

Abstract
N-Acetylglucosaminyltransferase-IV (GnT-IV) has two isoenzymes, GnT-IVa and GnT-IVb, which initiate the GlcNAcβ1-4 branch synthesis on the Manα1-3 arm of the N-glycan core thereby increasing N-glycan branch complexity and conferring endogenous lectin binding epitopes. To elucidate the physiological significance of GnT-IV, we engineered and characterized GnT-IVb-deficient mice and further generated GnT-IVa/-IVb double deficient mice. In wild-type mice, GnT-IVa expression is restricted to gastrointestinal tissues, whereas GnT-IVb is broadly expressed among organs. GnT-IVb deficiency induced aberrant GnT-IVa expression corresponding to the GnT-IVb distribution pattern that might be attributed to increased Ets-1, which conceivably activates the Mgat4a promoter, and thereafter preserved apparent GnT-IV activity. The compensative GnT-IVa expression might contribute to amelioration of the GnT-IVb-deficient phenotype. GnT-IVb deficiency showed mild phenotypic alterations in hematopoietic cell populations and hemostasis. GnT-IVa/-IVb double deficiency completely abolished GnT-IV activity that resulted in the disappearance of the GlcNAcβ1-4 branch on the Manα1-3 arm that was confirmed by MALDI-TOF MS and GC-MS linkage analyses. Comprehensive glycomic analyses revealed that the abundance of terminal moieties was preserved in GnT-IVa/-IVb double deficiency that was due to the elevated expression of glycosyltransferases regarding synthesis of terminal moieties. Thereby, this may maintain the expression of glycan ligands for endogenous lectins and prevent cellular dysfunctions. The fact that the phenotype of GnT-IVa/-IVb double deficiency largely overlapped that of GnT-IVa single deficiency can be attributed to the induced glycomic compensation. This is the first report that mammalian organs have highly organized glycomic compensation systems to preserve N-glycan branch complexity.
Keywords: Ets-1/glycomic compensation/GnT-IVa/GnT-IVb/MALDI-TOF MS
Introduction
Protein glycosylation is one of the most important post-translational modifications that produce a highly complex structural repertoire, which is indispensable for precise regulation of biological functions (Hartiwanger and Lowe 2004; Ohtsubo and Marth 2006). Glycans are constructed in an ordered sequential manner involving the distinct substrate specificities of glycosyltransferases and glycosidases in the endoplasmic reticulum (ER) and Golgi and occasionally on the cell surface. Protein N-glycosylation encompasses two major functions corresponding to the structural maturation stage of N-glycans. The high-mannose-type N-glycans appeared early in evolution and are conserved in both protozoa and metazoa. They play an important role in allowing only properly folded proteins to pass the quality control system in the endoplasmic reticulum and to move to the Golgi apparatus (Helenius and Aebi 2004). The N-glycans are subsequently further processed by Golgi-resident glycosyltransferases and glycosidases and acquire complex branch structures bearing a variety of glycan moieties, which can be ligands for endogenous lectins (Kornfeld and Kornfeld 1985; Taylor and Drickamer 2007; Garner and Baum 2008) and confer particular features to the glycoproteins that carry these glycans (Perillo et al. 1998; Crocker et al. 2007; Rabinovich et al. 2007; Garner and Baum 2008). Furthermore, mammalian glycans are formed by an endogenous portfolio of glycosyltransferases, glycosidases and substrates that reflect the cellular microenvironment and control the fine tuning of the glycan structures and the resulting cellular responses (Ohtsubo and Marth 2006; Taniguchi 2009). Thus, glycosylation defects should evoke a variety of abnormalities and disorders in specific cell types, tissues, and the whole body, reflecting the functional requirements. This is exemplified by comprehensive studies of glycosyltransferase-deficient mice. For example, the genetic inactivation of N-acetylglucosaminyltransferases (GnTs), which initiate the formation of particular branch structures on the mannose core of N-glycans, resulted in remarkable physiological dysregulations in vivo (Ioffe and Stanley 1994; Metzler et al. 1994; Priatel et al. 1997; Granovsky et al. 2000; Demetriou 2001; Wang et al. 2001; Bhattacharyya et al. 2002; Yang et al. 2003; Ohtsubo et al. 2005; Soleimani et al. 2008).
In the past decade, the integral assembly of genetic sequence information accelerated the exploring of novel glycan processing enzymes and elucidated that GnT-IV and GnT-V have isoenzymes, GnT-IVa and GnT-IVb; GnT-V and GnT-IX (GnT-Vb), respectively, with inherent tissue distribution patterns (Shoreibah et al. 1993; Yoshida et al. 1998; Yoshida et al. 1999; Inamori et al. 2003; Kaneko et al. 2003;). GnT-IV and GnT-V are required for the formation of multiantennary complex-type N-glycans bearing ligands for galectins. The galectins are a family of lectins which preferentially bind and cross-link glycoproteins bearing β-galactosides. The binding avidity depends on the number and the antennary arrangement of N-glycans on glycoproteins (Lau et al. 2007) suggesting the precise spatial regulation of N-glycan antennae development in response to an evolutionary advantage of galectin binding. Expression of glycosyltransferases is concomitantly controlled by multiple transcriptional regulations to respond to a variety of cellular signaling. For example, promoters of the Mgat2 gene (encoding GnT-II), and Mgat5 gene (encoding GnT-V) commonly have cis-elements for the tissue-specific transcription factor, Ets-1, which predominantly controls their expression (Kang et al. 1996; Zhang et al. 2000). The tumor-related GnT-V expression is considerably attributed to Ets-1-dependent trans-activation (Ko et al. 1999). Elevated expression of GnT-IVa, -IVb and -V was found in many types of carcinoma cells (Kobata 1988; Nan et al. 1998; Takamatsu et al. 1999; Ide et al. 2006) and is well correlated with tumor malignancy (Asada et al. 1997; D’Arrigo et al. 2005). In contrast, mice deficient in GnT-IVa or GnT-V demonstrated abnormalities arising from cellular dysfunctions evoked by the diminished cell surface residency of plasmalemmal glycoproteins, due to the impaired molecular interactions between N-glycan antennae and galectins on the cell surface (Demetriou et al. 2001; Partridge et al. 2004; Ohtsubo et al. 2005). These studies demonstrated that the formation of proper antennal complexity on N-glycans is crucial for maintaining biological processes in normal contexts.
In this study, we engineered and characterized GnT-IVb deficient mice and further generated GnT-IVa/-IVb double deficiency in conjunction with comprehensive glycomic analyses by MALDI-TOF MS profiling to elucidate the fundamental enzymological function and biological significance of GnT-IV isoenzymes.
Results
Genetic inactivation of Mgat4b in mouse embryonic stem cells and production of mice bearing the homozygote mutation
To investigate the biological significance of GnT-IVa and -IVb isoenzymes, we characterized enzymatic activity using an in vitro cell culture system expressing recombinant enzymes, and found that the GnT-IVb has about 30% activity of GnT-IVa against an asialo-agalacto-bi-antennary N-glycan acceptor substrate (supplementary Figure 1) as previously described (Oguri et al. 2006). These results suggest that each GnT-IV isoenzyme has particular in vivo functions, according to their temporal and spatial expression profiles. We have previously reported that Mgat4a exhibits a limited organ distribution pattern and is prominent in pancreas and gastrointestinal tissues (Ohtsubo et al. 2005). In contrast, Mgat4b exhibited a broad organ distribution pattern (Figure 1A). We have investigated the in vivo function of GnT-IVa by engineering and characterizing GnT-IVa-deficient mice and found that GnT-IVa deficiency evokes type 2 diabetes with impaired insulin secretion from pancreatic β cells that was due to GLUT2 misglycosylation coincident with reduced β cell surface residency (Ohtsubo et al. 2005). Extensive in vivo analyses of GnT-IV isoenzymes were required to elucidate the physiological relevance of GnT-IV isoenzymes. To address this subject, we generated GnT-IVb-deficient mice. Eliminating GnT-IVb from the mouse germline was initiated by conditional mutagenesis of the Mgat4b allele in mouse embryonic stem (ES) cells using the Cre-loxP recombination system (Figure 1B, C). Exon 2, 3, and 4 of Mgat4b were flanked with loxP sites (F[tkneo] allele). Following Cre recombination and Gancyclovir selection, the parental Mgat4bF[tkneo] allele was converted to the conditional mutant Mgat4bF allele or to the deletion mutant Mgat4bΔ allele. The genetic conversion of the Mgat4b allele was confirmed by southern blot analyses in conjunction with genetic and loxP-specific probes (Figure 1D). Genetic excision of exon 2, 3, and 4 causes a frame shift at the junction of exon 1 and 5, and newly generates a translational termination codon right after the junction (supplementary Figure 2A). Therefore, the Mgat4bΔ allele produces the truncated GnT-IVb lacking the catalytic domain, which should be enzymatically inactive (Minowa et al. 1998). This was confirmed by the GnT-IV enzyme assay of intact and mutant Mgat4b gene products (supplementary Figure 1). The ES cells bearing the Mgat4bF allele were subjected to blastocyst injection and the germline-transmitted mice were bred into the C57BL/6J background for more than six generations. Systematic Mgat4bΔ mutant mice were generated by breeding the Mgat4bF mice with ZP3-Cre transgenic mouse, which specifically expresses Cre recombinase in developing oocytes. Cre recombination excises the experimentally targeted DNA sequence between the two loxP sites on the Mgat4bF allele that results in a residual loxP site remaining in the target genetic locus as shown in Mgat4bΔ allele (Shafi et al. 2000; Figure 1E).
Fig. 1.

