biosciences), after which PBMC were collected and the rest of the cells were resuspended in 3% dextran for 60 min at 37°C and 5% CO2. The supernatants were then collected and centrifuged for 5 min at 1,600 rpm at 4°C. RBCs were lysed by hypotonic lysis. PMN purity was determined by Giemsa staining and by flow cytometry staining of CD11b+/CD14− and ranged between 97% and 100%. Isolated PMN were resuspended at 2.5 × 106/mL in complete RPMI 1640, plated in 24-well plates, and stimulated with formyl-Met-Leu-Phe-OH (fMLP; 1 μmol/L) or phorbol 12-myristate 13-acetate (PMA; 20 ng/mL) for 30 min at 37°C and 5% CO2. In some experiments, activated PMN were centrifuged over Ficoll-Hypaque.
Arginase activity
Plasma and cytoplasmic extracts from PBMC and from MDSC and PMN were tested for arginase activity by conversion of l-arginine to l-ornithine, as described elsewhere (6).
ELISA
Arginase I levels were tested in plasma of healthy volunteer donors and RCC patients using an ELISA kit (BioVendor). Levels of VEGF, gelatinase B [matrix metalloproteinase (MMP)-9], gelatinase A (MMP-2), and IFNγ were determined in plasma by ELISA kits from R&D Systems, following the vendor’s protocols. Myeloperoxidase (MPO) activity kit was determined using InnoZyme activity kit (Calbiochem/EMD Biosciences). Detection of nitrotyrosine in plasma was performed by using BIOXYTECH Nitrotyrosine-EIA Assay from Oxis International.
Arginase I mRNA detection by real-time PCR
Total mRNA was isolated from MDSC and PMN from RCC patients and PMN from normal controls by Trizol (Invitrogen). Two micrograms of RNA were converted into cDNA using the high-capacity RNA-to-cDNA kit from Applied Biosystems. Real-time PCR was done using probe-based detection from Applied Biosystems (Taqman Gene Expression Assays) for the human arginase I (Applied Biosystems). Housekeeping controls included the human large ribosomal protein (Applied Biosystems) and the human cyclophilin A (Applied Biosystems) and were detected using kits for the detection of endogenous controls (Taqman Endogenous Controls) from Applied Biosystems. The reaction contained 1 μg cDNA and 1× primers and probes in a final volume of 10 μL. The real-time PCR was conducted in a 7900 HT real-time PCR machine (Applied Biosystems) with an initial hold for 2 min at 50°C followed by a hold at 95°C during 10 min and 40 cycles of 95°C for 15 s and 60°C for 1 min. PCR results were analyzed with the Sequence Detection Software version 2.2 (Applied Biosystems) and are expressed as fold induction of arginase I mRNA in relation to the average cycle threshold of the two endogenous controls.
Western blots and immunoprecipitations
Cell lysates (30 μg) were electrophoresed in 10% Tris-glycine gels, transferred to polyvinylidene difluoride membranes, and immunobloted with anti-human arginase I, arginase II (Santa Cruz Biotechnology), and glyceraldehyde-3-phosphate dehydrogenase (RDI). One hundred microliters of plasma from RCC patients and controls were immunoprecipitated using antibodies against arginase II, following a protocol previously described (12). Arginase II–positive control included the murine renal cell line clone 19 (a kind gift from Dr. Robert Wiltrout, National Cancer Institute, Bethesda, MD).
l-arginine and l-ornithine serum levels
High-performance liquid chromatography (HPLC)-electrochemical detector was performed using an ESA CoulArray Model 540, with an 80 × 3.2 column with 120A pore size (13). Plasma was deproteinized and derivatized with 0.2 mol/L ophthalaldehyde/β-mercaptoethanol. Fifty microliters were injected per sample. Standards of l-arginine in methanol were run with each experiment.
Apoptosis assay
Expression of Annexin V, as a measurement of apoptosis, was tested in freshly isolated MDSC and PMN from RCC patients and PMN from normal controls. Positive controls for apoptosis were PMN isolated from normal controls and activated with PMA (20 ng/mL) for 30 min. PMN and tumor cell cocultures were done by culturing 1 × 106 human RCC tumor cell line 786-O cells in the bottom chamber with 5 × 106 PMN in the upper chamber of a Transwell system (0.4 μm pores; A Falcon-Becton Dickinson). Controls included PMN cultured without tumor cells. The expression of Annexin V/propidium iodide was tested after 6, 12, and 24 h using the Annexin V-FITC Apoptosis Detection kit (BD Pharmingen).
