

Abstract
The glmS catalytic riboswitch is part of the 5'-untranslated region of mRNAs encoding glucosamine-6-phosphate (GlcN6P) synthetase (glmS) in numerous Gram-positive bacteria. Binding of the cofactor GlcN6P induces site-specific self-cleavage of the RNA. However, detailed reaction mechanism as well as protonation state of glmS reactive form remains still elusive. To probe the dominant protonation states of key active site residues, we carried out explicit solvent molecular dynamic simulations involving various protonation states of three crucial active site moieties observed in the available crystal structures: (i) guanine G40 (following the T. tengcongensis numbering), (ii) the GlcN6P amino/ammonium group, and (iii) the GlcN6P phosphate moiety. We found that a deprotonated G40− seems incompatible with the observed glmS active site architecture. Our data suggest that the canonical form of G40 plays a structural role by stabilizing an in-line attack conformation of the cleavage site A-1(2'-OH) nucleophile, rather than a more direct chemical role. In addition, we observe weakened cofactor binding upon protonation of the GlcN6P phosphate moiety, which explains the experimentally observed increase of Km with decreasing pH. Finally, we discuss a possible role of cofactor binding and its interaction with the G65 and G1 purines in structural stabilization of the A-1(2'-OH) in-line attack conformation. Based on the identified dominant protonation state of the reaction precursor, we propose a hypothesis of self-cleavage mechanism, in which A-1(2'-OH) is activated as nucleophile by the G1(pro-Rp) non-bridging oxygen of the scissile phosphate, whereas the ammonium group of GlcN6P acts as the general acid protonating the G1(O5') leaving group.
Keywords: riboswitch, ribozyme, RNA structure, molecular dynamics simulations
INTRODUCTION
Riboswitches are RNA motifs embedded in messenger RNAs (mRNAs) that regulate gene expression in response to binding of a specific small molecule ligand.1–6 The glmS catalytic riboswitch (or ribozyme) is part of the 5'-untranslated region (5'-UTR) of mRNAs encoding glucosamine-6-phosphate synthetase (glmS), which catalyzes the conversion of glutamine and fructose-6-phosphate to glucosamine-6-phosphate (GlcN6P) and glutamate in numerous Gram-positive bacteria (Fig. 1).7–9 Riboswitches typically modulate gene expression on the transcriptional or translational level by undergoing structural rearrangements upon ligand binding that involve a transcription (anti-) terminator or ribosome binding site, respectively.10–12 However, GlcN6P binding to the glmS riboswitch does not lead to any detectable structural rearrangements.8,13–15 Instead, site-specific self-cleavage of the glmS riboswitch is activated directly by GlcN6P binding.16 Self-cleavage consigns the RNA to the bacterial degradation pathway, which relies on the action of RNase J1 and ultimately results in down-regulation of glmS expression and GlcN6P production in a negative feedback loop.17 The mechanism of the self-cleavage involves the nucleophilic attack of the A-1(2'-OH) hydroxyl group on its neighboring scissile phosphate of G1, thereby generating the 2',3'-cyclic and 5'-OH termini of the reaction products. The same general mechanism is found in all ribozymes classified as “small” with typically less than 100 nucleotides in the catalytic core, yet the details of how the reaction participants are activated differ substantially.18–23
FIGURE 1.

Structure of the glmS riboswitch from Thermoanareobacter tengcongensis. (A) Rear and front views of the three-dimensional structure of the glmS riboswitch. Double-helical stems are shown in different colors, base stacking modules are in yellow, and unstructured parts and GNRA tetraloop are in gray. (B) The sequence and secondary structure of the glmS riboswitch. The colors of structural elements match those in panel A. Base pairs are annotated using standard classification71.
The glmS catalytic riboswitch is unique in that it completely depends on an external ligand. Mechanistic studies proved that GlcN6P is absolutely required to activate the glmS riboswitch, accelerating the cleavage rate by more than 105-fold over background hydrolysis.8,16,24 The related compound glucose-6-phosphate, which contains a hydroxyl group in place of the 2-amine of GlcN6P (Scheme 1), is a competitive inhibitor of the self-cleavage reaction.16 Weak activity was observed for glucosamine, serinol, L-serine, tris(hydroxymethyl)aminomethane, and ethanolamine, suggesting that a primary amine and hydroxyl group in a vicinal position, is the structural feature required for all effectors activating glmS.16 The cleavage rate depends on the pKa of the amino group. Thus the GlcN6P ligand acts as a cofactor and its 2-amino group is directly involved in catalysis, while the vicinal hydroxyl group is presumably required for proper effector positioning.16
SCHEME 1.
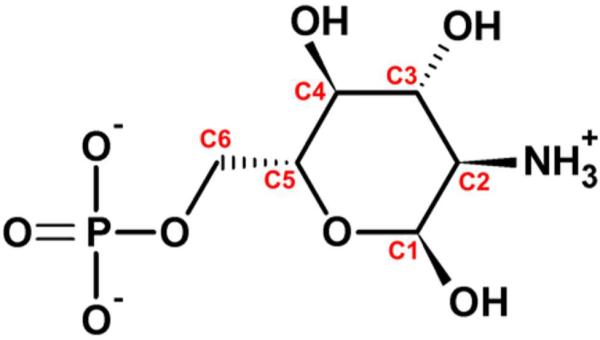
Structure and atom numbering of the cofactor GlcN6P.
Crystal structures of the glmS riboswitch from Thermoanaerobacter tengcongensis25–27 and Bacillus anthracis28 in various functional states show the RNA fold with a rigid core formed by a pseudoknot motif between the P2, P2.1, and P2.2 stems that is nested within another pseudoknot (Fig. 1). The global structure does not rearrange upon cofactor and/or inhibitor binding. In addition, the riboswitch conformation is identical over a large pH range (5.5 – 8.5).27 The pH-rate profiles show that the cleavage rate increases with pH,16 wherein the low activity at low pH originates mainly from a Km (cofactor binding affinity) rather than from a kcat (catalytic) effect.28 This observation can also explain why Ferré-D'Amaré and coworkers were unable to crystallographically observe the complex of GlcN6P with the glmS riboswitch at pH 5.5.26 These authors were, however, able to demonstrate that the P2.2 stem (Fig. 1) plays an important role in cofactor binding, with G1(N1) significantly contributing by interacting with the phosphate moiety of GlcN6P.27 The substitution of G1 with several purine analogs resulted in a switched cofactor specificity between GlcN6P and glucosamine at neutral pH, suggesting that G1 helps position the cofactor.27
G57C and G57A (G65C and G65A according to the T. tengcongensis numbering, Fig. 1B) mutations in the consensus-type glmS riboswitch resulted in minimal and no catalytic activity, respectively.29 Strobel and coworkers found that G57 (G65 in T. tengcongensis) directly interacts with the A-1 nucleobase and explained the abolished activity of the mutants by a loss of the proper in-line attack conformation of the A-1 nucleophile (Fig. 2).28 These authors also identified another substantial contact, G1(2'-OH)…G30(N7) (G1(2'-OH)…G37(N7) in T. tengcongensis), that stabilizes the reactive in-line attack conformation of A-1(2'-OH) relative to the scissile phosphate,28 consistent with the observed loss of activity upon 7-deazaguanine substitution of this guanosine.30 The active-site G40 (G33 in B. anthracis), which is in hydrogen bond distance to the A-1(2'-OH) nucleophile, was also identified as a nucleobase essential for riboswitch activity (Fig. 2).25 A G40A mutation reduces the cleavage rate constant ~105-fold, however, the crystal structure of the G40A mutant does not reveal any significant distortion of the active site (AS) compared to that of the wild-type;25 the only difference found is an increased distance between the A-1(2'-OH) nucleophile and the N1 nitrogen of A40 compared to A-1(2'-OH)…G40(N1) distance, but the in-line attack conformation of A-1(2'-OH) remains intact.25
FIGURE 2.

Stereo view of key nucleotides in the glmS riboswitch active site taken from the first snapshot of the G40/GlcN+6P2− simulation, showing a stabilized reactive in-line attack conformation of the nucleophile A-1(O2') and proper binding of GlcN6P by a specific hydrogen bond network (T. tengcongensis numbering). See supplemental Fig. S10 for enlarged but non-stereo version of this figure.