Tissue distribution pattern and mutagenesis of the Mgat4b-encoded GnT-IVb glycosyltransferase. (A) Expression profile of mouse Mgat4b RNA transcripts among total RNA samples from indicated tissues. The bottom panel represents the ethidium bromide-stained gel as a loading control of RNA samples. (B) Mouse genomic clone of Mgat4b bearing exons 2, 3, and 4 (black boxes) used for construction of the targeting vector with the pflox plasmid as indicated. Restriction enzyme sites are indicated: (AL) Apa LI; (Bg) Bgl II; (Bm) Bam HI; (E) Eco RI; (Nh) Nhe I; (S) Sal I; (Sp) Spe I; (Sh) Sph I; (Xb) Xba I; (Xh) Xho I. (C) Homologous recombination produces the Mgat4b F[tkneo] allele. Following Cre recombination and Gancyclovir selection, embryonic stem cell clones are isolated bearing the type 1 (Δ, deleted) and type 2 (F, floxed) alleles. Represented restriction enzyme sites are same as those in B. (D) Southern blot analysis of ES cell genomic DNA probed with a genomic or a loxP probe confirmed the predicted allelic structures. Allelic structures in parental R1 ES cell and derived ES cell clones (3-6D, 12A, 1E4, and 1A) are indicated as the position of the individual band on the blot with a genomic probe and the number of the loxP site on the blot with the loxP probe. (E) Adult mice genotypes with germline modifications to the Mgat4b gene, including Mgat4bF and Mgat4bΔ alleles. Mouse genotype is indicated as the position of the band on the blot with the genomic probe: A, wild type; B, Mgat4bΔ homozygote; C, Mgat4bΔ heterozygote; D, Mgat4bF heterozygote.
GnT-IVb deficiency induces aberrant Mgat4a expression to compensate for loss of GnT-IV enzymatic activity
Fundamental enzymatic contribution of GnT-IVb was elucidated by analyzing GnT-IV enzymatic activity in GnT-IVb-deficient mouse organs. Although the Mgat4b is broadly expressed among organs, only heart, testis, ovary, and bone marrow showed 30–50% reduction of GnT-IV enzymatic activity (Table I), and others did not show apparent enzymatic alterations. Extensive analyses of the gene expression profile of the Mgat4a in GnT-IVb deficiency revealed that the Mgat4a expression was aberrantly upregulated among organs corresponding to the Mgat4b distribution pattern (Figure 2). The transcription factor binding motif search of the Mgat4a promoter revealed that the Mgat4a promoter has putative Ets-1 cis-elements proximal to the transcription initiation site (TFBIND, http://tfbind.ims.u-tokyo.ac.jp/, Tsunoda and Takagi 1999; supplementary Figure 2B). We analyzed Ets-1 expression in pancreas of GnT-IVb-deficient mouse and detected an extremely upregulated expression level (wt 100 ± 11; GnT-IVb deficiency 1249 ± 12, p < 0.0001) that should result in the compensatory expression of the Mgat4a. The increased Ets-1 expression might consequently contribute to preserve the GnT-IV enzymatic activity in GnT-IVb-deficient mouse organs. Furthermore, the increased Ets-1 expression was coincident with enhanced Mgat5 expression (Wt 100 ± 9; GnT-IVb deficiency 444 ± 33, p = 0.0005) that should also contribute to preserving N-glycan branch complexity. Indeed, we could not detect any major structural alteration of N-glycans in GnT-IVb-deficient mouse organs using MALDI-TOF glycomic methodologies (data not shown).
Table I.
GnT-IV Activity (pmol/hr mg)
| Mgat4a/Mgat4b | |||
|---|---|---|---|
| Wild type | Mgat4b Null | double Null | |
| Brain | 47.30 ± 4.92 | 60.45 ± 10.45 | N.D. |
| Thymus | 82.45 ± 6.34 | 86.66 ± 1.73 | N.D. |
| Lung | 68.78 ± 3.74 | 51.96 ± 6.91 | N.D. |
| Heart | 18.30 ± 1.37 | 11.83 ± 1.14* | N.D. |
| Liver | 16.34 ± 1.21 | 11.88 ± 1.58 | N.D. |
| Pancreas | 214.73 ± 77.42 | 258.64 ± 48.69 | N.D. |
| Spleen | 116.15 ± 15.91 | 95.38 ± 10.99 | N.D. |
| Stomach | 142.50 ± 47.41 | 91.40 ± 21.11 | N.D. |
| Small intestine | 404.78 ± 228.44 | 463.40 ± 365.60 | N.D. |
| Kidney | 58.03 ± 3.54 | 52.11 ± 2.42 | N.D. |
| Adrenal gland | 22.76 ± 5.55 | 18.57 ± 2.91 | N.D. |
| Colon | 917.16 ± 33.98 | 937.53 ± 185.05 | N.D. |
| Testis | 61.11 ± 5.45 | 36.57 ± 4.43* | N.D. |
| Ovary | 33.52 ± 4.76 | 16.22 ± 0.16* | N.D. |
| Uterus | 80.51 ± 5.87 | 75.72 ± 6.01 | N.D. |
| Lymph nodes | 83.56 ± 9.34 | 84.00 ± 7.54 | N.D. |
| Bone marrow | 23.97 ± 2.23 | 16.39 ± 2.40 | N.D. |
| Muscle | 3.83 ± 0.25 | 2.91 ± 0.38 | N.D. |
Each result represented the mean ± SEM of four independent experiments.
*P < 0.05 versus Wt (unpaired Student's t-test).
N.D.: not detected.
Fig. 2.

Aberrant Mgat4a expression in GnT-IVb deficiency. Total RNAs obtained from mouse tissues of wild-type and Mgat4b null were subjected to electrophoresis on formaldehyde denaturing gel, and then blotted onto the nylon membrane. Mgat4a transcripts were detected by probing with Mgat4a-specific cDNA fragment. Each bottom panel represents the ethidium bromide-stained gel image that indicates similar loading of analyzed RNA. Ethidium bromide-stained reference gels represent the quality of RNA samples.
Blood chemistry and hematology of GnT-IVb-deficient mice
The segregation behavior of mutant alleles amongst offspring was normal and animals homozygous for either allele lacked overt physical, neurologic, or reproductive defects. Animals homozygous for the Mgat4bF allele were indistinguishable from wild-type littermates in all studies undertaken below (data not shown). Serological analyses of 8- to 12-week-old mice revealed that systematic inactivation of GnT-IVb evokes no major metabolic alterations, but a slightly elevated glucose and AST levels, slightly reduced ALT, and alkaline phosphatase levels, and moderately reduced LDL levels were detected (Table II). Hematological characterization of GnT-IVb-deficient mice showed altered peripheral hematopoietic cell populations that included reduced circulating neutrophil number and cellularity, and increased lymphocyte cellularity. Mean Corpuscular Volume and mean Corpuscular Hemoglobin of erythrocytes were increased and the red cell distribution width (RDW) was decreased in GnT-IVb deficiency (Table II).
Table II.
Serum chemistry and hematology of GnT-IVb deficiency and GnT-IVa/-IVb double deficiency
| Wild type (n = 36) | Mgat4b null (n = 35) | Mgat4a/Mgat4b double null (n = 10) | |
|---|---|---|---|
| Glucose (mg/dL) | 136.3 ± 5.0 | 145.9 ± 5.5 | 189.7 ± 6.80 (p = 0.002) |
| Creatinine (mg/dL) | 0.3 ± 0.1 | 0.3 ± 0.0 | 0.2 ± 0.1 |
| Bicarbonate (mEq/L) | 14.6 ± 0.3 | 14.3 ± 0.3 | 12.1 ± 2.9 |
| Chloride (mEq/L) | 108.1 ± 0.4 | 109.1 ± 0.4 | 107.6 ± 3.8 |
| Sodium (mEq/L) | 154.2 ± 0.3 | 154.6 ± 0.3 | 151.2 ± 3.1 |
| Potassium (mEq/L) | 4.9 ± 0.1 | 4.8 ± 0.1 | 3.8 ± 0.5 |
| Calcium (mg/dL) | 9.1 ± 0.4 | 9.2 ± 0.1 | 7.6 ± 1.0 |
| Phosphorus (mg/dL) | 10.3 ± 0.3 | 10.2 ± 0.3 | 7.3 ± 1.1 |
| Magnesium (mg/dL) | 3.7 ± 0.1 | 3.6 ± 0.1 | 2.5 ± 0.3 |
| Total protein (g/dL) | 4.2 ± 0.2 | 4.2 ± 0.0 | 3.9 ± 0.2 |
| Albumin (g/dL) | 1.3 ± 0.0 | 1.3 ± 0.0 | 1.2 ± 0.1 |
| AST (SGOT) (IU/L) | 66.9 ± 17.4 | 82.9 ± 7.6 | 84.4 ± 25.3 |
| ALT (SGPT) (IU/L) | 25.1 ± 3.0 | 19.1 ± 1.7 | 29.8 ± 9.7 |
| Alkaline phosphatase (IU/L) | 101.0 ± 7.0 | 89.6 ± 7.8 | 110.3 ± 28.6 |
| Cholesterol (mg/dL) | 56.4 ± 2.2 | 55.9 ± 2.2 | 60.6 ± 8.1 |
| HDL-cholesterol (mg/dL) | 46.7 ± 2.6 | 48.0 ± 2.5 | 52.9 ± 6.1 |
| LDL-choresterol (mg/dL) | 2.6 ± 0.7 | 0.9 ± 0.3 (p < 0.05) | 0.3 ± 0.6 |
| Triglycerides (mg/dL) | 50.1 ± 3.7 | 51.5 ± 4.6 | 60.0 ± 19.6 |
| Wild type (n = 48) | Mgat4b Null (n = 48) | Mgat4a/Mgat4b double Null (n = 15) | |
| (Leukocytes) | |||
| White blood cell Count (1000/μL) | 6.66 ± 0.35 | 6.43 ± 0.34 | 6.01 ± 2.69 |
| Neutrophil count (1000/μL) | 1.15 ± 0.10 | 0.88 ± 0.05 (p = 0.0181) | 0.91 ± 0.38 |
| Lymphocyte count (1000/μL) | 5.25 ± 0.27 | 5.30 ± 0.29 | 4.81 ± 2.32 |
| Monocyte count (1000/μL) | 0.23 ± 0.02 | 0.21 ± 0.01 | 0.25 ± 0.11 |
| Eosinophil count (1000/μL) | 0.03 ± 0.00 | 0.03 ± 0.00 | 0.03 ± 0.05 |
| Basophil count (1000/μL) | 0.01 ± 0.00 | 0.01 ± 0.00 | 0.01 ± 0.01 |
| Neutrophil% | 16.6 ± 0.7 | 13.9 ± 0.5 (p = 0.0052) | 15.5 ± 3.1 |
| Lymphocyte% | 79.2 ± 0.8 | 82.1 ± 0.6 (p = 0.0060) | 79.3 ± 3.7 |
| Monocyte% | 3.56 ± 0.28 | 3.37 ± 0.16 | 4.43 ± 1.43 |
| Eosinophil% | 0.48 ± 0.10 | 0.47 ± 0.10 | 0.56 ± 0.88 |
| Basophil% | 0.10 ± 0.01 | 0.12 ± 0.02 | 0.11 ± 0.19 |
| (Erythrocytes) | |||
| Red blood cell count (106/μL) | 9.57 ± 0.08 | 9.43 ± 0.07 | 9.79 ± 1.08 |
| Hemoglobin concentration (g/dL) | 12.9 ± 0.2 | 13.2 ± 0.1 | 12.9 ± 0.8 |
| Hematocrit of erythrocytes (%) | 48.1 ± 0.4 | 48.2 ± 0.3 | 57.0 ± 9.0 |
| Mean corpuscular volume (fL) | 50.3 ± 0.3 | 51.1 ± 0.2 (p = 0.0119) | 58.1 ± 5.8 |
| Mean corpuscular hemoglobin (pg) | 13.4 ± 0.1 | 13.9 ± 0.1 (p = 0.0337) | 13.3 ± 1.2 |
| Mean corpuscular hemoglobin concentration (g/dL) | 26.8 ± 0.3 | 27.1 ± 0.3 | 23.1 ± 3.9 |
| Red cell distribution width (%) | 18.7 ± 0.2 | 17.7 ± 0.1 (p < 0.0001) | 16.8 ± 0.8 |
| (Thrombocytes) | |||
| Platelet count (PLT) (1000/μL) | 765.4 ± 20.0 | 765.3 ± 22.8 | 1071.4 ± 174.1 |
| Mean platelet volume (fL) | 4.2 ± 0.0 | 4.2 ± 0.0 | 4.2 ± 0.3 |
Altered hemostasis and prolonged bleeding in GnT-IVb-deficient mice
We detected prolonged bleeding time among GnT-IVb-deficient mice (Table III) that implies altered hemostasis. Moreover, while GnT-IVb deficiency showed normal prothrombin time (PT), the activated partial thromboplastin time (APTT) was slightly prolonged in GnT-IVb deficiency. This might be a consequence of the altered coagulation factor levels or other blood hemostatic components, such as von Willebrand factor (vWF) caused by GnT-IVb deficiency. The altered platelet function also possibly changes hemostasis, even though GnT-IVb-deficient mice showed the normal platelet number (Table II). We further characterized coagulation factor activity levels in GnT-IVb-deficient mice and found a slight increase of coagulation inhibitors (protein C and antithrombin), and a remarkable reduction of coagulation factor levels (factor VIII and vWF: about 30% decreased; factors II, V, VII, IX, and XII: about 10–20% decreased). The prolonged APTT and bleeding time are consistent with and likely due to those alterations (Table III).