Statistics
Statistical analysis was done using GraphPad Prism 3.0 (GraphPad Software). Differences between the groups were determined by Students’ t test and ANOVA.
Results
Human MDSC in patients with RCC express markers of activated PMN
Our group and others (2, 3) have described that MDSC found in peripheral blood of patients with RCC and pancreatic carcinoma separate with PBMC when centrifuged over a Ficoll-Hypaque gradient and have the morphology of granulocytes. These MDSC are CD11b+ and CD15+, express high levels of arginase I, and are negative for or express low levels of the macrophage/monocyte marker CD14 (3). We have compared the expression of these and other markers, including CD10, CD11b, CD11c, CD13, CD15, CD16, CD24, CD33, CD62L, CD66b, and VEGFR1, between MDSC isolated from PBMC of RCC patients and PMN from normal controls. MDSC have high levels of CD11b, CD66b, and VEGFR1 (Flt-1) and low levels of CD62L and CD16 compared with normal granulocytes (Fig. 1A). This phenotype fits that of an activated PMN previously described by Elghetany (14). Autologous PMN (from RCC patients) that do not copurify with PBMC show a similar phenotype as PMN isolated normal controls (data not shown). We also found that MDSC and control PMN express similarly high levels of other granulocyte markers, such as CD15, CD33, CD13, CD11c, and CD24 (data not shown).
Figure 1.
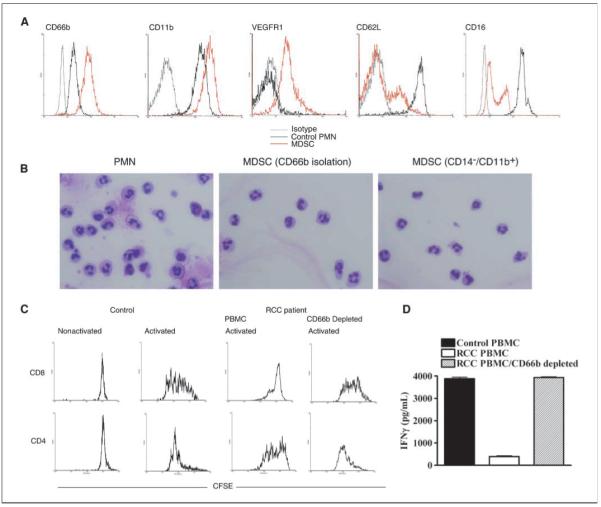
MDSC express markers of activated PMN and suppress CD8+ T-cell proliferation and IFNγ production. A, a representative flowcytometry analysis of MDSC from RCC patients (n = 10) compared with PMN from normal controls (n = 8) showing increased expression of CD66b, CD11b, and VEGFR1 and low expression of CD16 and CD62L in MDSC. B, a representative cytospin of PMN and MDSC (sorted using anti-CD66b or anti-CD11b+/CD14−) from peripheral blood of RCC patients (n = 10). C, 5 × 105 PBMC labeled with CFSE were activated with immobilized anti-CD3 (1 μg/mL) and anti-CD28 (0.1 μg/mL) antibodies. After 96 h of culture, T cells were labeled using antibodies against CD4 and CD8 and proliferation was determined by flow cytometry. D, supernatants were also collected 72 h after stimulation with anti-CD3/CD28 and measured for IFNγ production by ELISA. C and D, experiments were repeated thrice using samples from individual RCC patients and controls.
Cytospins comparing the morphology of MDSC (sorted by CD11b+/CD14− or by CD66b+) and autologous PMN from RCC patients showed a significant similarity (Fig. 1B), with a high percentage (~90%) of granulocytes with a segmented mature morphology and <10% of bands and other immature granulocytes cells.
MDSC suppress T-cell proliferation and IFNγ production
As has been previously shown (3), MDSC isolated from peripheral blood of RCC patients suppress T-cell proliferation and IFNγ production. Similarly, PBMC from RCC patients (which contain MDSC) stimulated with anti-CD3/CD28 antibodies showed a decreased proliferation compared with normal controls. This effect was completely reversed after depletion of MDSC (Supplementary Fig. S1A). Interestingly, the proliferation of CD8+ T cells in PBMC from RCC patients was more profoundly inhibited compared with CD4+ T cells (Fig. 1C). The depletion of CD66b+ MDSC completely restored CD8+ proliferation and enhanced CD4+ T-cell proliferation (Fig. 1C). Activated PBMC from RCC patients also failed to produce IFNγ after activation, an inhibitory effect that was also reversed by the depletion of CD66b+ MDSC (Fig. 1D). Because the depletion of CD66b+ cells in the PBMC of RCC patients may increase the proportion of T cells in the experiments, we normalized the increase in thymidine incorporation and IFNγ production by using a fixed number of T cells (1 × 105). Data obtained after this analysis are similar and confirm that the depletion of CD66b+ MDSC from PBMC of RCC patients restored proliferation and IFNγ production after activation with anti-CD3 plus anti-CD28 (Supplementary Fig. S1B and C).