Two mechanisms were suggested for glmS riboswitch self-cleavage (Fig. 3). The conserved G40 guanine (G33 in B. anthracis) was suggested to be deprotonated in the reactive state so that G40− acts as the general base accepting the proton from the A-1(2'-OH) nucleophile. In this mechanism the ammonium form of GlcN6P is bound to the AS to act as the general acid (Fig. 3A).20,26 Ferré-D'Amaré and coworkers proposed an alternative mechanism, in which amino form of GlcN6P is bound to the AS to act as the general base accepting the proton of the A-1(2'-OH) nucleophile via two tightly bound water molecules. Subsequently, the now protonated ammonium form of GlcN6P acts as the general acid by transferring its proton to the G1(O5') leaving group (Fig. 3B).25
FIGURE 3.
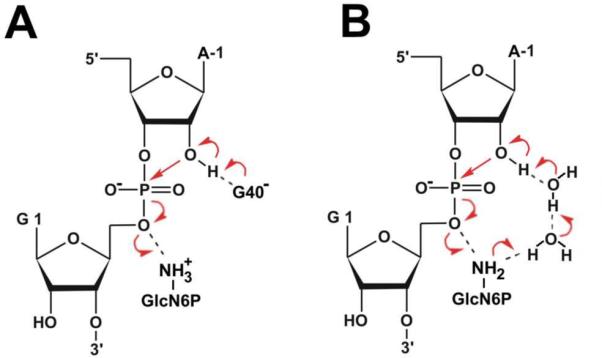
Proposed mechanisms for glmS riboswitch self-cleavage. (A) The conserved G40 is deprotonated and acts as the general base while GlcN6P acts as the general acid. (B) Glucosamine-6-phosphate acts both as the general base accepting a proton from the A-1(O2') nucleophile via two tightly bound waters and as the general acid transferring this proton to the leaving G1(O5') oxygen. Red arrows denote electron flow during the reaction.
The X-ray crystallography provides inherently static and averaged view and the crystal structure is not able to give direct information about protonation states (e.g., of key AS residues). However, the crystal structure can be effectively complemented by molecular dynamics (MD) providing structural and time-resolved information. MD with explicit inclusion of water molecules and ions is a computational tool that can be used to obtain direct atomic-resolution insights into the structural dynamics of an RNA. The applicability of the MD simulation technique is limited by the approximations of the underlying classical force field and the accessible timescale of simulations. When used wisely, however, MD can provide very valuable structural information.31–37 Among the most appropriate applications for MD simulations of RNA are the analysis of the basic structural dynamics and structural substates associated with experimentally observed, inherently averaged and static, atomic structures,38–41 the prediction of binding sites for monovalent ions,42–45 the description of specific hydration sites including the elucidation of the role of structured long-residency waters,46–48 and the prediction of structural and dynamic consequences of base modifications and substitutions including modifications of the protonation states.43,49–51 Although classical MD simulations cannot be used to directly address catalysis, they can provide plausible mechanistic hypotheses and starting structures for direct investigation of catalysis by hybrid quantum chemical/molecular mechanical methods.18,52–54
Here we apply MD simulations to the glmS riboswitch from T. tengcongensis with the native cofactor GlcN6P and different protonation states of the essential residues in the AS. MD is suitable to suggest the protonation states of the critical nucleobases corresponding to the crystalline conditions. This was previously shown, e.g., for protonated cytosines in HDV ribozyme and frameshifting pseudoknot.50,55 The aim of our simulations is to suggest dominant protonation states of AS residues, to describe the cofactor binding and to study dynamic behavior of AS in order to get ideas about plausible reaction state. In particular, our data indicate that G40 is not deprotonated and likely plays a structural role in stabilizing the in-line attack conformation of the cleavage site A-1(2'-OH). We propose that A-1(2'-OH) could be activated as nucleophile by the G1(pro-Rp) non-bridging oxygen of the scissile phosphate, whereas the ammonium group of GlcN6P acts as the general acid that neutralizes the leaving group 5'-oxygen (to avoid any confusion, we will further use terms pro-Sp for O1P and pro-Rp for O2P non-bridging oxygens according to IUPAC terminology). The presented results should be considered within the context of common limitations (mentioned above) of the contemporary simulation methods.
METHODS
Preparation of starting structures
The starting geometries were based on the currently available crystal structures. The native structure of glmS riboswitch precursor from T. tengcongensis resolved at 2.9 Å (PDB ID 2HO7) was used as a template.26 This structure was obtained with the competitive inhibitor glucose-6-phosphate (Glc6P) bound in the AS. Structure of the glmS riboswitch precursor from B. anthracis with native cofactor GlcN6P bound in the AS and inhibited by 2'-O-methyl substitution on A-1 residue (PDB ID 2NZ4) show that the competitive inhibitor Glc6P and native cofactor have the same binding pattern.28 Thus, the 2NZ4 and 2HO7 structures were superimposed and modified to derive the native glmS structure with bound native cofactor. The coordinates of the native cofactor as well as two Mg2+ ions bound to the phosphate of the cofactor with their first solvation shells were pasted from the 2NZ4 structure into the 2HO7 structure. A structural Mg2+ ion with three inner-shell contacts to the phosphates of C2, G36, and G37 in the 2HO7 structure was retained in the starting MD structure as the third divalent ion. The classical empirical non-polarizable force fields describe divalents inaccurately while divalents sample poorly in simulations.31,37,42,55,56 The three retained Mg2+ ions in the glmS structure represent specific binding patterns supported by unequivocal experimental data.26,28 However, other Mg2+ ions seen in the X-ray structure do not appear to be of any functional importance and thus were not considered in the simulations, to avoid force field artifacts arising from the inaccurate description of divalents. Finally, the system was neutralized by sodium ions as described below.
Seven starting structures differing in protonation of the important acid-base groups in the AS were prepared on the basis of this structure. These include systems with both amino and ammonium form in combination with both singly-protonated mono-charged phosphate and deprotonated double-charged phosphate of GlcN6P. The abbreviation GlcN06P2− denotes the amino form of GlcN6P with deprotonated double-charged phosphate, while GlcN+6P2− and GlcN+6P− stand for the ammonium form of GlcN6P with deprotonated double-charged and singly-protonated mono-charged phosphate, respectively. The abbreviation GlcN6P generally represents the glucosamine-6-phosphate without specification of its protonation states. Two simulations were carried out with deprotonated guanine G40− in the AS while the remaining five were executed with a canonical G40. Thus the following simulations were prepared (Tab. 1): G40−/GlcN+6P2−, G40−/GlcN+6P−, G40/GlcN06P2−, G40/GlcN+6P2−, and three independent simulations of G40/GlcN+6P− differing in the starting orientation of the hydroxyl group at phosphate moiety of the GlcN6P cofactor. The simulation in which the hydroxyl group was initially oriented toward the G1 nucleobase is labeled as G40/GlcN+6P−(G1). G40/GlcN+6P−(P2.2) represents the simulation with the hydroxyl group oriented toward the P2.2 stem, whereas the hydroxyl group of simulation G40/GlcN+6P−(bulk) was initially exposed to the bulk solvent (Supplementary Materials, Fig. S3). In addition, one reference structure was prepared with a ligand-free AS and a canonical G40 (G40/free). The structural triple inner-shell bound Mg2+ ion was retained while the other two Mg2+ ions neighboring the cofactor were deleted.
TABLE 1.
Overview of the MD simulations performed here. The presence of ligand in the active site and the charge of the acid-base groups differing in protonation state are indicated.
| GlcN6P | GlcN6P | Simulation length [ns] | |||
|---|---|---|---|---|---|
| Simulation name | cofactor | G40 | amino group | phosphate | |
| G40−/GlcN+6P2− | yes | −1 | +1 | −2 | 20 |
| G40−/GlcN+6P− | yes | −1 | +1 | −1 | 20 |
| G40/GlcN06P2− | yes | 0 | 0 | −2 | 20 |
| G40/GlcN+6P2− | yes | 0 | +1 | −2 | 50 |
| G40/GlcN+6P−(G1)a | yes | 0 | +1 | −1 | 20 |
| G40/GlcN+6P−(P2.2)a | yes | 0 | +1 | −1 | 20 |
| G40/GlcN+6P−(bulk)a | yes | 0 | +1 | −1 | 20 |
| G40/free | no | 0 | - | - | 50 |
The labels “G1”, “P2.2”, or “bulk” in parentheses indicate initial orientation of the hydroxyl of the GlcN+6P− phosphate toward G1, P2.2 stem and bulk solvent, respectively.