Table III.
Hemostasis and coagulation of GnT-IVb deficient mice
| Wild type | Mgat4b null | |
|---|---|---|
| (n = 30) | (n = 30) | |
| Bleeding time (s) | 41 ± 16 | 63 ± 41 |
| PT (s) | 10.7 ± 0.2 | 10.7 ± 0.2 |
| APTT (s) | 26.2 ± 1.9 | 27.7 ± 1.8** |
| Antithrombin (% of wt BL/6) | 130 ± 13 | 143 ± 17* |
| Ptotein C (% of wt BL/6) | 98 ± 18 | 117 ± 23* |
| Protein S (% of wt BL/6) | 200 ± 94 | 202 ± 64 |
| Plasminogen (% of wt BL/6) | 113 ± 17 | 120 ± 17 |
| Alpha-2 antiplasmin (% of wt BL/6) | 114 ± 13 | 111 ± 9 |
| Fibrinogen (mg/dL) | 158 ± 35 | 145 ± 37 |
| Factor II (% of wt BL/6) | 129 ± 28 | 109 ± 25* |
| Factor V (% of wt BL/6) | 95 ± 13 | 85 ± 15 |
| Factor VII (% of wt BL/6) | 119 ± 23 | 93 ± 31* |
| Factor VIII (% of wt BL/6) | 147 ± 65 | 97 ± 24*** |
| Factor IX (% of wt BL/6) | 77 ± 32 | 60 ± 18* |
| Factor X (% of wt BL/6) | 76 ± 7 | 71 ± 12 |
| Factor XI (% of wt BL/6) | 55 ± 22 | 45 ± 19 |
| Factor XII (% of wt BL/6) | 66 ± 9 | 60 ± 11* |
| vWF (% of wt BL/6) | 145 ± 34 | 106 ± 31*** |
Results are expressed as mean ± SD (*p < 0.05, **p < 0.01, ***p < 0.001).
PT: prothrombin time; APTT: activated partial thromboplastin time; vWF: von Willebrand factor.
Complete elimination of GnT-IV enzymatic activity in GnT-IVa/-IVb double deficient mice
To elucidate the in vivo function of GnT-IV isoenzymes and their products, we generated GnT-IVa/-IVb double deficient mice by crossbreeding of GnT-IVa- and -IVb-deficient mice. The GnT-IV enzyme assay revealed that GnT-IVa/-IVb double deficiency completely abolished GnT-IV enzymatic activity in 18 tissues, which encompass all major organ systems in the mouse (Table I). This indicates that no other GnT-IV isoenzymes are involved in the GnT-IV enzymatic activity in all the organs examined.
Comprehensive MS analysis of N-glycan structures in GnT-IVa/-IVb double deficient mouse pancreas
The results reported above indicated that only GnT-IVa and -IVb are responsible enzymes for forming the β1,4-GlcNAc branch structure on the α1,3-Man arm of N-glycans in various organs. Nevertheless it has recently been reported that a putative Mgat4c gene encodes a third GnT-IV isoenzyme (NCBI NM_001162368.1, NP_001155840.1). To confirm the functional relevance of GnT-IV isoenzymes to the protein N-glycosylation, we performed glycomic profiling of pancreas, kidney, small intestine, and testis from GnT-IVa/-IVb double deficient mice and their littermate controls by MALDI-TOF-TOF MS/MS fingerprinting and sequencing plus GC-MS linkage analysis of N-glycans isolated from tissue glycoproteins, as previously described (Haslam et al. 2006).
The glycomic profile of the permethylated N-glycans of the wild-type pancreas was characterized by high-mannose, truncated and complex-type structures (Figure 3A and B upper panels). The assignments shown in the figure were arrived at by integrating sugar compositional information, derived from the displayed MS profile, with information from collisional activation MS/MS experiments carried out on representative molecular ions (Figure 4 and data not shown), together with the results of linkage analysis experiments (supplementary Table I). The majority of the complex-type N-glycans were found to be core fucosylated, nonbisected, and corresponded to bi-, tri-, and tetra-antennary structures, which are mostly endcapped with Galα1-3Gal epitopes although a minority of antennae are fucosylated. A great number of the tetra-antennary structures are extended with polylactosamine (Figure 3A and B upper panels). Glycans bearing sialic acid caps were very minor and had only two or three short antennae (m/z 2852, 3693, and 4084).
Fig. 3.
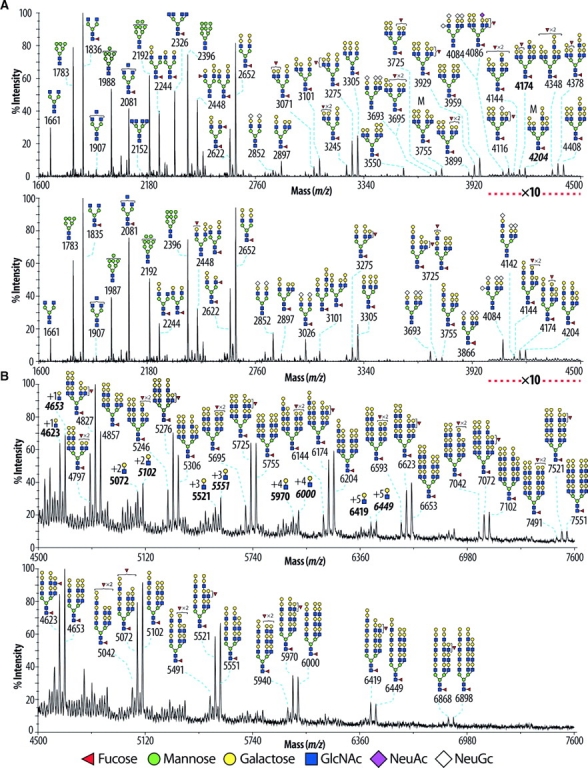
MALDI-TOF mass spectra of permethylated N-glycans derived from mouse pancreas. Glycomic profiles of wild-type (A and B upper panels) and GnT-IVa/-IVb double deficient (A and B lower panels) pancreas were obtained from the 50% MeCN fraction from a C18 Sep-Pak (Material and methods). For clarity major ions are shown. Cartoon structures are according to the Consortium for Functional Glycomics (http://www.functionalglycomics.org) guidelines. All molecular ions are [M+-Na]+-. Putative structures based on composition, tandem mass spectrometry, and the literature are shown. Structures that show sugars outside a bracket have not been unequivocally defined. The signals at m/z 4623, 5072, 5521, 5970, and 6419, designated with bold characters, correspond to structure at m/z 4174 extended with LacNAc repeats as indicated in the corresponding signals. The signals at m/z 4653, 5102, 5551, 6000, and 6449, designated with bold italic characters, correspond to structure at m/z 4204 extended with LacNAc repeats as indicated in the corresponding signals.
Fig. 4.