MDSC are activated PMN
Activation of normal PMN in vitro and in vivo leads to an increased expression of CD66b, which is mobilized from intracellular granules to the cell membrane (15, 16). In fact, MDSC from RCC patients had an increased expression of CD66b compared with autologous PMN and PMN from controls (P = 0.0272; Fig. 2A). The increase in CD66b expression was not the result of nonspecific activation of PMN during sorting procedures because PMN from RCC patients and normal controls did not have an increase in CD66b expression after using dextran and/or CD14 and CD11b magnetic beads (data not shown).
Figure 2.

Activation of PMN induces MDSC characteristics. A, MDSC were isolated from RCC patients (n = 15) by sorting of CD11b+/CD14− cells, whereas PMN (from patients and controls) were isolated over dextran. The expression of CD66b was measured as MFI by flowcytometry. B, resting PMN from normal individuals were activated with fMLP and PMA for 30 min, after which the cells were tested for CD66b expression, changes in density after centrifugation over Ficoll-Hypaque (C), and levels of arginase I in the tissue culture medium (D). B to D, columns, mean of three different experiments; bars, SD.
In an effort to understand the origin of MDSC, PMN from normal donors were activated with fMLP or PMA for 30 minutes. Similar to MDSC, the activated PMN increased the expression of CD66b (P = 0.0003; Fig. 2B) and separated in the mononuclear layer during centrifugation over Ficoll-Hypaque (Fig. 2C). We also found high levels of arginase I in the supernatants of activated PMN but not in supernatants from nonactivated PMN (P = 0.0013; Fig. 2D). These results suggested that MDSC from RCC patients could be a subset of highly activated mature PMN that degranulate, releasing arginase I into plasma.
MDSC release arginase I into the circulation of RCC patients
l-arginine depletion by murine MDSC occurs as the result of an increased uptake through CAT-2B transporter (6). In contrast, human MDSC do not express CAT-2B and therefore do not uptake l-arginine (3), which suggests that l-arginine reduction by human MDSC may be the result of arginase I release. Indeed, we found a high arginase activity (P = 0.0001; Fig. 3A) and high levels of arginase I (P = 0.0008; Fig. 3B) in the plasma of RCC patients compared with normal controls. Furthermore, the increased levels of arginase I in plasma of RCC patients was associated with low plasma levels of l-arginine (P = 0.0091; Fig. 3C). To determine the possible role of arginase II, we tested its presence in plasma by immunoprecipitation using an anti-arginase II antibody because there are no existing ELISA assays for arginase II. Minimal and similar amounts of arginase II were detected in the plasma of both RCC patients and normal controls (Fig. 3D).
Figure 3.
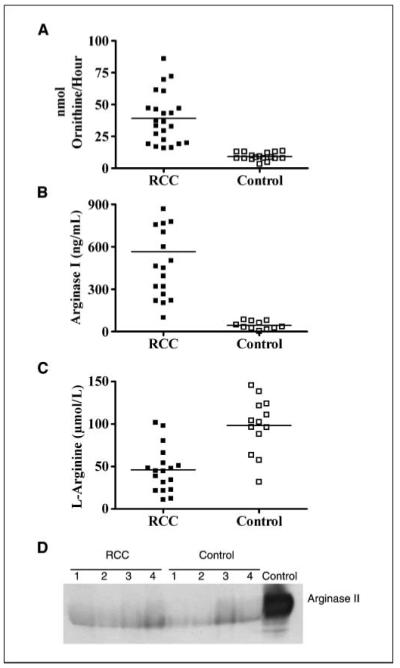
High arginase activity and arginase I protein are found in the plasma of RCC patients. Plasma obtained from RCC patients (n = 23) and controls (n = 13) was tested for arginase activity (A), arginase I protein by ELISA (B), and l-arginine levels by HPLC (C). D, arginase II protein expression in plasma of RCC patients and controls was tested by immunoprecipitation.