Molecular dynamics simulations
All structures were neutralized with Na+ counter ions (Na+ radius 1.868 Å and well depth 0.0028 kcal/mol) that were iteratively placed into the minima of the electrostatic potential calculated on a grid with spacing 1 Å using the program Leap of AMBER 9.0.57 The structures with ligand in the AS contained 3 Mg2+ ions and 136–138 Na+ ions depending on the charge of AS residues. The reference structure contained one structural Mg2+ ion and 140 Na+ ions. All structures were immersed in a rectangular box with at least 10-Å thick layer of TIP3P water molecules around the solute. The size of the boxes was ~130×80×70 Å3. The complete structures contained ~55,000 atoms, including ~6,500 water molecules. The overall concentration of monovalent ions was ~0.33 mol/l, which is entirely sufficient to provide stable RNA simulation trajectories with realistic local counter ion accumulation around the solute molecule. The whole RNA-solvent system was minimized prior to the simulations as follows. Minimization of the riboswitch hydrogen atoms was followed by minimization of counter ions and water molecules. Subsequently, the riboswitch was constrained and solvent molecules with ions were allowed to move during a 10 ps long MD run. The nucleobases were allowed to relax in several minimization runs with decreasing force constants applied to the backbone phosphate atoms. After full relaxation, each system was slowly heated to 298.15 K over 100 ps using 2-fs time steps and the NpT conditions. The simulations were evolved under periodic boundary conditions in the NpT ensemble (298.15 K, 1 atm) with 2-fs time steps. The particle-mesh Ewald method was used to calculate electrostatic interactions and a 9.0-Å cutoff was applied for Lennard–Jones interactions. The SHAKE algorithm was applied to all bonds containing hydrogen atoms. The PMEMD module of AMBER 9.057 with the Cornell et al. force field parm9958,59 was used for all simulations. The length of each simulation was 20 ns except of the G40/GlcN+6P2− and G40/free simulations that were expanded to 50 ns.
The parameters of all non-standard residues were determined by the RESP procedure of Cornell et al.60 The ab-initio calculations required for the parametrization of GlcN6P in various protonation states and deprotonated guanine were carried out using Gaussian03 (see Supplementary Materials for details and parameters).61,62
RESULTS
We carried out MD simulations of the glmS riboswitch to characterize cofactor binding and to study the dynamic behavior of the glmS riboswitch as a whole. Nonetheless, the main aim was to elucidate the dominant protonation states of the key AS residues and to obtain insights about plausible reaction mechanisms. Three different groups in the AS were identified as of uncertain protonation state: (i) The N1 nitrogen of guanine G40 that was suggested to be deprotonated in precursor state to act as the general base.26,28 (ii) The amino group of GlcN6P that was suggested to act either as general base26 or general acid28 during cleavage, and (iii) the phosphate moiety of GlcN6P (see Methods and Table 1 for details).
Basic structural dynamics shows extremely rigid pseudoknot core of glmS riboswitch
The structural dynamics and flexibility of the glmS riboswitch were monitored as B-factors of the glmS nucleotides. The pseudoknot core formed by the P2, P2.1, and P2.2 stems and including the AS was extremely rigid in all simulations. This is in agreement with the experimentally observed low B-factors in the crystal structures.26,28 By contrast, the P1, P3, P3.1 and P4 stems represent more flexible regions of the RNA fold with correspondingly high B-factors (Fig. 4). The rigidity of the pseudoknot core likely originates from a structural stabilization of the core arrangement by the coaxial P4 and P4.1 stems. They stabilize the pseudoknot fold by two interactions: i) a ribose zipper motif63 formed between the GNRA tetraloop64 closing P4.1 and the P1 stem, which contains a type I A-minor interaction65 between A117 and the C10=G31 base pair, and ii) the oblique interaction of the G128|A127|A104|A105|A106 purine stack with the minor groove of P2.1 (Fig. 5).26 This salient tertiary interaction is entirely stable and very rigid in all simulations and might represent the key stabilizing interaction of the P2.1 stem fold. The flexibility of the P1 stem is non-uniform. The ribose zipper motif between the GNRA tetraloop and P1 stem buttresses the P1 segment adjacent to the pseudoknot core up to the U12-A29 base pair, making it as rigid as the pseudoknot core itself. The remainder of the P1 stem is significantly more flexible (Fig. 4). The pseudoknot core is also stabilized by the structural Mg2+ ion. As noted above, inclusion of divalent ions into simulations is hampered by considerable limitations.31,56,66 The cation dynamics is in detail explained in Supplementary Materials).
FIGURE 4.

(A) The superimposed snapshots of G40/GlcN+6P2− simulation taken at each nanosecond demonstrate flexibility of P1, P3, P3.1 and P4 stems and rigidity of the pseudoknot core. (B) The thermal B-factors of the backbone atoms of each residue calculated from MD simulation (for comparison with X-ray B-factors see Supplementary Materials). The coloring of the stripes at the top of the plot matches the colors of stems (shown above).
FIGURE 5.

The front and rear view of the oblique interaction of G128|A127|A104|A105|A106 purine stack with minor groove of the P2.1 stem.
Four stacking modules were found in glmS riboswitch structure (a term stacking module will be further used for stacks within a single-stranded region): the abovementioned G128|A127|A104|A105|A106 stacking module between P4 and P4.1 stems, G94|G138 stacking module connecting the P3 and P4 stems, and two G65|G66 and G39|G40|U67|G41 stacking modules occurring in the AS. These stacking modules form base-zipper motif or partial base-zipper motifs associated with a crossover of single strands.67–70 We found that these stacking modules are involved in cofactor binding, AS preorganization and structural stabilization of the overall fold (see Supplementary Materials for details). Furthermore, the stacking module regions are often accompanied by non-Watson-Crick (non-WC) and base-phosphate (BPh) interactions (see Fig. 1, Supplementary Materials for analysis of non-WC and BPh interactions, and refs. 71 and 72 for their classification).
Only certain protonation states of specific active site residues are consistent with experimental active site architecture
An initial insight to the structural integrity of the AS with different protonation states was assessed by monitoring the mass-weighted root mean square deviations (RMSD) of three AS segments: (i) The sugar-phosphate backbone between the A-1(C'4) and G1(C'4) atoms encompassing the scissile phosphate; (ii) the preceding segment with the G40 base and (iii) the preceding segment including the cofactor molecule with exception of its phosphomethyl group (Fig. 6). The geometry of each segment was fit with its reference crystal structure conformation and RMSD value was calculated. Subsequently, all the structural deviations from the X-ray structure were analyzed in detail by monitoring of key hydrogen bond interactions and sugar-phosphate backbone torsions in the AS.
FIGURE 6.

The RMSDs of the selected active site region: (A) Time dependence of the RMSD from the initial crystal structure calculated for three different regions of the glmS riboswitch active site as depicted in (B) for the green line, (C) for the red line and (D) for the black line. The specific atoms used for the RMSD calculation are shown in stick representation. The lowest RMSD of ~0.5 Å is characteristic for structures with no geometrical changes compared to the starting X-ray geometries. RMSD values above ~0.5 Å indicate changes from the starting crystal structure (for detailed description see Supplementary Materials).
The AS conformation remained close to the crystal structure throughout the entire G40/GlcN+6P2− and G40/GlcN+6P−(bulk) simulations. Backbone atoms neighboring the scissile phosphate and G40 base fluctuated near the crystal structure geometry (Fig. 6, green and red lines, respectively), and the cofactor was bound tightly (Fig. 6, black line).