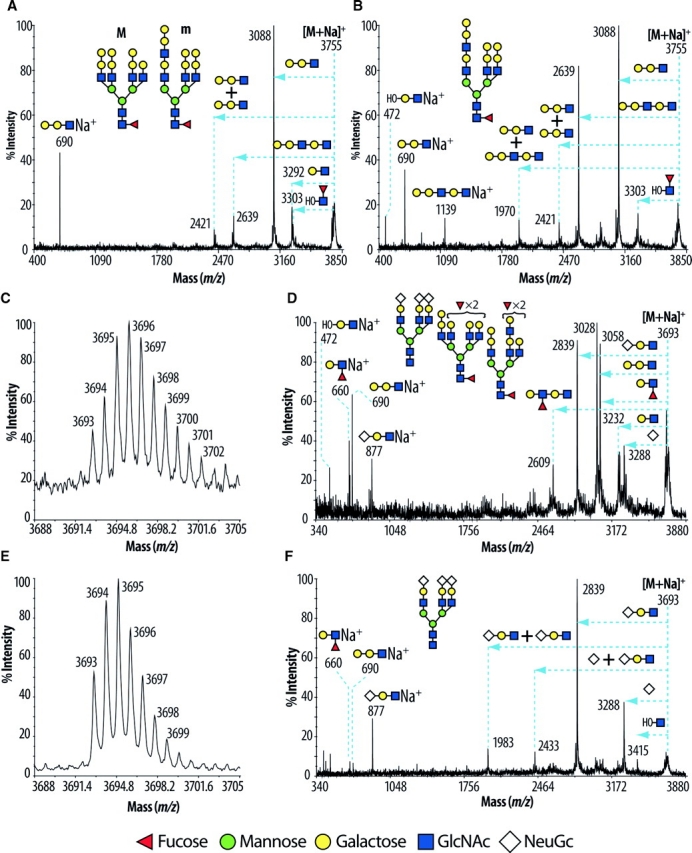
MALDI-TOF-TOF spectra of the [M+-Na]+- molecular ions from mouse pancreas. Signals present at m/z 3755 of wild-type and GnT-IVa/-IVb double deficiency in Figure 3 were subjected to tandem MS and the results are shown in (A) and (B), respectively. Structures labeled with “M” and “m” indicate major and minor abundances, respectively. The horizontal arrows on the spectra indicate losses from the molecular ion [M+-Na]+- of the designated N-glycan sequences in insets. (A) In the wild-type pancreas, the ion m/z 690 established the presence of Galα1-3Gal epitope, while the ions at m/z 3292, 3088, and 2639 corresponded to losses of HexHexNAc, Hex2HexNAc, and Hex3HexNAc2 respectively. Moreover, the signal at m/z 3303 corresponded to the loss of FucHexNAc from the reducing side of the glycan, indicative of core fucosylation. (B) In the GnT-IVa/-IVb double deficient pancreas, the spectrum was characterized by substantial differences suggesting structural alterations; compare the ratios of the ions at m/z 2639 and 3088 between the wild-type and GnT-IVa/-IVb double deficiency pancreas. Signals present at m/z 3695 (wild-type) and 3693 (GnT-IVa/-IVb double deficiency) in Figure 3 were zoomed in (C) and (E), respectively. Structures that show sugars outside a bracket have not been unequivocally defined. Those ions were subjected to tandem MS and the results are represented in (D) and (F), respectively. MALDI-TOF-TOF MS/MS of the above two ions established the presence of a fucosylated terminated antenna (m/z 660), of a Galα1-3Gal epitope (m/z 690), and of terminated NeuGc antenna (m/z 877). (D) In the wild-type mouse pancreas, the spectrum was characterized by dominant fragment ions at m/z 2839, 3028, and 3058 corresponding to losses of HexHexNAcNeuGc, Hex2HexNAc, and FucHexHexNAc respectively. The fragment ions at m/z 3232 and 3288 corresponded to losses of HexHexNAc and NeuGc respectively. The above data taken together indicated the presence of three different structures as indicated in the inset of Figure 4D. (F) In the GnT-IVa/-IVb double deficient pancreas, the spectrum was characterized by losses (m/z 1983, 2433, 2839, and 3288) and fragment ions (m/z 877) corresponding mainly to the structure indicated in Figure 4F.
Prominent structural alterations of N-glycans found in GnT-IVa/-IVb double deficient mouse pancreas were (i) significantly reduced branching complexity, and (ii) increased abundance of the NeuGc-capped structures (Figure 3A and B lower panels). The detected complex structures were core fucosylated, nonbisected, and corresponded to bi- or tri-antennary N-glycans bearing polylactosamine structures. The absence of tetra-antennary N-glycans was confirmed by the linkage analysis data which lacked 2,4-linked mannose (supplementary Table I). The branch extension with polylactosamine was considerably facilitated on tri-antennary N-glycans (Figure 3A and B, lower panel). The abundance of the fraction of sialic acid bearing N-glycans was substantially increased with bi- and tri-antennary glycans, without and with core fucose, carrying up to four sialic acids, being observed (m/z 2852, 3026, 3693, 3866, and 4084; Figure 3A, lower panel). MALDI-TOF-TOF-MS/MS provided evidence for the above structural alterations. (i) The N-glycan at m/z 3755 from wild-type and GnT-IVa/-IVb double deficiency was subjected to tandem MS and the results are depicted in Figure 4A and B, respectively. The extensive fragment ion analyses revealed that the ion at m/z 3755 of the wild-type pancreas corresponded mainly to tetra-antennary, and to a lesser extent, tri-antennary N-glycans with a lactosamine extended unit, and that of the GnT-IVa/-IVb double deficient mouse pancreas corresponded to a tri-antennary N-glycan with a lactosamine extended unit as indicated in the insets of Figure 4A and B. (ii) Compositional analyses consistent with the isotopic distribution of the zoomed scan of the m/z 3693–3695 molecular ion cluster of wild-type pancreas (Figure 4C), complemented by the fragment ion information arising from collisional activation (Figure 4D), indicated that this cluster corresponded mainly to a core fucosylated tetra-antennary structure bearing two fucosylated antennae, and, to a lesser extent, triantennary structures capped with either NeuGc or a combination of Galα1-3Gal and fucosylated epitopes (Figure 4C and D). In contrast the GnT-IVa/-IVb double deficiency data corresponded almost exclusively to the triantennary glycan capped with NeuGc and lacking core fucose (Figure 4E and F). The molecular ion cluster detected at m/z 4084–4086 in wild-type and GnT-IVa/-IVb double deficiency was similarly confirmed to be a triantennary NeuGc-sialylated glycan in the GnT-IVa/-IVb double deficiency, but was mostly a core fucosylated tetra-antennary glycan capped with Galα1-3Gal, Fuc, and NeuAc in the wild-type (see annotations on Figure 3; data not shown). The profiling of glycosyltransferase expression levels in pancreas revealed that the increased branch extension with polylactosamine and sialylated glycans could be attributed to the increased expression of UDP-GlcNAc:β-Galactoside β-1,3- N-acetylglucosaminyltransferase-1 (β3GnT1) (Sasaki et al. 1997) (wt 100 ± 13; IVa/IVb deficiency 220 ± 26, p = 0.0040), β3GnT2 (Zhou et al. 1999; Shiraishi et al. 2001) (wt 100 ± 5; IVa/IVb deficiency 228 ± 6, p < 0.0001), UDP-Gal:βGlcNAc β-1,4- galactosyltransferase (β4GalT)-I (wt 100 ± 4; IVa/IVb deficiency 165 ± 8, p = 0.0018), β4GalT-II (wt 100 ± 9; IVa/IVb deficiency 151 ± 14, p = 0.0399), β4GalT-III (wt 100 ± 9; IVa/IVb deficiency 253 ± 9, p = 0.0003), β-galactoside α-2,6-sialyltransferase (ST6Gal)-I (wt 100 ± 6; IVa/IVb deficiency 400 ± 45, p = 0.0027), and β-galactoside α-2,3-sialyltransferase (ST3Gal)-IV (wt 100 ± 7; IVa/IVb deficiency 189 ± 10, p = 0.0021). Increased expression of fucosyltransferase (Fut)-4 (wt 100 ± 10; IVa/IVb deficiency 215 ± 24, p = 0.0113), and Fut-7 (wt 100 ± 2; IVa/IVb deficiency 180 ± 72, not statistical significant) were also detected, but they were not reflected to the aspect of the glycomic profile of pancreas.
Glycomic analysis and GC-MS linkage analysis of kidney, small intestine, and testis of GnT-IVa/-IVb double deficiency also showed the complete loss of tetra-antennary glycans and the absence of 2,4-linked mannose (supplementary Figures 3–5, supplementary Table I and data not shown). Like the pancreas, the loss of tetra-antennary structures is frequently accompanied by concomitant increases in polylactosamine-containing bi- and tri-antennary glycans of the same overall compositions. This is most clearly exemplified by the LewisX (LeX)-rich glycans giving molecular ions at m/z 4127, 4301, 4533, and 4751 in the kidney data which are of comparable abundance in GnT-IVa/-IVb double deficiency and wild-type, despite having very different branching arrangements (supplementary Figure 3, panels A and B). Notably the LeX aggregate is broadly similar in the GnT-IVa/-IVb double deficiency and wild-type because antennae consisted of tandem repeats of this epitope are, on average, longer in the former.
The O-glycomic profile of all examined wild-type and GnT-IVa/-IVb double deficient mice tissues did not reveal any significant difference between them (data not shown). Therefore it was assumed, as expected, that the GnT-IV enzyme activity did not play any role in O-glycans biosynthesis. The above results indicate that GnT-IVa and GnT-IVb are the sole responsible enzymes for producing the β1,4-GlcNAc branch structure on N-glycans and contributing to maintaining the N-glycan branch complexity in vivo.
Blood chemistry and hematology of GnT-IVa/-IVb double deficient mice
GnT-IVa/-IVb double deficient mice showed significantly elevated blood glucose and slightly increased AST relative to wild-type mice. These levels are similar to those in GnT-IVa-deficient mice (Ohtsubo et al. 2005). Although no other major alterations were detected in the serum chemistry of GnT-IVa/-IVb double deficiency, some factors reflecting renal function that are possibly symptomatic of diabetic nephropathy were moderately changed: creatinine, calcium, phosphorus, magnesium, and total protein (Table II). However more extensive analyses regarding kidney function in GnT-IVa/-IVb double deficiency are required to clarify the role of hyperglycemia in the development of diabetic nephropathy.
GnT-IVa/-IVb double deficient mice were indistinguishable from their littermates on general hematological analyses (Table II).