If arginase I is released from MDSC, one should expect a decrease in the levels of intracellular arginase I in MDSC. In fact, MDSC purified from RCC patients had lower arginase protein activity (Fig. 4A) and expression (Supplementary Fig. S2A) compared with autologous PMN or granulocytes from normal controls. Interestingly, arginase I mRNA expression was increased in MDSC compared with autologous PMN and PMN from normal controls (P = 0.0109), suggesting a compensatory increase in arginase I transcription. Previous reports have suggested that arginase I in PMN could be stored in primary granules (17) and tertiary or gelatinase granules (18). Therefore, to confirm the degranulation of MDSC in RCC patients, we tested MPO activity (located in primary granules) and the levels of gelatinase B (located in gelatinase or tertiary granules) in the plasma of RCC patients. We found high levels of both MPO activity (P = 0.0043; Fig. 4C) and gelatinase B (P = 0.0008; Fig. 4D) in the plasma of RCC patients compared with control individuals. In addition, the levels of gelatinase A, which is not produced by PMN (19), were similar in the plasma from RCC patients and controls (Supplementary Fig. S2B). Under limiting amounts of l-arginine, reactive oxygen species may be transformed into peroxynitrites (ONOO2), a highly reactive oxidizing agent that nitrosylates proteins and induces T-cell apoptosis (20). This affects the conformational flexibility of the TCR and its interaction with MHC by inducing nitrosylation of TCR proteins in CD8+ cells (21). However, we did not find significant differences in the levels of nitrotyrosine in the plasma of RCC patients compared with normal controls (Supplementary Fig. S2C).
Figure 4.

Degranulation and release of arginase I decreases intracellular arginase I in MDSC. Arginase activity (A) and arginase I mRNA expression (B) were determined in MDSC and autologous PMN from RCC patients (n = 19) and PMN from controls (n = 13). PMN from RCC patients were isolated from the fraction of cells that did not separate with PBMC over Ficoll-Hypaque. Plasma from RCC patients (n = 5) and controls (n = 6) was tested for the levels of MPO activity (C) and gelatinase B (D) by ELISA.
MDSC and apoptosis
After activation and degranulation, PMN normally undergo apoptosis, which is fundamental for their removal and the resolution of inflammation (22, 23). Interestingly, there was no increase in the expression of Annexin V in MDSC collected from RCC patients compared with the autologous PMN or to PMN from normal donors (Fig. 5A). In contrast, normal PMN activated with PMA expressed high levels of Annexin V. Furthermore, apoptosis mRNA arrays did not show an increase in the expression of proapoptotic or a decrease in antiapoptotic genes in MDSC (data not shown). We tested whether tumor microenvironment might protect PMN from undergoing spontaneous apoptosis. For this, we cocultured a human RCC cell line (786-O) with PMN from normal controls using Transwells. As shown in Fig. 5B, PMN cocultured with the RCC cell line had lower expression of Annexin V after 6 hours of culture compared with PMN cultured alone. It is, however, unclear what tumor-derived factor(s) is responsible for this antiapoptotic effect.
Figure 5.
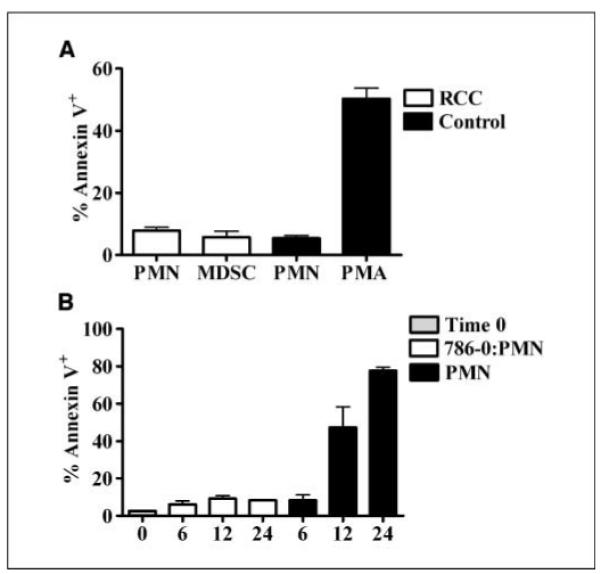
MDSC have decreased levels of Annexin V. A, apoptosis was tested by the expression of Annexin V in MDSC and autologous PMN from RCC patients (n = 11) and PMN from normal controls (n = 13). Positive controls were PMN from healthy donors activated in vitro with 20 ng/mL PMA for 1 h. B, 1 × 106 human RCC tumor cell line 786-O cells were cocultured in Transwells (0.4 μm pores) with 5 × 106 PMN and Annexin V expression was tested after 6, 12, and 24 h. Tumor cells were in the lower chamber, whereas PMN were in the upper chamber. Controls included PMN cultured alone for the same time period.