A rearrangement of the AS took place in both the G40/GlcN+6P−(G1) and G40/GlcN+6P−(P2.2) simulations (Figs. 6 and 7) representing the same protonation state as does the G40/GlcN+6P−(bulk) simulation, but with different initial orientation of the hydroxyl group of GlcN6P phosphate moiety (Methods and Supplemental Fig. S3). The structural changes in the AS in the G40/GlcN+6P−(P2.2) simulation were initiated by an α/γ flip of the G41 nucleobase from gauche(−)/gauche(+) to trans/trans conformation, rather than by the protonation state of the cofactor phosphate moiety (Supplemental Fig. S2). This α/γ flip caused a disruption of the G40(2'-OH)…U67(O2') hydrogen bond and subsequent rearrangement of the G40 base. The G40(2'-OH)…U67(O2') hydrogen bond was not reestablished during the remainder of the simulation even after the α/γ torsions of G41 flipped back (see Supplementary Materials). It cannot be ruled out that the α/γ flip may be related to the use of parm99 force field. This force field has been recently rendered outdated for DNA simulations due to the accumulation of pathological γ-trans substates with concomitant B-DNA degradation in longer simulations, and has been replaced by the parmbsc0 force field.73 However, the α/γ flip issue of parm99 is assumed to be irrelevant for RNA simulations where the α/γ t/t substates do occur in X-ray structures and do not accumulate during MD simulations in a manner that would destabilize the RNA structure.42,74,75 Nevertheless, we still cannot rule out that the parm99 force field modestly exaggerates the propensity to adopt the γ-trans substate for this particular nucleotide. Note that even an application of the parmbsc0 force field would not necessarily provide an ultimate answer in this case, since parmbsc0 may instead overstabilize the canonical backbone conformation and thus eliminate correct sampling of γ-trans substates.76
FIGURE 7.

Time evolution of key hydrogen bonds in our MD simulations of the glmS riboswitch, documenting cofactor binding and stability of the active site architecture. (A) Glucosamine-6-phosphate bound in the glmS active site with its hydrogen bonding network. (B) Time evolution of the hydrogen bonds of panel A. (C) Part of the active site including the scissile phosphate and two neighboring guanines with their network of hydrogen bonds. (D) Time evolution of the hydrogen bonds of panel C (the G65(N2)…G1(pro-Rp) distance marked in red is shown in panel A).
The disruption of the AS architecture in the G40/GlcN+6P−(G1) simulation was clearly initiated by a rotation of the GlcN6P phosphate moiety. The phosphate's hydroxyl group originally pointing toward the N1 and N2 nitrogen atoms of the G1 base was repelled early after 0.38 ns, which resulted in a rupture of the stabilizing contact between G65 and A-1 (Fig. 2) and subsequent AS rearrangement (see Supplementary Materials). This structural change is evidently caused by an unfavorable contact between GlcN6P phosphate's hydroxyl group and G1 in the starting structure.
RMSD analysis indicated large AS rearrangements in both simulations with deprotonated guanine (G40−/GlcN+6P2− and G40−/GlcN+6P−), primarily due to changes in the relative position of G40− and the backbone around the scissile phosphate, with some internal reconfiguration of the backbone (Fig. 6). The loss of the hydrogen bond between A-1(2'-OH) and G40−(N1), and subsequent expulsion of G40− from the AS was observed in both simulations (Fig. 7D). The G41 residue (or any other residue in proximity to AS) does not undergo any α/γ flip and thus the shift of G40− away from the scissile phosphate is unambiguously caused by the effect of G40 deprotonation (see Supplementary Materials). The G40− is not compatible with the experimental crystal structures.
Slight and almost insignificant increase in RMSD was observed in the G40/GlcN06P2− simulation (Fig. 6). However, the detailed structural analysis showed that the cofactor amino group was not able to establish a stable hydrogen bond with the U51(O4) carbonyl, in contrast to the simulations involving the protonated ammonium form of the cofactor, in which this hydrogen bond was stable over the entire simulation (Fig. 7B). The weak binding between the GlcN06P2−(N1) amino group and U51(O4) in the G40/GlcN06P2− simulation caused increased flexibility of the amino group with subsequent disruption of the G65(N2)…G1(pro-Rp) hydrogen bond. Simultaneously, U51 shifted out of the AS and influenced the conformation of the functionally important G40 base. Ultimately, also the interaction between G39(N1,N2) and the G1 (pro-Sp) non-bridging oxygen (Fig. 2) was disrupted, the backbone around the scissile phosphate rearranged and the AS remained distorted over the remainder of the simulation (see Supplementary Materials).
Cofactor binding is weakened by protonation of GlcN6P phosphate
The binding of the GlcN6P cofactor in the AS was stable in all simulations except for G40−/GlcN+6P2−, where a shift of the cofactor toward U51 and G39 and a disruption of the GlcN+6P2−(N2)…G1(O5') hydrogen bond were observed at 9 ns (Fig. 7B), accompanying the overall structural destabilization of the AS in the presence of the deprotonated G40−. In all other simulations carrying ammonium form of GlcN6P, the cofactor remained in the conformation corresponding to its crystallographic binding pattern with stable a hydrogen bond between the ammonium group of the cofactor and the G1(O′5) backbone oxygen. This hydrogen bond is assumed to be important in self-cleavage of the glmS riboswitch.16 In addition, the GlcN6P ammonium group was stabilized in its position by a hydrogen bond to the U51(O4) carbonyl (Fig. 7B), while the third hydrogen of the ammonium group was bound to a water molecule.
The stabilizing interaction between U51(O4) and the cofactor was absent in the G40/GlcN06P2− simulation (Fig. 7B), where the cofactor bore the unprotonated amino group incapable of such a hydrogen bond, causing a shift of the amino group toward the non-bridging oxygens of the scissile phosphate and subsequent AS rearrangement.
A firm and stable hydrogen bond was observed between GlcN6P C1-OH hydroxyl group and the G1(pro-Rp) oxygen in all simulations. This interaction was supported by two additional G65(N1)…GlcN6P(C1-OH) and G63(N2)…G1(pro-Rp) oxygen hydrogen bonds (Fig. 7B) which, however, were completely lost in simulation G40−/GlcN+6P2− and partially in simulations G40−/GlcN+6P− and G40/GlcN06P2− accompanied by a rearrangement of the AS. The local interaction of G65, the scissile phosphate and the C1-OH hydroxyl group of GlcN6P represents a compact binding pattern, which stabilizes the contact between the cofactor ammonium group and G1(O5').
A strong bifurcated hydrogen bond between G1(N1,N2) and the cofactor phosphate group was observed in all simulations with deprotonated double-charged phosphate. The same interaction was observed in simulations with singly-protonated mono-charged GlcN6P phosphate (one with deprotonated G40− and three with canonical G40). However, an early reorientation of the cofactor phosphate group was observed in the G40−/GlcN+6P− and G40/GlcN+6P−(G1) simulations that started with phosphate hydroxyl group oriented toward the G1 base. This hydroxyl group was exposed to the bulk solvent during the first nanosecond of the respective simulations, i.e., the hydroxyl group switched to the conformation corresponding to the starting structure of the G40/GlcN+6P−(bulk) simulation. G40/GlcN+6P−(P2.2) was the only simulation, in which the cofactor phosphate hydroxyl group was not oriented towards the bulk solvent, but rather interacted directly with the G64 or G65 bases, while in other simulations the GlcN6P phosphate interacted with guanines G64 and/or G65 via solvated Mg2+ ion.
Partially occupied hydrogen bonds were observed between the cofactor hydroxyl groups C3-OH and C4-OH and the U51(pro-Rp) non-bridging oxygen. These hydrogen bonds are fluctuating and do not appear to be essential for GlcN6P binding (Fig. 7).
In summary, both a deprotonated G40− and an unprotonated amino form of cofactor (GlcN06P instead of GlcN+6P) result in significant destabilization of cofactor binding. Further, protonation of the cofactor phosphate moiety (GlcN6P− instead of GlcN6P2−) likely negatively affects the stability of the interaction between cofactor phosphate and G1 nucleobase. More specifically, we note that the GlcN+6P− with the phosphate hydroxyl group oriented toward G1 represents an instable binding motif. By contrast, the cofactor was firmly bound in the active site when the GlcN+6P− phosphate hydroxyl group was oriented toward the bulk solvent. Thus, protonation of the GlcN6P phosphate moiety does not prevent cofactor binding for appropriate geometry but restricts a conformational variability of the cofactor, which should be accompanied with an entropic penalty for cofactor binding.
Structural dynamics of the glmS riboswitch in the absence of cofactor show that A-1(2'-OH) in-line attack conformation requires cofactor binding
Reference 50ns-long simulation of the ligand-free glmS riboswitch (without GlcN6P) was started from the crystal structure geometry (i.e., structure with bound GlcN6P) and revealed profound changes of the AS. This indicates that cofactor binding is needed for properly structuring the AS. The two-step rearrangement of the AS is described in Supplementary Materials.