Discussion
In the past decade, understanding of the biological significance of protein N-glycosylation has been considerably enhanced, so it has been revealed that precise N-glycosylation of specific proteins is required for regulating dynamic cell responses to properly maintain the biological systems (Priatel et al. 1997; Demetriou et al. 2001; Wang et al. 2001; Ohtsubo et al. 2005). To date, isoenzymes have been identified for two glycosyltransferases (GnT-IV and GnT-V) that play unique and essential roles in the biosynthesis of multiantennary complex N-glycans (Gleeson and Schachter 1983). Because expression of these isoenzymes is spatially regulated (Figure 1A, Inamori et al. 2003, Kaneko et al. 2003, Ohtsubo et al. 2005), the tissue and cellular locations of N-glycan complexity may be important for maintaining normal biological functions. Indeed, Mgat4a expression was upregulated in GnT-IVb-deficient mouse organs, and the apparent total GnT-IV enzymatic activities were preserved at comparable levels to wild type (Figure 2B, and Table I). This aberrant expression of GnT-IVa should compensate for the lost GnT-IV enzymatic activity arising from GnT-IVb deficiency thereby ameliorating any systemic abnormalities that might be expected to result from GnT-IVb deficiency. Conceivably, this compensation is genetically controlled by Ets-1-dependent trans-activation of the Mgat4a gene. Ets-1 is known as a key regulator of cell growth and development (Wasylyk et al. 1993), which binds and trans-activates the Mgat5 promoter (Kang et al. 1996; Ko et al. 1999). In agreement with elevated Ets-1 expression in GnT-IVb deficiency, Mgat5 expression was significantly upregulated. These findings suggest that the GnT-IVb deficiency evokes Ets-1 expression and considerably induces expression of GnT-IVa and -V that should contribute to increase and preserve N-glycan branch complexity. In contrast to GnT-IVb deficiency, GnT-IVa deficiency did not induce Mgat4b expression in any tissues (data not shown). These findings suggest that GnT-IVb-dependent glycosylation is a molecular switch to maintain the whole protein N-glycosylation profile, while GnT-IVa-dependent glycosylation is not. This might be attributed to the different enzymological characteristics between GnT-IVa and GnT-IVb. Although these enzymes have identical glycan acceptor substrate specificities (Oguri et al. 2006), they may differ in their preferences for the proteins that carry the glycans. Indeed, GnT-IVa-deficient mice showed a diabetic phenotype due to the misglycosylation of GLUT2 in pancreatic β cells (Ohtsubo et al. 2005), even though Mgat4b is expressed. While in GnT-IVb deficiency, the aberrant induction of the GnT-IVa-dependent protein N-glycosylation could compensate for the GnT-IVb-deficient phenotype and, moreover, might induce some cellular defects in organs relevant to hematopoiesis and hemostasis. This speculation was supported by the observation that GnT-IVa/-IVb double deficient mice were normal in hematopoiesis (Table II).
GnT-IVb deficiency evidently showed impaired hemostasis as prolonged APTT and bleeding time that should be consequences of the increased antithrombin and protein C levels and decreased factor II, V, VII, VIII, IX, XI, XII, and vWF levels (Table III). APTT is affected by factor II, V, and X levels in a common pathway and particularly by factor VIII, IX, and XII in the intrinsic pathway. Therefore, the decreased factor II, V, VIII, IX, and XII levels must result in prolonged APTT in GnT-IVb deficiency. Bleeding time is generally controlled by fibrinogen, factor II, V, VII, IX, X, XI, XII levels, and platelet number and function. The decreased factor II, V, VII, VIII, IX, XI, and XII levels contribute to the prolonged bleeding time in GnT-IVb deficiency. The elevated antithrombin and protein C levels also contribute to the observed prolonged bleeding time. Liver is a major organ to produce coagulation factors, such as fibrinogen, coagulation factor II, V, VII, IX, X, and XI, and anticoagulants, such as protein C and antithrombin. Furthermore, liver plays an important role to dispose circulating plasma proteins. Therefore, these findings imply that the N-glycan complexity of specific glycoproteins in the liver is crucial for the production, stability, and clearance of specific coagulation factors and anticoagulants in circulation. However, the detailed pathogenesis of these abnormalities in GnT-IVb deficiency awaits future research.
The biological significance of GnT-IVb in the human disease process still remains unknown. However, it has recently been reported that the GnT-IVb is dominantly expressed in pancreatic cancer tissues, while GnT-IVa is dominantly expressed in surrounding normal tissues (Ide et al. 2006). This suggests that GnT-IVb is involved in tumorigenesis and cancer progression.
N-Glycosylation confers glycan ligands for endogenous lectins on various proteins. The formation of plasmalemmal glycoprotein–endogenous lectin complex prevents endocytosis and prolongs cell surface residency of the glycoprotein, that is essential for proper cellular responses to the fluctuating micro-environmental factors (Garner and Baum 2008). The galectins are a family of lectins that preferentially bind and crosslink glycoproteins bearing β-galactoside. The binding avidity depends on the number and the complexity of the antennae of N-glycans (Lau et al. 2007). Hence, GnT-IV isoenzymes fundamentally contribute to the galectin-mediated glycoprotein clustering on the cell surface. We previously reported that the β cell glucose transporter 2 (GLUT2) is glycosylated by GnT-IVa, which confers tetra-antennary N-glycan as the ligand for galectin-9 (Ohtsubo et al. 2005). In the present study, we performed glycomic profiling of N-glycans derived from organs of wild-type and GnT-IVa/-IVb double deficient mice by employing ultrahigh sensitive MALDI-TOF-TOF instrumentation. The glycomic profiling of N-glycans in wild-type mouse organs revealed that tetra-antennary N-glycan structures are abundantly expressed in pancreas and small intestine, which have strong GnT-IV enzymatic activities (Figure 3, supplementary Figure 4, and Table I). The genetic elimination of GnT-IVa and GnT-IVb completely abolished GnT-IV enzymatic activity that resulted in the disappearance of the GlcNAcβ1-4 branch on the α1-3 Man arm of N-glycans in all mouse organs examined (Figure 3, supplementary Figures 3–5 and data not shown). Furthermore, the N-glycan antennae were elongated with linear polylactosamine structures (Figure 3) that can be a suitable galectin ligand (Hirabayashi et al. 2002). In GnT-IVa/-IVb double deficiency, therefore, the loss of the GlcNAcβ1-4 branch structure should consequentially diminish the avidity to galectin-9.
It has been reported that the N-terminal carbohydrate recognition domain (NCRD) of mouse galectin-9 preferentially binds to α2,3-sialylated N-glycans (Nagae et al. 2008). Interestingly, the abundance of the sialic acid-capped N-glycans was apparently increased in GnT-IVa/-IVb double deficient pancreas (Figure 3) that should increase the binding avidity of some fractions of glycoproteins to galectin-9 and might compensate the lost GlcNAcβ1-4 branch function in GnT-IVa/-IVb double deficiency. In GnT-IVa/-IVb double deficiency, the defect in the cell surface residency of receptor glycoproteins might alter the cellular signaling and change glycosyltransferase expression profile including increased expressions of β3GnT-1,-2, β4GalT-I, -II, -III, ST3Gal-IV, ST6Gal-I, and Fut-4, -7 that conceivably contribute to increase polylactosamine structure and to preserve the abundance of terminal glycan epitopes on N-glycans.
Similar compensation of glycan ligand formation was observed in kidney. The obtained glycomic profile of kidney was consistent with previous reports that the presence of LewisX (LeX) epitopes in bisected and core fucosylated bi- (m/z 2663 and 2837), tri- (m/z 3286 and 3461) and tetra-antennary (m/z 3910 and 4084) complex structures (supplementary Figure 3A and C) (Chui et al. 2001; Comelli et al. 2006). However, the greater sensitivity achieved with the current MALDI-TOF-TOF instrumentation, compared with the mass spectrometers used in the previous work, has enabled the detection in the kidney of families of high-molecular-weight glycans carrying polylactosamine antennae. Some of these antennae are capped with NeuGc but the majority carry tandemly repeated LeX epitopes (supplementary Figure 3A and B, m/z 4000–5500). In GnT-IVa/-IVb double deficiency, the GlcNAcβ1–4 branch had disappeared, while the molecular ion abundances in the MALDI-TOF spectra were not substantially altered. This was due to the significantly increased LeX epitope expression on tandemly repeated LacNAc on the N-glycan branches (supplementary Figure 3B and D). Therefore, the overall density of the LeX epitope was preserved in GnT-IVa/-IVb double deficiency. The LeX epitope is also called CD15 and SSEA-1 (stage-specific embryonic antigen 1), which is expressed on neutrophils and mediates phagocytosis and chemotaxis (Kerr and Stocks 1992), and also represents a marker for murine pluripotent stem cells, in which it plays an important role in adhesion and migration of the cells in the preimplantation embryo (Muramatsu T and Muramatsu H 2004). Although the role of the LeX epitope on N-glycans in kidney still remains unknown, the preserved expression of the LeX epitope in GnT-IVa/-IVb double deficiency implies that abundant expression of the LeX epitope on N-glycans is indispensable for maintaining kidney functions.
Since mouse Mgat4a and Mgat4b are separately localized at the chromosomal position 1B and 11B1.3, respectively, the mutant alleles should independently segregate and the natality of offspring from the Mgat4a/Mgat4b heterozygote breeding should be consonant with Mendelian inheritance manner. Nevertheless, the natality of offspring harboring mutant Mgat4b homozygote was reduced by combination with mutant Mgat4a allele(s), (supplementary Figure 6). The degree of reduction was correlated with the number of mutant Mgat4a alleles. This is well explained by the abolition of enzymatic compensation attributed to the aberrant Mgat4a expression in GnT-IVb deficiency. These results suggest that the GnT-IV-dependent glycosylation in reproductive organs and/or in embryo is important for maintaining normal prenatal ontogeny.
This is the first report that mammalian organs have highly organized glycomic compensation systems to preserve the N-glycan branch complexity. The molecular mechanism still remains unknown, but it is anticipated that more extensive analyses of the GnT-IVa/-IVb double deficient mice should contribute to revealing the detail.
Material and methods
Mgat4b expression and mutagenesis
Mouse Mgat4b cDNA clones, GT4F7/GT4R6 and GT4F3/GB5, were obtained by RT-PCR with primers: GT4F7, ATCGGGGGAGATGAGGCTCCGCAATGGCAC; GT4R6, GTGGTCCAGCAGCCAGTCAATGGGCT; GT4F3, CTTCAGCCCGCCGTACAGATTGGC; GB5, CAGTAGACTTCACAAGAGGTAGTGTTCA, and were used as probes to isolate Mgat4b genomic DNA clones from a 129/SvJ genomic DNA bacteriophage library (Stratagene, San Diego, CA). Chimeric mice were generated from C57BL/6 blastocyst-stage embryos and ES cells bearing the conditional (F, type 2) Mgat4b mutation (Figure 1A, B). Female mice bearing a germline Mgat4bF allele and the Zp3-Cre transgene (Shafi et al. 2000) were bred to acquire offspring containing the Mgat4bΔ allele.