Bevacizumab treatment does not decrease the percentage of MDSC or change the levels of arginase I
Previous results have suggested that high serum levels of VEGF increase the numbers of immature dendritic cells in patients with cancer (7, 8). We tested the effect of anti-VEGF antibody, bevacizumab, in the accumulation of MDSC and arginase activity in peripheral blood of RCC patients. Peripheral blood was collected from RCC patients 1 day before treatment with bevacizumab. Two weeks later, blood was collected before patients received a second dose of bevacizumab and started IL-2 (day 15). A final sample was collected 2 weeks after that (day 29). Bevacizumab treatment effectively decreased VEGF levels in the plasma of RCC patients (Fig. 6A); however, it did not change the accumulation of MDSC in peripheral blood, nor the levels of arginase I and l-arginine in plasma (Fig. 6B–D). In fact, an increased percentage of MDSC (P = 0.01) and higher levels of free arginase I (P = 0.03) were noted in the peripheral blood of patients after receiving the second dose of bevacizumab plus IL-2 (Fig. 6B and C). This resulted in slightly lower levels of l-arginine (Fig. 6D). in vitro culture of normal PMN with VEGF and/or IL-2, however, failed to increase CD66b expression (data not shown), suggesting that the increase in MDSC numbers in RCC patients might not be a direct effect of VEGF or the treatment with IL-2 but a result of the release of other factor(s) from cells activated by IL-2.
Figure 6.

Treatment with anti-VEGF antibody bevacizumab does not prevent the accumulation of MDSC in peripheral blood of RCC patients. RCC patients (n = 23) receiving bevacizumab followed by bevacizumab/IL-2 therapy were tested for levels of VEGF (A), number of MDSC in peripheral blood (B), arginase I (C), and l-arginine concentrations in plasma (D). Normal control samples were included in each measurement (n = 12). A, levels of VEGF were tested by ELISA. B, the percentage of MDSC (CD66b+) in the PBMC of RCC patients was measured by flowcytometry. C, the levels of arginase I were determined by ELISA. D, the levels of l-arginine in plasma were determined by HPLC.
Discussion
Tumor-induced T-cell anergy/tolerance can severely impair the potential therapeutic benefit of immunotherapy. Among the possible mechanisms leading to anergy/tolerance is the depletion of the amino acids tryptophan and l-arginine (24). These mechanisms are usually triggered by the interaction of T cells with tumor cells or myeloid cells (macrophages, immature dendritic cells, and granulocytes). Several reports have described the presence of a highly suppressive subpopulation of myeloid cells infiltrating mouse tumors, which has an increased arginase activity, depletes l-arginine, and blocks T-cell proliferation and cytokine production (6, 25, 26). Murine MDSC infiltrating s.c. 3LL lung carcinoma tumors are mature monocytes/macrophages expressing arginase I (6). Blocking arginase eliminated the MDSC suppressor function in vitro and induced an antitumor effect in vivo. In contrast, human MDSC found in the peripheral blood of patients with advanced RCC and pancreatic cancer were morphologically similar to granulocytes rather than mononuclear cells (2, 3). Human MDSC from RCC patients express markers of mature activated PMN, including high levels of CD66b and low levels of CD62L and CD16. Neutrophils are known to contribute to innate and adaptive immune responses, but their role in tumors has been unclear. PMN producing H2O2 and copurifying with PBMC have been previously reported as a mechanism to explain T-cell dysfunction in cancer patients (2, 27). In our studies, the addition of H2O2 scavenger catalase (up to 1,000 units/mL) did not restore CD4+ or CD8+ T-cell proliferation nor IFNγ production (data not shown).
High production of arginase I by MDSC is not unique to cancer. Activated PMN producing arginase I accumulate in the peripheral blood and placenta of pregnant women, suggesting that arginase I plays a role in suppressing the maternal immune responses against the fetus (28). In addition, researchers have shown a correlation between low levels of CD3ζ chain and high arginase activity in PBMC of patients with active pulmonary tuberculosis (29). Furthermore, granulocyte activation and degranulation has been described in abscesses of patients where pus (which has a large number of activated PMN) induces a profound suppression of T-cell proliferation and cytokine synthesis (17). After granulocyte activation, PMN normally undergo apoptosis, which is critical for the resolution of the inflammation. Surprisingly, we did not detect an increase in apoptosis in MDSC from RCC patients. Therefore, although activation and degranulation of PMN is important in controlling acute infections, it may become a powerful inhibitor of T-cell function in a chronic inflammatory microenvironment where the offending agent (progressively growing malignant cells) is not eliminated.