In contrast to the AS occupied by cofactor, the stable ligand-free AS arrangement lacks the G65(N2)… G1(pro-Rp) hydrogen bond and the in-line attack conformation of A-1(2'-OH) (Supplemental Fig. S5 and Tab. 2). On the other hand the base-phosphate interaction between G39(N1) and the G1(pro-Sp) non-bridging oxygen of the scissile phosphate as well as the G65(N2)…A-1(N3) and G65(2'-OH)…A-1(N1) hydrogen bonds were preserved (Supplemental Fig. S4). It seems that the position of the G1 base controls the conformation of the backbone between A-1 and G1. We suggest that cofactor binding can affect the position of G1 and thus is vital for a proper local conformation of the scissile phosphate. Furthermore, cofactor binding can help to establish the interaction between the scissile phosphate and G65 and thus stabilizes the A-1 nucleotide in a way that supports the A-1(2'-OH) in-line conformation.
TABLE 2.
The mean values of A-1(2'-O)…G1(P)-G1(O5') in-line attack angle (IAA) in degrees with standard deviations in all presented simulations.
| simulation | IAA | simulation | IAA |
|---|---|---|---|
| G40−/GlcN+6P2−a,b | 155±10 | G40/GlcN+6P−(G1)a,c | 135±10 |
| G40−/GlcN+6P−a | 155±10 | G40/GlcN+6P−(P2.2)a | 165±5 |
| G40/GlcN06P2−a,b | 160±10 | G40/GlcN+6P−(bulk) | 170±5 |
| G40/GlcN+6P2− | 170±5 | G40/freed | 130±10 |
Poor or missing interaction between G40 and A-1(2'-OH).
Poor or missing interaction between G39 and G1(pro-Sp).
Disruption of G65(N2)…A-1(N3) hydrogen bond.
Missing cofactor binding.
DISCUSSION
We have carried out a set of explicit solvent MD simulations of the glmS riboswitch with the aim to elucidate its overall structural dynamics, the arrangement of its AS in the reactive state, and its major protonation state.
As the X-ray crystallography is not able to identify proton positions, the MD simulation can be employed to assign a protonation state, which corresponds to the state reflected in the X-ray structure. Here, such identification is based on structural deviations of AS residues from the X-ray structure in MD simulation applying various protonations states of AS residues. It can be expected that the protonation state corresponding to that observed in the X-ray structure (here resolved at pHs ranging form 5.5–8.5) would exhibit the smallest structural deviation in MD. This represents an indirect way, how to assign a protonation state that likely corresponds to the experimentally observed crystal structure. A direct theoretical method addressing the protonation state and estimating pKa of a given titrable group is a constant-pH molecular dynamics.77–79
However, the constant-pH MD methods that are used and widely tested for proteins77–80 typically utilize implicit solvent models, which reasonably limits their applicability in case of nucleic acids. We used the constant-pH MD as implemented in AMBER57,77 to estimate pKa values of three discussed titrable group of glmS riboswitch (see Supplementary Materials). Unfortunately, we observed a rapid and massive degradation of glmS riboswitch global fold during the first ~10ps of thermalization phase in simulations using implicit solvent (see Supplementary Materials). Thus, we restrained the structure of glmS riboswitch to its crystal-like geometry and estimated pKa using constant-pH MD. The sign of the observed pKa shifts were in agreement with the conclusions presented in this work, however, the absolute value of pKa shifts were severely overestimated (see Supplementary Materials). The overestimated pKa shifts together with the rapid degradation of RNA structure in implicit solvent show that the implicit solvent methods (at least in current implementation) are not able to efficiently screen out the electrostatics of complex folded RNA molecules. It seems that the RNA molecule remains a challenge for MD simulations in combination with implicit solvent methods.
Overall dynamics and stability
Our simulations highlight a significant rigidity of the pseudoknot core of the glmS riboswitch, formed by the P2, P2.1, and P2.2 stems, which is contrasted by the flexible outer parts including the top of the P1 stem and the pseudoknot formed by stems P3 and P3.1. The core appears to be rigidified by interactions with coaxial P4 and P4.1 stems, namely oblique interaction of the G128|A127|A104|A105|A106 stacking module with the minor groove of the P2.1 stem26 and the interaction of the GNRA tetraloop at the tip of P4.1 stem with the minor groove of the P1 stem that forms a ribose-zipper motif with class I A-minor interaction. Our simulations further suggest that the stacking modules made by stacked single strands and/or base-zipper motifs play an important role in structural stabilization, AS preorganization, and cofactor binding. There are several base-phosphate interactions that are stable in MD simulations. These interactions are often connected with the non-canonical segments of the glmS riboswitch, especially the stacking modules, and likely play an important role in the structural stabilization of its non-canonical parts. All interactions above are very stable in MD simulations.
Protonation state and proposed functional role of G40
Simulations of the glmS riboswitch were used to identify the likely dominant protonation state of three critical moieties in the AS. G40 was the first moiety of uncertain protonation state, which was previously identified by biochemical data to play an essential role in glmS self-cleavage.24,27 It was further suggested that the deprotonated G40− acts as the general base during self-cleavage.25,26,28,81 However, expulsion of the G40− base from the AS and subsequent AS distortion were observed in both simulations with a deprotonated G40−. The deprotonated G40− appears to be incompatible with the glmS AS architecture found in all available crystallographic studies. The same has been observed in simulations with deprotoned form of G8, which was suggested to be involved in the hairpin ribozyme self-cleavage (Otyepka M., Mlýnský V., unpublished data). A structural rather than catalytic role of G8 in transition state stabilizing the self-cleavage reaction in the hairpin ribozyme was suggested by York et al. who proposed that the tautomeric form of the active site guanine G8 is not likely involved in the reaction chemistry of hairpin ribozyme.52,82 We found that G40 structurally stabilizes the A-1(2'-OH) in-line attack conformation relative to the scissile phosphate and thus we suggest that G40 is important for structural and/or electrostatic stabilization of reactive state rather than directly acting in the catalytic reaction as the general base.
Still, it is possible that the deprotonated form of G40− may be involved in catalysis, however, in such a case it would likely correspond to a low-populated, transient reactive state. In the 20ns+ scale simulations, the AS of systems with G40− is entirely unstable and rearranges swiftly to very different geometries. Assuming that the contact between A-1(O2') and G40− was disrupted at the beginning of the simulation and was not reestablished for even one single 2-ps-long snapshot, a rough estimate would yield an occupation of the deprotonated G40− in the reactive state of less than 0.01%. The corresponding ΔG correction for the low-populated reactive state would be higher than 6 kcal/mol. In addition, the unperturbed pKa of guanine of 9.283 suggests a free energy correction for formation of a guanine anion of 3 kcal/mol at pH 7. Thus the reaction mechanism with G40− acting as the general base would have to be chemically favored in transition state stabilization by at least 9 kcal/mol over other alternative pathways (with significantly populated reactive state). We cannot ultimately rule out that longer simulations may rearrange the AS into a suitable, more populated geometry involving G40− that would be separated from the presently sampled geometries by an energy barrier and thus not immediately accessible. However, we have no indication what type of geometry that would be and consider this possibility as less likely. In addition, it is common in the hydrolysis of the sugar-phosphate backbone that the departure of the O5'-alcoholate from the pentahedral intermediate is the rate-limiting step, rather than the 2'-OH nucleophilic attack.84,85 Thus it appears unlikely that the increased basicity of G40− affects the reaction rate sufficiently to counteract the 9 kcal/mol penalty in ΔG arising from a low-populated G40− reactive state and rare protonation state of G40−.