Northern blot analysis
Total RNA was prepared from mouse tissues according to a single-step procedure. Total RNA (15 μg) was denatured in the RNA sample-loading buffer (Sigma Aldrich Corp., St. Louis, MO) followed by electrophoresis in 1% agarose/formaldehyde gel. The separated RNA was then transferred to a Hybond N+- nylon membrane (Amersham Biosciences, Piscataway, NJ). Mgat4b and Mgat4a cDNA probes were prepared by random oligonucleotide priming with [α-32P] dATP (Perkin Elmer) and the Klenow fragment of DNA polymerase (Prime-It II Random Primer Labeling Kit, Stratagene). The template cDNA fragments were amplified with PCR using the following primers: Mgat4b sense, TTATCCGGTTCCGCTTCTTCCAGC; Mgat4b anti-sense, GACCCACACCGGGGAGT-CAGTCTG; Mgat4a sense, TTATCCGGTTCCGCTTCTTCCAGC; Mgat4a anti-sense, GACCCACACCGGGGAGTCAGTCTG. The blots were hybridized with each probe in the Rapid-hyb buffer (Amersham Biosciences) at 65°C for 5 h. Next, the filters were washed three times in 2× SSC/1.0% SDS at 65°C for 20 min, once in 0.5× SSC/0.5% SDS at 65°C for 30 min and three times in 0.1× SSC/0.1% SDS at 65°C for 20 min. Then, the relative emissions of Mgat4b or Mgat4a signals were detected using a BioMax MS X-ray film (Kodak, Rochester, NY).
Quantitative PCR
Mouse tissue samples for real-time quantitative PCR (RT-Q-PCR) were collected from about 5 months old mouse in each genotype. Total RNA was extracted using RNAiso-plus (Takara, Ohtsu, Japan). cDNA was synthesized by using the Transcriptor First Strand cDNA Synthesis Kit (Roche) as manufacturer's instructions. Amplification of specific PCR products was carried out with THUNDERBIRD SYBR qPCR mix (TOYOBO, Tsuruga, Japan). Primers were used at 300 nM each in 20 μL reactions. Cycle parameters were as follows: a denaturation step at 95°C for 3 min, and 40 cycles composed of 15 s denaturation step at 95°C, 10 s annealing at 59°C, and 25 s polymerization at 72°C. Total RNA from each sample was analyzed in triplicate for each target RNA in separate wells. RT-Q-PCR was performed on the Mx3000P Real-Time QPCR System (Stratagene). We used the following oligonucleotide sequences: RPL4 forward primer, GTTCAAAGCTCCCATTCGAC; RPL4 reverse primer, AATTCACTGACGGCATAGGG; Fut-4 forward primer, CGTGGACGATTTCCCTAATG; Fut-4 reverse primer, TTGCTCATCCCAGAAAGAGG; Fut-7 forward primer, CCACCTACGAGGCTTTTGTG; Fut-7 reverse primer, CATTCATGCTGACGAGGAAG; ST6Gal-I forward primer, TAAGGAAGGCGTTCAGATCC; ST6Gal-I reverse primer, CAGCCTGGGGTTAAGTTTTG; ST3Gal-IV forward primer, AGCCTCGAAAGATCAAGCAG; ST3Gal-IV reverse primer, GATAGCCAAAGCCAGCAATG; B4GalT-I forward primer, TGGCAAAGAAGAACCCAGAG; B4GalT-I reverse primer, GGTTACGGAATGGGATGATG; B4GalT-II forward primer, TCAACGTGGGCTTCCTAGAG; B4GalT-II reverse primer, AGATTGCGGTCATCCATAGG; B4GalT-III forward primer, TACTTTGGCGGAGTTTCAGC; B4GalT-III reverse primer, TGGTAGCAATGTCGTCATCC; β3GnT-1 forward primer, GCTGAACGAAGGTTTTCTGG; β3GnT-1 reverse primer, GTTTGAACTGGCGGTAAAGG; β3GnT-2 forward primer, CGTGTTTGTGAACACCCATC; β3GnT-2 reverse primer, AGCATTGTGGATCACGTCAC; Ets-1 forward primer, ATCTCGAGCTTTTCCCTTCC; Ets-1 reverse primer, GTCGCTGCTGTTCTTTTGTG; Mgat5 forward primer, ACAACAGCTTTCTGGGCTTC; Mgat5 reverse primer, TTGCCATACACAAGGGACTG. Expression levels of the genes of interest were normalized to ribosomal protein L4 and calculated based on the ΔΔCT method (Livak and Schmittgen, 2001). The results were expressed as the percent ratio to RPL4.
Mouse breeding and maintenance
The Mgat4bΔ and Mgat4bF alleles were bred into the C57BL/6 background at least six generations prior to producing offspring for study. Mice were housed in specific pathogen-free conditions. Animals were ordinarily used between 6 and 10 weeks of age and in compliance with standards and procedures approved by the University of California, San Diego Institutional Animal Care and Use Committee.
Determination of GnT-IV activity
Assays of GnT-IV activity were measured in tissue homogenates by reverse-phase HPLC according to the method of Oguri et al. 1997, which was a modification of the method of Nishikawa et al. (1990). The substrate Gn2(2′,2) core-PA [GlcNAcβ1–2 Manα1–6(GlcNAc β1–2Man α1–3) Manβ1–4GlcNAc β1–4GlcNAc-PA] was prepared as reported previously (Tokugawa et al. 1996). In briefly, 70 μg of enzyme solution was incubated at 37°C for 1 h or 4 h with the 125 mM MOPS buffer, pH 7.3, containing 0.2 mM substrate, 20 mM UDP-GlcNAc, 7.5 mM MnCl2, 200 mM GlcNAc, 0.5% (w/v) Triton X-100, 10% glycerol, and 5 mg mL−1 bovine serum albumin in total volume of 20 μL. Twenty microliters of water was added after the incubation, and the enzyme reaction was stopped by boil for 2 min. The specific activity of enzyme was expressed as moles of products per hour of incubation per milligram of protein.
Serum Chemistry
Mice were anesthetized with inhalation of a mixture of 3.5% isoflurane with oxygen in an induction chamber. Blood samples were collected from orbital sinus and allowed to clot in Serum Separator Tubes (Becton Dickinson, Mountainview, CA) for at least 3 h at room temperature, and then the serum was collected by centrifugation at 7000 × g for 5 min. Serum chemistry analysis was performed using a Beckman CX-7 automated chemistry analyzer with a general coefficient of variation of <5%.
Hematology
Blood from the orbital sinus bleed of anesthetized mice with inhalation of a mixture of 3.5% isoflurane with oxygen in an induction chamber were collected into EDTA-containing polypropylene microtubes (Becton Dickinson, NJ). Whole EDTA blood samples are analyzed in duplicate for CBC with leukocyte differential and platelet count on a Hemavet 950 Multi Species Hematology System (DREW Scientific Inc.) programmed with mouse hematology settings.
Bleeding time
Mice are anesthetized by inhalation of a mixture of 3.5% isoflurane with oxygen in an induction chamber and then maintained with a nose cone in a warm brass cone restraint. Each tail of horizontally restrained mouse is transected 2 mm from tip with razor blade, and then immersed vertically 2 cm below the surface of 37°C saline. Time until bleeding stop for at least 10 s is recorded. After that, the tail is withdrawn from saline and the tip is cauterized to stop bleeding.
Coagulation
Up to 1 mL of blood was quickly collected by cardiac puncture of mouse anesthetized as for hematology using a 1 mL plastic syringe with 25 gauge needle containing 30 mL buffered citrate (0.06 mole/L sodium citrate, 0.04 mole/L citric acid, pH 7.4). Blood was poured into a plastic tube containing sufficient additional citrate to achieve the final ratio of nine parts whole blood to one part citrate, and immediately mixed well. Citrated platelet poor plasma is prepared from citrated blood by centrifugation twice at 2000 × g for 15 min at 22°C. VWF analysis was performed as described previously. Factor VIII and other factors were analyzed as described elsewhere.
Homogenization of mouse tissues
Mouse tissues were homogenized as described previously (Sutton-Smith and Dell 2006). Briefly kidney, pancreas, small intestine, and testes tissues were homogenized using a 130 W Vibra-Cell ultrasonic processor (VC 130 PB, Sonics & Materials Inc.) within a sound-abating enclosure in the extraction buffer (25 mM Tris, 150 mM NaCl, 5 mM EDTA, and 1% (3-[(3-Cholamidopropyl)dimethyl-ammonio]-1-propane sulfonate, CHAPS at pH 7.4). The homogenized tissues were then dialyzed against 4 × 4.5 L of ammonium bicarbonate (50 mM, pH 8.5) at 4°C for 48 h. After dialysis, the samples were lyophilized.
Reduction and carboxymethylation
Reduction and carboxymethylation were carried out using an established procedure (Dell et al. 1993). Each homogenized tissue was reduced in 2 mL 4 M guanidine–HCl (Pierce, Cramlington, Northumberland, UK), 0.6 M Tris–HCl buffer, pH 8.5 (adjusted with HCl), containing 2 mg mL−1 dithiothreitol at 50°C for 2 h. Carboxymethylation was carried out by the addition of 2 mL of iodoacetic acid (5-fold molar excess over dithiothreitol), and the reaction was allowed to proceed at room temperature in the dark for 2 h. Carboxymethylation was terminated by dialysis against 4 × 4.5 L of ammonium bicarbonate (50 mM, pH 8.5) at 4°C for 48 h. After dialysis, each sample was lyophilized.
Tryptic digestion
The reduced carboxymethylated proteins were digested with TPCK pre-treated bovine pancreas trypsin (EC 3.4.21.4, Sigma), for 24 h at 37°C in the ammonium bicarbonate buffer (50 mM, pH 8.4). The products were purified by C18-Sep-Pak (Waters Corp., Hertfordshire, UK) as described previously (Dell et al. 1993).