The primary mechanism of l-arginine depletion by murine MDSC is the increased uptake of l-arginine through CAT-2B (6). In contrast, human MDSC do not express CAT-2B and therefore do not uptake l-arginine. Instead, human MDSC release arginase I from intracellular granules into the microenvironment, where it depletes l-arginine and induces T-cell dysfunction. There is controversy on whether arginase I is stored in primary or in the gelatinase granules of PMN (17, 18). We found an increase in both MPO and gelatinase B in the plasma of RCC patients compared with normal controls. Gelatinase A, which is not produced by PMN (19), was similarly expressed in plasma of RCC patients and controls. Although these results do not answer where arginase is stored, they confirm the degranulation of MDSC in RCC patients. In addition, one would expect to see a decrease in the intracellular arginase I following degranulation. Indeed, arginase I protein levels and arginase activity were lower in MDSC compared with autologous PMN. In contrast, arginase I mRNA expression was higher in MDSC compared with autologous PMN and PMN from normal controls. This could be interpreted as an increase in arginase I mRNA transcription in MDSC as a response to the increased arginase I protein release or that arginase protein is decreased by posttranscriptional or translational regulations in MDSC.
The mechanisms of accumulation of human MDSC in peripheral blood are still unclear. Previous results have suggested an association between VEGF levels and an increase of immature dendritic cells that suppressed T-cell function (30). We therefore hypothesized that VEGF could also mediate the accumulation of MDSC in the peripheral blood of RCC patients. However, although treatment with anti-VEGF antibody decreased VEGF levels, it did not decrease the number of MDSC in peripheral blood. This contrasts with a recent report by Kusmartsevand colleagues (31), where the use of anti-VEGF antibody prevented the accumulation of murine MDSC in athymic mice injected with human renal cell line Caki. These discrepancies may be the result of the timing in which anti-VEGF antibody is used (i.e., the RCC patients had advanced tumors and high numbers of MDSC before receiving anti-VEGF), whereas mice injected with RCC tumor received anti-VEGF during early stages of tumor development. Interestingly, the addition of IL-2 to anti-VEGF antibody treatment resulted in a marked increase in the percentage of MDSC in blood and the levels of arginase I in plasma. The mechanism for this effect by IL-2 is unknown. Activation of normal PMN with IL-2 alone or with VEGF alone failed to induce activation of PMN in vitro, which suggests that this effect was not directly induced by these cytokines. It is possible that other soluble factors produced by IL-2–activated natural killer cells, macrophages, T lymphocytes, or tumor cells may play a role in the induction of arginase I release by IL-2. The effect of IL-2 on granulocytes is complex. An increased number of granulocytes was previously shown to be associated with poor responses to IL-2 treatment in patients with RCC (32). In patients treated with high-dose IL-2, neutrophil chemotaxis is profoundly inhibited, although superoxide production, bactericidal activity, and the secretion of neutrophil granule constituents remained normal or increased throughout IL-2 treatment (33, 34).
In summary, tumors seem to hijack a protective acute inflammatory process primarily mediated by PMN, converting it instead into a chronic inflammatory event that results in T-cell anergy/tolerance and facilitates tumor growth. Approaches that inhibit this chronic inflammatory event mediated by MDSC may enhance the therapeutic efficacy of immunotherapy.
Supplementary Material
Acknowledgments
Grant support: National Cancer Institute, NIH, grants CA82689 and CA107974. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
We thank the following physicians for their important contribution in collecting samples from patients: Dr. David McDermott (Beth Israel Deaconess Medical Center), Dr. Joseph I. Clark (Loyola University), Dr. Lawrence E. Flaherty (Wayne State University), Dr. Geoffrey R. Weiss (University of Texas), Dr. Theodore F. Logan (Indiana University), Dr. John M. Kirkwood (University of Pittsburgh), Dr. Michael S. Gordon (University of Arizona), Dr. Jeffrey A. Sosman (Vanderbilt University), Dr. Christopher P.G. Tretter (Dartmouth Medical Center), Dr. Walter J. Urba (Robert W. Childs Cancer Center), Dr. Kim A. Margolin (City of Hope Cancer Center), Dr. Jared A. Gollob (Duke University), and Dr. Janice P. Dutcher (Our Lady of Mercy Cancer Center).