Mechanistic and structural experiments have previously found that a G40A mutation abrogates most of the catalytic activity of the glmS riboswitch but does not extensively affect the arrangement of the AS in both T. tengcongensis and B. anthracis.25,81 The only difference between G40A and wild-type AS conformation is a slight increase in distance between N1 of residue 40 and the A-1(O2') nucleophile, whereas the in-line conformation remains intact.25,81 One explanation is that G40 is deprotonated and acts as the general base.25,26,28,81 Alternatively, donation of the hydrogen bond by canonical G40 to A-1(O2') could be critical for the precise positioning of the nucleophile to reactive in-line attack conformation and/or for electrostatic stabilization of the attacking A-1(2'-OH), while an in-line like position in G40A mutant is non-productive. Preliminary MD simulations of the G40A mutant (data not shown) suggest that the mutation, consistently with the crystal structures, causes only slight structural changes in the AS. The A40 remains locked in the same position as G40 of the wild type, but A-1(O2') does not establish any hydrogen bond with the A40(N1). Furthermore, the lack of a stabilizing G40(N1)…A-1(O2') hydrogen bond allows tighter binding of A-1(O2') to the G1(pro-Rp) non-bridging oxygen compared to the wild type MD simulations.
On the basis of abovementioned arguments we suggest that G40 is not directly involved in the chemical reaction but rather plays a role in electrostatic stabilization of the attacking A-1(2'-OH) nucleophile (i.e. in electrostatic transition state stabilization). The same role has been proposed also for the G8 involved in the hairpin ribozyme self-cleavage.49,52,82 However, further clarification of the inhibition effect of G40A mutation is still required.
GlcN6P - Amino or ammonium group?
The second chemical group in the AS with uncertain protonation state is the amino/ammonium group of GlcN6P. The hydrogen bond between the cofactor ammonium group and U51(O4) was stable in all simulations with ammonium form of GlcN6P, however, this hydrogen bond was missing in the simulation where the cofactor contained the (uncharged) amino group, consistently with the reduced proton donor capability of the amino compared to the ammonium group. As the hydrogen bond contact between U43(O4) (U51(O4) in T. tengcongensis) and GlcN6P nitrogen was observed in crystal structures of B. anthracis with native GlcN6P cofactor,28 the loss of this hydrogen bond detected in our MD simulations with amino form of GlcN6P shows that amino form of GlcN6P is not consistent with X-ray structures. Since the crystal structures of B. anthracis were obtained at pH 6.828 and the unperturbated pK of the GlcN6P equals to 8.2,29 our data do not support significant pKa shift of this amino/ammonium group in the active site. The simulations suggest that the ammonium form of the cofactor is preferably bound to the AS at physiological pH and is bound more tightly compared to the amino form. Thus the ammonium form of the cofactor is capable of tightly binding G1(O5') and is in a suitable position to act as general acid of the reaction.
The protonation of GlcN6P phosphate weakened cofactor binding
The third moiety of uncertain protonation state is the phosphate of GlcN6P. From the presented simulations it was not possible to clearly suggest whether the phosphate group of the cofactor bound to the AS is deprotonated (double-charged) or singly-protonated (mono-charged). Both protonation forms appear to remain bound to the AS, yet tighter binding of the cofactor phosphate moiety with G1 base was observed in the deprotonated double-charged compared to the singly-protonated mono-charged form. In addition, the binding of singly-protonated phosphate may be disadvantageous because the phosphate hydroxyl group cannot bind to the G1 base and instead prefers the orientation to the bulk solvent. This conformational restriction is likely associated with an additional entropic penalty for binding the GlcN6P with protonated phosphate moiety. The increase in Km with decreasing pH28 supports our observation that GlcN6P with protonated phosphate binds more weakly compared to the double-charged deprotonated phosphate of GlcN6P. A recent kinetic analysis shows that the pH dependence of cofactor binding is consistent with the pKa of the GlcN6P phosphate moiety, suggesting that the protonation of the phosphate moiety of GlcN6P may inhibit cofactor binding.81 This experimental finding together with the MD data lend support to the notion that GlcN6P with a fully deprotonated phosphate binds most efficiently to the glmS riboswitch.
Possible role of G65
Biochemical analysis has suggested an essential role for the G65 base (or equivalent G57 in B. anthracis) in catalysis.28,29 G65 was suggested to stabilize a proper conformation of the A-1 base and to neutralize the negative charge on the G1(pro-Rp) non-bridging oxygen of the scissile phosphate. We found that both these interactions are indeed required for stabilization of the AS in a reactive A-1(2'-OH) in-line attack conformation. In addition, we suggest that a further crucial role of G65 is to stabilize the hydrogen bond between the C1-OH group of GlcN6P and the G1(pro-Rp) oxygen that in turn helps maintain the hydrogen bond between the cofactor ammonium group and G1(O5'). The GlcN6P(C1-OH)…G1(pro-Rp) contact is the most rigid hydrogen bond in the AS of the glmS riboswitch. The G65 base stabilizes this hydrogen bond and forms the core of the GlcN6P binding motif. Our findings are consistent with the observation that ethanolamine is the minimal motif binding to the AS of the glmS riboswitch to activate self-cleavage.16 In addition, based on the simulation with ligand-free AS, we suggest that cofactor binding is crucial for establishing the interactions between the scissile phosphate, the cofactor and G65 that structurally support the A-1(2'-OH) in-line attack conformation. In other words, we suggest that there are two roles of the GlcN6P cofactor: (i) The A-1(2'-OH) in-line attack conformation is induced by cofactor binding, and (ii) the ammonium group of the cofactor acts as the general acid in the self-cleavage reaction.
Hypothesis of the reaction mechanism based on the suggested active site protonation state
Taken together, presented MD simulations suggest a plausible AS conformation of the reactive state including the protonation states of key AS residues that are compatible with X-ray structures. Based on this reactive conformation we can obtain an insight to possible mechanism of the self-cleavage reaction. Ribozymes are thought to employ four strategies to achieve catalysis:86,87 (i) They can stabilize the in-line attack conformation of the nucleophile toward the scissile phosphate. (ii) They can activate the nucleophile by deprotonation of the cleavage site 2'-hydroxyl either prior to or simultaneously with the nucleophile attack. (iii) They can neutralize the increased electron density of the scissile phosphate during catalysis, making it more susceptible to nucleophilic attack. (iv) They can help to protonate the leaving 5'-O alcoholate group. Here we argue that canonical G40 plays the role in structural and/or electrostatic stabilization of transition state rather than acting as the general base in the deprotonated form (G40−). We consequently suggest that the proton of the A-1(2'-OH) group could be transferred to the G1(pro-Rp) non-bridging oxygen of the scissile phosphate (Fig. 8). It is worth noting that the non-perturbed pKa of the non-bridging oxygen equals to ~1,83 however, the pKa of the non-bridging oxygen of the phosphorane intermediate equals ~6.584 so that the basicity of the non-bridging oxygens of the scissile phosphate is increasing during the course of the advancing nucleophilic attack. Thus, the non-bridging oxygen is capable to act as the general base during the reaction and accept the proton from the 2'-OH nucleophile. A similar reaction scenario has been proposed for the hairpin ribozyme.52,88 Simultaneously with the transfer of the proton to the G1(pro-Rp) oxygen, the electron density located on the oxygen atom would be polarized toward the incoming hydrogen making the scissile phosphate even more susceptible to nucleophilic attack. In addition to this effect, the 5BPh interaction of G39 with scissile phosphate and strong hydrogen bond pattern between C1-OH hydroxyl of GlcN6P, G65 and the G1(pro-Rp) non-bridging oxygen further draws electron density from the scissile phosphate. Finally, the ammonium group of the GlcN6P cofactor is perfectly positioned to act as general acid, protonating the leaving G1(O5') group (Fig. 8).
FIGURE 8.

Proposed mechanism for glmS riboswitch self-cleavage based on presented MD simulations. Simultaneously with the nucleophilic attack the G1(pro-Rp) non-bridging oxygen acts as the general base accepting proton from A-1(O2') nucleophile and the GlcN6P acts as the general acid to donate its proton to the leaving oxygen G1(O5'). Red arrows denote electron flow during the reaction.
CONCLUSIONS
MD simulations suggest that a deprotonated G40− is incompatible with the active site architecture observed in all glmS riboswitch crystal structures. We therefore propose that canonical G40 stabilizes the A-1(2'-OH) in-line conformation and/or plays the key role in electrostatic stabilization of transition state rather than directly participating in reaction chemistry. We suggest a possibility that the A-1(2'-OH) nucleophile is activated and deprotonated by the G1(pro-Rp) non-bridging oxygen of the scissile phosphate.
The simulations reveal that the protonated ammonium form of the cofactor is bound in the AS more tightly and is more consistent with crystal structures than its uncharged amino form. In addition, the ammonium group is in a suitable position to act as the general acid.