PNGase digestion of glycopeptides
PNGase F (EC 3.5.1.52, Roche Applied Science, Burgess Hill, UK) digestion was carried out in 250 μL of ammonium bicarbonate (50 mM, pH 8.5) for 24 h at 37°C using 3 U of enzyme. A second aliquot of enzyme (3 U) was added after 12 h. The reaction was terminated by lyophilization and the released N-glycans were separated from peptides and O-glycopeptides by Sep-Pak C18 (Waters Corp, Hertfordshire, UK) as described previously (Dell et al. 1993).
Reductive elimination
O-Glycans were released by reductive elimination in 400 μL of 0.1 M potassium borohydride (54 mg/mL of potassium hydroxide in water) solution at 45°C for 24 h. The reaction was terminated by the dropwise addition of glacial acetic acid, followed by Dowex 50W-X8 (H) 50–100 mesh (Sigma) chromatography and borate removal by co-evaporation with 10% (v/v) acetic acid in methanol (Dell et al. 1993; Sutton-Smith and Dell 2006).
Permethylation
Permethylation was performed using the sodium hydroxide procedure as described previously (Dell et al. 1993; Sutton-Smith and Dell 2006).
Mass spectrometry
MS and MS/MS data were acquired using a 4800 MALDI-TOF-TOF (Applied Biosystems, Darmstadt, Germany) mass spectrometer. Permethylated samples were dissolved in 10 μL of methanol and 1 μL of dissolved sample was premixed with 1 μL of matrix (20 mg mL−1 2,5-dihydroxybenzoic acid (DHB) in 70% (v/v) aqueous methanol), spotted onto a target plate (2 × 0.5 μL), and dried under vacuum. The collision energy was set to 1 kV, and argon was used as collision gas. The 4700 Calibration standard kit, calmix (Applied Biosystems), was used as the external calibrant for the MS mode and [Glu1] fibrinopeptide B human (Sigma-Aldrich) was used as an external calibrant for the MS/MS mode.
Analyses of MALDI data
The MS and MS/MS data were processed using Data Explorer 4.9 Software (Applied Biosystems, Warrington, UK). The spectra were subjected to manual assignment and annotation with the aid of the glycobioinformatics tool GlycoWorkBench (Ceroni et al. 2008). The proposed assignments for the selected peaks were based on 12C isotopic composition together with knowledge of the biosynthetic pathways. The proposed structures were then confirmed by data obtained from MS/MS and linkage analysis experiments.
Gas-chromatography –mass spectrometry (GC-MS) linkage analysis
Partially methylated alditol acetates (PMAA) were prepared as previously described (Dell et al. 1993; Sutton-Smith and Dell 2006). Linkage analysis of PMAA was performed on a Perkin Elmer Clarus 500 instrument fitted with a RTX-5 fused silica capillary column (30 m × 0.32 mm i.d.; Restek Corp.). All samples were dissolved in 50–250 μL of hexanes (Sigma) and injected manually (1–2 μL) to the injector set at 250°C. A linear gradient oven temperature was set as follows: initially the oven was heated at 65°C for 1 min, and then heated to 290°C at a rate of 8°C per min, held at 290°C for 5 min and finally heated to 300°C at a rate of 10°C per min.
Standards for linkage analysis
The standards compounds C1160 and L604 (Dextra Laboratories, UK) were used to confirm the retention time and the fragmentation spectra of the 2,4,6-linked mannose and 3,6-linked galactose residues respectively. Both standard compounds were permethylated and then subjected to PMAA analysis as stated previously in this section.
Statistical analysis
Data were plotted as the mean of the number (n) of samples analyzed ± the standard error unless otherwise indicated. The student's t-test was used to calculate indicated p values.
Supplementary Material
Glossary
Abbreviations
- ALT
alanine transaminase
- APTT
activated partial thromboplastin time
- AST
aspartate transaminase
- β3GnT
UDP-GlcNAc:β-Galactoside β-1,3-N-acetylglucosaminyltransferase
- β4GalT
UDP-Gal:βGlcNAc β-1,4-galactosyltransferase
- ER
endoplasmic reticulum
- Fut
fucosyltransferase
- GnT
N-acetylglucosaminyltransferase
- GnT-IV
UDP-N-acetylglucosamine:α-1,3-d-mannoside β-1,4-N-acetylglucosaminyl-transferase IV
- GnT-V
UDP-N-acetylglucosamine:α-1,6-d-mannoside β-1,6-N-acetylglucosaminyltransferase V
- GLUT2
glucose transporter 2
- LacNAc
N-acelyllactosamine (Galβ1-4GlcNAc)
- LeX
LewisX
- LDL
low-density lipoprotein
- MALDI-TOF MS
matrix-assisted laser desorption/ionization–time of flight mass spectrometry
- Mgat4a
mannosyl (alpha-1,3-)-glycoprotein beta-1,4-N-acetylglucosaminyltransferase, isozyme A
- Mgat4b
mannosyl (alpha-1,3-)-glycoprotein beta-1,4-N-acetylglucosaminyltransferase, isozyme B
- NeuAc
α-d-N-acetylneuraminic acid
- NeuGC
α-d-N-glycolylneuraminic acid
- PT
prothrombin time
- RDW
red cell distribution width
- SSEA-1
stage-specific embryonic antigen1
- ST3Gal
β-galactoside α-2,3-sialyltransferase
- ST6Gal
β-galactoside α-2,6-sialyltransferase
- vWF
von Willebrand factor
Supplementary Data
Supplementary data for this article is available online at http://glycob.oxfordjournals.org/.
Funding
National Institute of Health (DK48247 to J.D.M); an Investigator award from the Howard Hughes Medical Institute (to J.D.M.); the Biotechnology and Biological Sciences Research Council for support (B19088 and BBF008309 to A.D., S.M.H. and H.R.M.); Mochida Memorial Foundation for Medical and Pharmaceutical Research (to K.O.); Astellas Foundation for Research on Metabolic Disorders (to K.O.); Japan Diabetes Foundation (to K.O.); ONO Medical Research Foundation (to K.O.); and Suntory Institute for Bioorganic Research (to K.O.).
References
- Asada M, Furukawa K, Segawa K, Endo T, Kobata A. Increased expression of highly branched N-glycans at cell surface is correlated with the malignant phenotypes of mouse tumor cells. Cancer Res. 1997;57:1073–1080. [PubMed] [Google Scholar]
- Bhattacharyya R, Bhaumik M, Raju TS, Stanley P. Truncated, inactive N-acetylglucosaminyltransferase III (GlcNAc-TIII) induces neurological and other traits absent in mice that lack GlcNAc-TIII. J Biol Chem. 2002;277:26300–26309. doi: 10.1074/jbc.M202276200. [DOI] [PubMed] [Google Scholar]
- Ceroni A, Maass K, Geyer H, Geyer R, Dell A, Haslam SM. GlycoWorkbench: A tool for the computer-assisted annotation of mass spectra of glycans. J Proteome Res. 2008;7(4):1650–1659. doi: 10.1021/pr7008252. [DOI] [PubMed] [Google Scholar]
- Chui D, Sellakumar G, Green RS, Sutton-Smith M, McQuistan T, Marek KW, Morris HR, Dell A, Marth JD. Genetic remodeling of protein glycosylation in vivo induces autoimmune disease. Proc Natl Acad Sci USA. 2001;98(3):1142–1147. doi: 10.1073/pnas.98.3.1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comelli EM, Head SR, Gilmartin T, Whisenant T, Haslam SM, North SJ, Wong NW, Kudo T, Narimatsu H, Esko JD, et al. A focused microarray approach to functional glycomics: Transcriptional regulation of the glycome. Glycobiology. 2006;16(2):117–131. doi: 10.1093/glycob/cwj048. [DOI] [PubMed] [Google Scholar]
- Crocker P, Paulson JC, Varki A. Siglecs and their roles in the immune system. Nature Rev Immunol. 2007;7(4):255–266. doi: 10.1038/nri2056. [DOI] [PubMed] [Google Scholar]
- D’Arrigo A, Belluco C, Ambrosi A, Digito M, Esposito G, Bertola A, Fabris M, Nofrate V, Mammano E, Leon A, et al. Metastatic transcriptional pattern revealed by gene expression profiling in primary colorectal carcinoma. Int. J. Cancer. 2005;115:256–262. doi: 10.1002/ijc.20883. [DOI] [PubMed] [Google Scholar]
- Dell A, Khoo K.-H, Panico M, McDowell RA, Etienne AT, Reason AJ, Morris HR.1993FAB-MS and ES-MS of glycoproteins Fukuda M, Kobata A.editors Glycobiology: A Practical ApproachOxford (UK)Oxford University Press; p. 187–222 [Google Scholar]
- Demetriou M, Granovsky M, Quaggin S, Dennis J. Negative regulation of T-cell activation and autoimmunity by Mgat5 N-glycosylation. Nature. 2001;409:733–739. doi: 10.1038/35055582. [DOI] [PubMed] [Google Scholar]
- Garner OB, Baum LG. Galectin–glycan lattices regulate cell-surface glycoprotein organization and signaling. Biochem Soc Trans. 2008;36:1472–1477. doi: 10.1042/BST0361472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleeson PA, Schachter H. Control of glycoprotein synthesis. J Biol Chem. 1983;258:6162–6173. [PubMed] [Google Scholar]
- Granovsky M, Fata J, Pawling J, Muller WJ, Khokha R, Dennis JW. Suppression of tumor growth and metastasis in Mgat5-deficient mice. Nat Med. 2000;6:306–312. doi: 10.1038/73163. [DOI] [PubMed] [Google Scholar]
- Haltiwanger RS, Lowe JB. Role of glycosylation in development. Annu Rev Biochem. 2004;73:491–537. doi: 10.1146/annurev.biochem.73.011303.074043. [DOI] [PubMed] [Google Scholar]
- Haslam SM, North SJ, Dell A. Mass spectrometric analysis of N- and O-glycosylation of tissues and cells. Curr Opin Struct Biol. 2006;16(5):584–591. doi: 10.1016/j.sbi.2006.08.006. [DOI] [PubMed] [Google Scholar]