Footnotes
Disclosure of Potential Conflicts of Interest
A. Ochoa is a consultant for NewLink Genetics Inc. The other authors disclosed no potential conflicts of interest.
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
References
- 1.McDermott DF, Regan MM, Clark JI, et al. Randomized phase III trial of high-dose interleukin-2 versus subcutaneous interleukin-2 and interferon in patients with metastatic renal cell carcinoma. J Clin Oncol. 2005;23:133–41. doi: 10.1200/JCO.2005.03.206. [DOI] [PubMed] [Google Scholar]
- 2.Schmielau J, Finn OJ. Activated granulocytes and granulocyte-derived hydrogen peroxide are the underlying mechanism of suppression of T-cell function in advanced cancer patients. Cancer Res. 2001;61:4756–60. [PubMed] [Google Scholar]
- 3.Zea AH, Rodriguez PC, Atkins MB, et al. Arginase-producing myeloid suppressor cells in renal cell carcinoma patients: a mechanism of tumor evasion. Cancer Res. 2005;65:3044–8. doi: 10.1158/0008-5472.CAN-04-4505. [DOI] [PubMed] [Google Scholar]
- 4.Gabrilovich DI, Bronte V, Chen SH, et al. The terminology issue for myeloid-derived suppressor cells. Cancer Res. 2007;67:425. doi: 10.1158/0008-5472.CAN-06-3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rodriguez PC, Zea AH, DeSalvo J, et al. l-arginine consumption by macrophages modulates the expression of CD3ζ chain in T lymphocytes. J Immunol. 2003;171:1232–9. doi: 10.4049/jimmunol.171.3.1232. [DOI] [PubMed] [Google Scholar]
- 6.Rodriguez PC, Quiceno DG, Zabaleta J, et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res. 2004;64:5839–49. doi: 10.1158/0008-5472.CAN-04-0465. [DOI] [PubMed] [Google Scholar]
- 7.Almand B, Resser JR, Lindman B, et al. Clinical significance of defective dendritic cell differentiation in cancer. Clin Cancer Res. 2000;6:1755–66. [PubMed] [Google Scholar]
- 8.Almand B, Clark JI, Nikitina E, et al. Increased production of immature myeloid cells in cancer patients: a mechanism of immunosuppression in cancer. J Immunol. 2001;166:678–89. doi: 10.4049/jimmunol.166.1.678. [DOI] [PubMed] [Google Scholar]
- 9.Gabrilovich D, Ishida T, Oyama T, et al. Vascular endothelial growth factor inhibits the development of dendritic cells and dramatically affects the differentiation of multiple hematopoietic lineages in vivo. Blood. 1998;92:4150–66. [PubMed] [Google Scholar]
- 10.Presta LG, Chen H, O’Connor SJ, et al. Humanization of an anti-vascular endothelial growth factor monoclonal antibody for the therapy of solid tumors and other disorders. Cancer Res. 1997;57:4593–9. [PubMed] [Google Scholar]
- 11.Yang JC, Haworth L, Sherry RM, et al. A randomized trial of bevacizumab, an anti-vascular endothelial growth factor antibody, for metastatic renal cancer. N Engl J Med. 2003;349:427–34. doi: 10.1056/NEJMoa021491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rodriguez PC, Quiceno DG, Ochoa AC. l-arginine availability regulates T-lymphocyte cell-cycle progression. Blood. 2007;109:1568–73. doi: 10.1182/blood-2006-06-031856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tcherkas YV, Kartsova LA, Krasnova IN. Analysis of amino acids in human serum by isocratic reversed-phase high-performance liquid chromatography with electrochemical detection. J Chromatogr A. 2001;913:303–8. doi: 10.1016/s0021-9673(00)01206-1. [DOI] [PubMed] [Google Scholar]
- 14.Elghetany MT. Surface antigen changes during normal neutrophilic development: a critical review. Blood Cells Mol Dis. 2002;28:260–74. doi: 10.1006/bcmd.2002.0513. [DOI] [PubMed] [Google Scholar]
- 15.Kormoczi GF, Wolfel UM, Rosenkranz AR, Horl WH, Oberbauer R, Zlabinger GJ. Serum proteins modified by neutrophil-derived oxidants as mediators of neutrophil stimulation. J Immunol. 2001;167:451–60. doi: 10.4049/jimmunol.167.1.451. [DOI] [PubMed] [Google Scholar]
- 16.Sengelov H, Follin P, Kjeldsen L, Lollike K, Dahlgren C, Borregaard N. Mobilization of granules and secretory vesicles during in vivo exudation of human neutrophils. J Immunol. 1995;154:4157–65. [PubMed] [Google Scholar]
- 17.Munder M, Mollinedo F, Calafat J, et al. Arginase I is constitutively expressed in human granulocytes and participates in fungicidal activity. Blood. 2005;105:2549–56. doi: 10.1182/blood-2004-07-2521. [DOI] [PubMed] [Google Scholar]