GlcN+6P− with a singly-protonated phosphate binds to the G1 nucleobase more weakly compared to the double-charged, deprotonated phosphate of GlcN+6P2−, consistently with experimental data.28,81
We suggest that alongside a role in structural stabilization of the A-1(2'-OH) in-line attack conformation, G65 might play a crucial role in cofactor binding by stabilizing the hydrogen bond between C1-OH hydroxyl of GlcN6P and the G1(pro-Rp) oxygen that, in turn, helps maintain the hydrogen bond between the cofactor ammonium group and G1(O5'). Thus two roles of the GlcN6P cofactor in self-cleavage are suggested: (i) The A-1(2'-OH) in-line attack conformation is induced by the cofactor binding and (ii) the ammonium group of the cofactor acts as the general acid in the reaction.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by grants LC512, LC06030, and MSM6198959216 from the Ministry of Education of the Czech Republic, grants 203/09/1476 and 203/09/H046 from the Grant Agency of the Czech Republic, grants IAA400040802 and 1QS500040581 from the Grant Agency of the Academy of Sciences of the Czech Republic, grants AV0Z50040507 and AV0Z50040702 from the Academy of Sciences of the Czech Republic, and NIH grant GM62357 (to NGW). We thank S.R. Das for helpful comments and discussions.
Footnotes
Supplementary Materials: The content of supporting information includes force field parameters of non-standard residues, a detailed analysis of the structural dynamic of glmS riboswitch active site, details of the structural dynamics of the glmS riboswitch without cofactor, analysis of constant-pH MD simulations using implicit solvent methods, and some other material. This information is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).Mandal M, Boese B, Barrick JE, Winkler WC, Breaker RR. Cell. 2003;113:577. doi: 10.1016/s0092-8674(03)00391-x. [DOI] [PubMed] [Google Scholar]
- (2).Tucker BJ, Breaker RR. Curr. Opin. Struct. Biol. 2005;15:342. doi: 10.1016/j.sbi.2005.05.003. [DOI] [PubMed] [Google Scholar]
- (3).Winkler WC, Breaker RR. Annu. Rev. Microbiol. 2005;59:487. doi: 10.1146/annurev.micro.59.030804.121336. [DOI] [PubMed] [Google Scholar]
- (4).Henkin TM. Gen. Dev. 2008;22:3383. doi: 10.1101/gad.1747308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Coppins RL, Hall KB, Groisman EA. Curr. Opin. Microbiol. 2007;10:176. doi: 10.1016/j.mib.2007.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Winkler WC. Curr. Opin. Chem. Biol. 2005;9:594. doi: 10.1016/j.cbpa.2005.09.016. [DOI] [PubMed] [Google Scholar]
- (7).Barrick JE, Corbino KA, Winkler WC, Nahvi A, Mandal M, et al. Proc. Natl. Acad. Sci. 2004;101:6421. doi: 10.1073/pnas.0308014101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Winkler WC, Nahvi A, Roth A, Collins JA, Breaker RR. Nature. 2004;428:281. doi: 10.1038/nature02362. [DOI] [PubMed] [Google Scholar]
- (9).Milewski S. BBA. 2002;1597:173. doi: 10.1016/s0167-4838(02)00318-7. [DOI] [PubMed] [Google Scholar]
- (10).Batey RT, Gilbert SD, Montange RK. Nature. 2004;432:411. doi: 10.1038/nature03037. [DOI] [PubMed] [Google Scholar]
- (11).Corbino KA, Barrick JE, Lim J, Welz R, Tucker BJ, et al. Genome Biology. 2005;6 doi: 10.1186/gb-2005-6-8-r70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Grundy FJ, Henkin TM. Crit. Rev. Biochem. 2006;41:329. doi: 10.1080/10409230600914294. [DOI] [PubMed] [Google Scholar]
- (13).Hampel KJ, Tinsley MM. Biochemistry. 2006;45:7861. doi: 10.1021/bi060337z. [DOI] [PubMed] [Google Scholar]
- (14).Fedor MJ. Annu. Rev. Biophys. 2009;38:271. doi: 10.1146/annurev.biophys.050708.133710. [DOI] [PubMed] [Google Scholar]
- (15).Tinsley RA, Furchak JRW, Walter NG. RNA. 2007;13:468. doi: 10.1261/rna.341807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).McCarthy TJ, Plog MA, Floy SA, Jansen JA, Soukup JK, et al. Chem. Biol. 2005;12:1221. doi: 10.1016/j.chembiol.2005.09.006. [DOI] [PubMed] [Google Scholar]
- (17).Collins JA, Irnov I, Baker S, Winkler WC. Gen. Dev. 2007;21:3356. doi: 10.1101/gad.1605307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Banas P, Rulisek L, Hanosova V, Svozil D, Walter NG, et al. J. Phys. Chem. B. 2008;112:11177. doi: 10.1021/jp802592z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Bevilacqua PC, Yajima R. Curr. Opin. Chem. Biol. 2006;10:455. doi: 10.1016/j.cbpa.2006.08.014. [DOI] [PubMed] [Google Scholar]
- (20).Cochrane JC, Strobel SA. Acc. Chem. Res. 2008;41:1027. doi: 10.1021/ar800050c. [DOI] [PubMed] [Google Scholar]
- (21).Strobel SA, Cochrane JC. Curr. Opin. Chem. Biol. 2007;11:636. doi: 10.1016/j.cbpa.2007.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Lilley DMJ, Eckstein F. Ribozymes and RNA Catalysis. The Royal Society of Chemistry; Cambridge: 2008. [Google Scholar]
- (23).Walter NG. Mol. Cell. 2007;28:923. doi: 10.1016/j.molcel.2007.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Roth A, Nahvi A, Lee M, Jona I, Breaker RR. RNA. 2006;12:607. doi: 10.1261/rna.2266506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Klein DJ, Been MD, Ferre-D'Amare AR. J. Am. Chem. Soc. 2007;129:14858. doi: 10.1021/ja0768441. [DOI] [PubMed] [Google Scholar]
- (26).Klein DJ, Ferre-D'Amare AR. Science. 2006;313:1752. doi: 10.1126/science.1129666. [DOI] [PubMed] [Google Scholar]
- (27).Klein DJ, Wilkinson SR, Been MD, Ferre-D'Amare AR. J. Mol. Biol. 2007;373:178. doi: 10.1016/j.jmb.2007.07.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Cochrane JC, Lipchock SV, Strobel SA. Chem. Biol. 2007;14:95. doi: 10.1016/j.chembiol.2006.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Soukup GA. Nucleic Acids Res. 2006;34:968. doi: 10.1093/nar/gkj497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Jansen JA, McCarthy TJ, Soukup GA, Soukup JK. Nat. Struct, Mol. Biol. 2006;13:517. doi: 10.1038/nsmb1094. [DOI] [PubMed] [Google Scholar]
- (31).Banas P, Jurecka P, Walter NG, Sponer J, Otyepka M. Methods. 2009;49:202. doi: 10.1016/j.ymeth.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).McDowell SE, Spackova N, Sponer J, Walter NG. Biopolymers. 2007;85:169. doi: 10.1002/bip.20620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Auffinger P, Hashem Y. Curr. Opin. Struct. Biol. 2007;17:325. doi: 10.1016/j.sbi.2007.05.008. [DOI] [PubMed] [Google Scholar]
- (34).Hall KB. Curr. Opin. Chem. Biol. 2008;12:612. doi: 10.1016/j.cbpa.2008.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Sponer J, Lankas F. Computational studies of RNA and DNA. Springer; 2006. [Google Scholar]
- (36).Cheatham TE. Curr. Opin. Struct. Biol. 2004;14:360. doi: 10.1016/j.sbi.2004.05.001. [DOI] [PubMed] [Google Scholar]
- (37).Ditzler MA, Otyepka M, Sponer J, Walter NG. Acc. Chem. Res. 2010;42:40. doi: 10.1021/ar900093g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Razga F, Koca J, Mokdad A, Sponer J. Nucleic Acids Res. 2007;35:4007. doi: 10.1093/nar/gkm245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Almlof M, Ander M, Aqvist J. Biochemistry. 2007;46:200. doi: 10.1021/bi061713i. [DOI] [PubMed] [Google Scholar]