- 18.Jacobsen LC, Theilgaard-Monch K, Christensen EI, Borregaard N. Arginase 1 is expressed in myelocytes/metamyelocytes and localized in gelatinase granules of human neutrophils. Blood. 2007;109:3084–7. doi: 10.1182/blood-2006-06-032599. [DOI] [PubMed] [Google Scholar]
- 19.Opdenakker G, Van den Steen PE, Dubois B, et al. Gelatinase B functions as regulator and effector in leukocyte biology. J Leukoc Biol. 2001;69:851–9. [PubMed] [Google Scholar]
- 20.Kusmartsev S, Gabrilovich DI. STAT1 signaling regulates tumor-associated macrophage-mediated T cell deletion. J Immunol. 2005;174:4880–91. doi: 10.4049/jimmunol.174.8.4880. [DOI] [PubMed] [Google Scholar]
- 21.Nagaraj S, Gupta K, Pisarev V, et al. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat Med. 2007;13:828–35. doi: 10.1038/nm1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Borregaard N, Sorensen OE, Theilgaard-Monch K. Neutrophil granules: a library of innate immunity proteins. Trends Immunol. 2007;28:340–5. doi: 10.1016/j.it.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 23.Theilgaard-Monch K, Porse BT, Borregaard N. Systems biology of neutrophil differentiation and immune response. Curr Opin Immunol. 2006;18:54–60. doi: 10.1016/j.coi.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 24.Rabinovich GA, Gabrilovich D, Sotomayor EM. Immunosuppressive strategies that are mediated by tumor cells. Annu RevImmunol. 2007;25:267–96. doi: 10.1146/annurev.immunol.25.022106.141609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bronte V, Apolloni E, Cabrelle A, et al. Identification of a CD11b(+)/Gr-1(+)/CD31(+) myeloid progenitor capable of activating or suppressing CD8(+) T cells. Blood. 2000;96:3838–46. [PMC free article] [PubMed] [Google Scholar]
- 26.Kusmartsev S, Nefedova Y, Yoder D, Gabrilovich DI. Antigen-specific inhibition of CD8+ T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. J Immunol. 2004;172:989–99. doi: 10.4049/jimmunol.172.2.989. [DOI] [PubMed] [Google Scholar]
- 27.Schmielau J, Nalesnik MA, Finn OJ. Suppressed T-cell receptor ζ chain expression and cytokine production in pancreatic cancer patients. Clin Cancer Res. 2001;7:933–9s. [PubMed] [Google Scholar]
- 28.Kropf P, Baud D, Marshall SE, et al. Arginase activity mediates reversible T cell hyporesponsiveness in human pregnancy. Eur J Immunol. 2007;37:935–45. doi: 10.1002/eji.200636542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zea AH, Culotta KS, Ali J, et al. Decreased expression of CD3ζ and nuclear transcription factor κB in patients with pulmonary tuberculosis: potential mechanisms and reversibility with treatment. J Infect Dis. 2006;194:1385–93. doi: 10.1086/508200. [DOI] [PubMed] [Google Scholar]
- 30.Gabrilovich DI, Chen HL, Girgis KR, et al. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat Med. 1996;2:1096–103. doi: 10.1038/nm1096-1096. [DOI] [PubMed] [Google Scholar]
- 31.Kusmartsev S, Eruslanov E, Kubler H, et al. Oxidative stress regulates expression of VEGFR1 in myeloid cells: link to tumor-induced immune suppression in renal cell carcinoma. J Immunol. 2008;181:346–53. doi: 10.4049/jimmunol.181.1.346. [DOI] [PubMed] [Google Scholar]
- 32.Donskov F, Bennedsgaard KM, Hokland M, et al. Leukocyte orchestration in blood and tumour tissue following interleukin-2 based immunotherapy in metastatic renal cell carcinoma. Cancer Immunol Immunother. 2004;53:729–39. doi: 10.1007/s00262-004-0525-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Klempner MS, Noring R, Mier JW, Atkins MB. An acquired chemotactic defect in neutrophils from patients receiving interleukin-2 immunotherapy. N Engl J Med. 1990;322:959–65. doi: 10.1056/NEJM199004053221404. [DOI] [PubMed] [Google Scholar]
- 34.Mier JW, Vachino G, Klempner MS, et al. Inhibition of interleukin-2-induced tumor necrosis factor release by dexamethasone: prevention of an acquired neutrophil chemotaxis defect and differential suppression of interleukin-2-associated side effects. Blood. 1990;76:1933–40. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.