- (40).Villa A, Wöhnert J, Stock G. Nucleic Acids Res. 2009;37:4774. doi: 10.1093/nar/gkp486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Lee TS, Lopez CS, Giambasu GM, Martick M, Scott WG, et al. J. Am. Chem. Soc. 2008;130:3053. doi: 10.1021/ja076529e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Krasovska MV, Sefcikova J, Reblova K, Schneider B, Walter NG, et al. Biophys. J. 2006;91:626. doi: 10.1529/biophysj.105.079368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Reblova K, Spackova N, Stefl R, Csaszar K, Koca J, et al. Biophys. J. 2003;84:3564. doi: 10.1016/S0006-3495(03)75089-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Auffinger P, Bielecki L, Westhof E. J. Mol. Biol. 2004;335:555. doi: 10.1016/j.jmb.2003.10.057. [DOI] [PubMed] [Google Scholar]
- (45).Lee TS, Giambasu GM, Sosa CP, Martick M, Scott WG, et al. J. Mol. Biol. 2009;388:195. doi: 10.1016/j.jmb.2009.02.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Martick M, Lee TS, York DM, Scott WG. Chem. Biol. 2008;15:332. doi: 10.1016/j.chembiol.2008.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Rhodes MM, Reblova K, Sponer J, Walter NG. Proc. Natl. Acad. Sci. 2006;103:13380. doi: 10.1073/pnas.0605090103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Razga F, Koca J, Sponer J, Leontis NB. Biophys. J. 2005;88:3466. doi: 10.1529/biophysj.104.054916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Ditzler MA, Sponer J, Walter NG. RNA. 2009;15:560. doi: 10.1261/rna.1416709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Csaszar K, Spackova N, Stefl R, Sponer J, Leontis NB. J. Mol. Biol. 2001;313:1073. doi: 10.1006/jmbi.2001.5100. [DOI] [PubMed] [Google Scholar]
- (51).Lee TS, York DM. J. Am. Chem. Soc. 2008;130:7168. doi: 10.1021/ja711242b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Nam KH, Gao JL, York DM. J. Am. Chem. Soc. 2008;130:4680. doi: 10.1021/ja0759141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Trobro S, Aqvist J. Proc. Natl. Acad. Sci. 2005;102:12395. doi: 10.1073/pnas.0504043102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Trobro S, Aqvist J. Mol. Cell. 2007;27:758. doi: 10.1016/j.molcel.2007.06.032. [DOI] [PubMed] [Google Scholar]
- (55).Krasovska MV, Sefcikova J, Spackova N, Sponer J, Walter NG. J. Mol. Biol. 2005;351:731. doi: 10.1016/j.jmb.2005.06.016. [DOI] [PubMed] [Google Scholar]
- (56).Gresh N, Sponer JE, Spackova N, Leszczynski J, Sponer J. J. Phys. Chem. B. 2003;107:8669. [Google Scholar]
- (57).Case DA, Darden TA, T.E. Cheatham I, Simmerling CL, Wang J, et al. AMBER 9. University of California; San Francisco: 2006. [Google Scholar]
- (58).Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, et al. J. Am. Chem. Soc. 1995;117:5179. [Google Scholar]
- (59).Wang JM, Cieplak P, Kollman PA. J. Comput. Chem. 2000;21:1049. [Google Scholar]
- (60).Cornell WD, Cieplak P, Bayly CI, Kollman PA. J. Am. Chem. Soc. 1993;115:9620. [Google Scholar]
- (61).Dennington RI, Keith T, Millam J, Eppinnett K, Hovell WL, et al. GaussView. Version 3.0 Semichem, Inc.; Shawnee Mission KS: 2003. [Google Scholar]
- (62).Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, et al. Gaussian 03. Revision C.02 Gaussian, Inc.; Wallingford CT: 2004. [Google Scholar]
- (63).Tamura M, Holbrook SR. J. Mol. Biol. 2002;320:455. doi: 10.1016/s0022-2836(02)00515-6. [DOI] [PubMed] [Google Scholar]
- (64).Hsiao C, Mohan S, Hershkovitz E, Tannenbaum A, Williams LD. Nucleic Acids Res. 2006;34:1481. doi: 10.1093/nar/gkj500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Nissen P, Ippolito JA, Ban N, Moore PB, Steitz TA. Proc. Natl. Acad. Sci. 2001;98:4899. doi: 10.1073/pnas.081082398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Sponer J, Sabat M, Gorb L, Leszczynski J, Lippert B, et al. J. Phys. Chem. B. 2000;104:7535. [Google Scholar]
- (67).Chou SH, Zhu LM, Reid BR. J. Mol. Biol. 1994;244:259. doi: 10.1006/jmbi.1994.1727. [DOI] [PubMed] [Google Scholar]
- (68).Shepard W, Cruse WBT, Fourme R, de la Fortelle E, Prange T. Structure. 1998;6:849. doi: 10.1016/s0969-2126(98)00087-2. [DOI] [PubMed] [Google Scholar]
- (69).Spackova N, Berger I, Sponer J. J. Am. Chem. Soc. 2000;122:7564. doi: 10.1021/ja002656y. [DOI] [PubMed] [Google Scholar]
- (70).Zimmermann GR, Jenison RD, Wick CL, Simorre JP, Pardi A. Nat. Struct. Biol. 1997;4:644. doi: 10.1038/nsb0897-644. [DOI] [PubMed] [Google Scholar]
- (71).Leontis NB, Stombaugh J, Westhof E. Nucleic Acids Res. 2002;30:3497. doi: 10.1093/nar/gkf481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Zirbel CL, Sponer JE, Sponer J, Stombaugh J, Leontis NB. J. Biol. Struct. Dyn. 2009;26:819. [Google Scholar]
- (73).Perez A, Marchan I, Svozil D, Sponer J, Cheatham TE, et al. Biophys. J. 2007;92:3817. doi: 10.1529/biophysj.106.097782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Reblova K, Lankas F, Razga F, Krasovska MV, Koca J, et al. Biopolymers. 2006;82:504. doi: 10.1002/bip.20503. [DOI] [PubMed] [Google Scholar]
- (75).Besseova I, Otyepka M, Reblova K, Sponer J. Phys. Chem. Chem. Phys. 2009;11:10701. doi: 10.1039/b911169g. [DOI] [PubMed] [Google Scholar]
- (76).Fadrna E, Spackova N, Sarzynska J, Koca J, Orozco M, et al. J. Chem. Theory Comput. 2009;5:2514. doi: 10.1021/ct900200k. [DOI] [PubMed] [Google Scholar]
- (77).Mongan J, Case DA, McCammon JA. J. Comput. Chem. 2004;25:2038. doi: 10.1002/jcc.20139. [DOI] [PubMed] [Google Scholar]
- (78).Khandogin J, Brooks CL. Biophys. J. 2005;89:141. doi: 10.1529/biophysj.105.061341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Lee MS, Salsbury FR, Brooks CL. Prot. Struct. Func. Bioinf. 2004;56:738. doi: 10.1002/prot.20128. [DOI] [PubMed] [Google Scholar]
- (80).Meng Yilin, E. RA. J. Chem. Theory Comput. 2010 in press. [Google Scholar]
- (81).Cochrane JC, Lipchock SV, Smith KD, Strobel SA. Biochemistry. 2009;48:3239. doi: 10.1021/bi802069p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Nam K, Gao JL, York DM. RNA. 2008;14:1501. doi: 10.1261/rna.863108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Bevilacqua PC, Brown TS, Nakano S, Yajima R. Biopolymers. 2004;73:90. doi: 10.1002/bip.10519. [DOI] [PubMed] [Google Scholar]
- (84).Perreault DM, Anslyn EV. Angew. Chem. Int. Ed. 1997;36:432. [Google Scholar]
- (85).Zhou DM, Taira K. Chem. Rev. 1998;98:991. doi: 10.1021/cr9604292. [DOI] [PubMed] [Google Scholar]
- (86).Emilsson GM, Nakamura S, Roth A, Breaker RR. RNA. 2003;9:907. doi: 10.1261/rna.5680603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Breaker RR, Emilsson GM, Lazarev D, Nakamura S, Puskarz IJ, et al. RNA. 2003;9:949. doi: 10.1261/rna.5670703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Liu L, Cottrell JW, Scott LG, Fedor MJ. Nat. Chem. Biol. 2009;5:351. doi: 10.1038/nchembio.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.