- Helenius A, Aebi M. Roles of N-linked glycans in the endoplasmic reticulum. Annu Rev Biochem. 2004;73:1019–1049. doi: 10.1146/annurev.biochem.73.011303.073752. [DOI] [PubMed] [Google Scholar]
- Hirabayashi J, Hashidate T, Arata Y, Nishi N, Nakamura T, Hirashima M, Urashima T, Oka T, Futai M, Muller WE, et al. Oligosaccharide specificity of galectins: A search by frontal affinity chromatography. Biochim Biophys Acta. 2002;1572:232–254. doi: 10.1016/s0304-4165(02)00311-2. [DOI] [PubMed] [Google Scholar]
- Ide Y, Miyoshi E, Nakagawa T, Gu J, Tanemura M, Nishida T, Ito T, Yamamoto H, Kozutsumi Y, Taniguchi N. Aberrant expression of N-acetylglucosaminyltransferase-IVa and IVb (GnT-IVa and b) in pancreatic cancer. Biochem Biophys Res Commun. 2006;341:478–482. doi: 10.1016/j.bbrc.2005.12.208. [DOI] [PubMed] [Google Scholar]
- Inamori K, Endo T, Ide Y, Fujii S, Gu J, Honke K, Taniguchi N. Molecular cloning and characterization of human GnT-IX, a novel beta 1, 6-N-acetylglucosaminyltransferase that is specifically expressed in the brain. J Biol Chem. 2003;278:43102–43109. doi: 10.1074/jbc.M308255200. [DOI] [PubMed] [Google Scholar]
- Ioffe E, Stanley P. Mice lacking N-acetylglucosaminyltransferase I activity die at mid-gestation, revealing an essential role for complex or hybrid N-linked carbohydrates. Proc Natl Acad Sci USA. 1994;91:728–732. doi: 10.1073/pnas.91.2.728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko M, Alvarez-Manilla G, Kamar M, Lee I, Lee JK, Troupe K, Zhang W, Osawa M, Pierce M. A novel beta(1,6)-N-acetylglucosaminyltransferase V (GnT-VB) FEBS Lett. 2003;554:515–519. doi: 10.1016/s0014-5793(03)01234-1. [DOI] [PubMed] [Google Scholar]
- Kang R, Saito H, Ihara Y, Miyoshi E, Koyama N, Sheng Y, Taniguchi N. Transcriptional regulation of the N-acetylglucosaminyltransferase V gene in human bile duct carcinoma cells (HuCC-T1) is mediated by Ets-1. J Biol Chem. 1996;271:26706–26712. doi: 10.1074/jbc.271.43.26706. [DOI] [PubMed] [Google Scholar]
- Kerr MA, Stocks SC.1992The role of CD15-(Le(X))-related carbohydrates in neutrophil adhesion. Histochem J. 24811–826. [DOI] [PubMed] [Google Scholar]
- Ko JH, Miyoshi E, Noda K, Ekuni A, Kang R, Ikeda Y, Taniguchi N. Regulation of the GnT-V promoter by transcription factor Ets-1 in various cancer cell lines. J Biol Chem. 1999;274:22941–22948. doi: 10.1074/jbc.274.33.22941. [DOI] [PubMed] [Google Scholar]
- Kobata A. Structures, function, and transformational changes of the sugar chains of glycohormones. J Cell Biochem. 1988;37:79–90. doi: 10.1002/jcb.240370108. [DOI] [PubMed] [Google Scholar]
- Kornfeld R, Kornfeld S. Assembly of asparagine-linked oligosaccharides. Ann Rev Biochem. 1985;54:631–664. doi: 10.1146/annurev.bi.54.070185.003215. [DOI] [PubMed] [Google Scholar]
- Lau KS, Partridge EA, Grigorian A, Silvescu CI, Reinhold VN, Demetriou M, Dennis JW. Complex N-glycan number and degree of branching cooperate to regulate cell proliferation and differentiation. Cell. 2007;129:123–134. doi: 10.1016/j.cell.2007.01.049. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the DDCT method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Metzler M, Gertz A, Sarkar M, Schachter H, Schrader JW, Marth JD. Complex asparagine-linked oligosaccharides are required for morphogenic events during post-implantation development. EMBO J. 1994;13:2056–2065. doi: 10.1002/j.1460-2075.1994.tb06480.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minowa MT, Oguri S, Yoshida A, Hara T, Iwamatsu A, Ikenaga H, Takeuchi M. cDNA cloning and expression of bovine UDP-N-acetylglucosamine: alpha1,3-d-mannoside beta1,4-N-acetylglucosaminyltransferase IV. J Biol Chem. 1998;273:11556–11562. doi: 10.1074/jbc.273.19.11556. [DOI] [PubMed] [Google Scholar]
- Muramatsu T, Muramatsu H. Carbohydrate antigens expressed on stem cells and early embryonic cells. Glycoconj J. 2004;21:41–45. doi: 10.1023/B:GLYC.0000043746.77504.28. [DOI] [PubMed] [Google Scholar]
- Nagae M, Nishi N, Nakamura-Tsuruta S, Hirabayashi J, Wakatsuki S, Kato R. Structural analysis of the human galectin-9 N-terminal carbohydrate recognition domain reveals unexpected properties that differ from the mouse orthologue. J Mol Biol. 2008;375:119–135. doi: 10.1016/j.jmb.2007.09.060. [DOI] [PubMed] [Google Scholar]
- Nan BC, Shao DM, Chen HL, Huang Y, Gu JX, Zhang YB, Wu ZG. Alteration of N-acetylglucosaminyltransferases in pancreatic carcinoma. Glycoconj J. 1998;15:1033–1037. doi: 10.1023/a:1006950311937. [DOI] [PubMed] [Google Scholar]
- Nishikawa A, Gu J, Fujii S, Taniguchi N. Determination of N-acetylglucosaminyltransferase III, IV, V in normal and hepatoma tissues of rats. Biochem Biophys Acta. 1990;1035:313–318. doi: 10.1016/0304-4165(90)90094-d. [DOI] [PubMed] [Google Scholar]
- Oguri S, Minowa MT, Ihara Y, Taniguchi N, Ikenaga H, Takeuchi M. Purification and characterization of UDP-N-acetylglucosamine: alpha1,3-d-mannoside beta1,4-N-acetylglucosaminyltransferase (N-acetylglucosaminyltransferase-IV) from bovine small intestine. J Biol Chem. 1997;272:22721–22727. doi: 10.1074/jbc.272.36.22721. [DOI] [PubMed] [Google Scholar]
- Oguri S, Yoshida A, Minowa MT, Takeuchi M. Kinetic properties and substrate specificities of two recombinant human N-acetylglucosaminyltransferase-IV isozymes. Glycoconj J. 2006;23:473–480. doi: 10.1007/s10719-006-6216-3. [DOI] [PubMed] [Google Scholar]
- Ohtsubo K, Marth JD. Glycosylation in cellular mechanisms of health and disease. Cell. 2006;126:855–867. doi: 10.1016/j.cell.2006.08.019. [DOI] [PubMed] [Google Scholar]
- Ohtsubo K, Takamatsu S, Minowa MT, Yoshida A, Takeuchi M, Marth JD. Dietary and genetic control of glucose transporter 2 glycosylation promotes insulin secretion in suppressing diabetes. Cell. 2005;123:1307–1321. doi: 10.1016/j.cell.2005.09.041. [DOI] [PubMed] [Google Scholar]
- Partridge EA, Le Roy C, Di Guglielmo GM, Pawling J, Cheung P, Granovsky M, Nabi IR, Wrana JL, Dennis JW. Regulation of cytokine receptors by Golgi N-glycan processing and endocytosis. Science. 2004;306:120–124. doi: 10.1126/science.1102109. [DOI] [PubMed] [Google Scholar]
- Perillo NL, Marcus ME, Baum LG. Galectins: versatile modulators of cell adhesion, cell proliferation, and cell death. J Mol Med. 1998;76(6):402–412. doi: 10.1007/s001090050232. [DOI] [PubMed] [Google Scholar]
- Priatel JJ, Sarkar M, Schachter H, Marth JD. Isolation, characterization and inactivation of the mouse Mgat3 gene: The bisecting N-acetylglucosamine in asparagines-linked oligosaccharides appears dispensable for viability and reproduction. Glycobiology. 1997;7:45–56. doi: 10.1093/glycob/7.1.45. [DOI] [PubMed] [Google Scholar]
- Rabinovich GA, Toscano MA, Jackson SS, Vasta GR. Functions of cell surface galectin–glycoprotein lattices. Curr Opin Struct. Biol. 2007;17(5):513–520. doi: 10.1016/j.sbi.2007.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki K, Kurata-Miura K, Ujita M, Angata K, Nakagawa S, Sekine S, Nishi T, Fukuda M. Expression cloning of cDNA encoding a human beta-1,3-N-acetylglucosaminyltransferase that is essential for poly-N-acetyllactosamine synthesis. Proc Natl Acad Sci USA. 1997;94:14294–14299. doi: 10.1073/pnas.94.26.14294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafi R, Iyer SPN, Ellies LG, O’Donnell N, Marek KW, Chui D, Hart GW, Marth JD. The O-GlcNAc transferase gene resides on the X chromosome and is essential for embryonic stem cell viability and mouse ontogeny. Proc Natl Acad Sci USA. 2000;97:5735–5739. doi: 10.1073/pnas.100471497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiraishi N, Natsume A, Togatachi A, Endo T, Akashima T, Yamada Y, Imai N, Nakagawa S, Koizumi S, Sekine S, et al. Identification and characterization of three novel beta 1,3-N-acetylglucosaminyltransferase structurally related to the beta 1,3-galactosyltransferase family. J Biol Chem. 2001;276:3498–3507. doi: 10.1074/jbc.M004800200. [DOI] [PubMed] [Google Scholar]
- Shoreibah M, Perng GS, Adler B, Weinstein J, Basu R, Cupples R, Wen D, Browne JK, Buckhaults P, Fregien N, et al. Isolation, characterization, and expression of a cDNA encoding N-acetylglucosaminyltransferase V. J Biol Chem. 1993;268:15381–15385. [PubMed] [Google Scholar]
- Soleimani L, Roder JC, Dennis JW, Lipina T. Beta N-acetylglucosaminyltransferase V (Mgat5) deficiency reduces the depression-like phenotype in mice. Genes Brain Behav. 2008;7:334–343. doi: 10.1111/j.1601-183X.2007.00358.x. [DOI] [PubMed] [Google Scholar]
- Sutton-Smith M, Dell A.2006Analysis of carbohydrates/glycoproteins by mass spectrometry Celis JE.editor Cell Biology: A Laboratory Handbook San Diego (CA)Academic; p. 425–435 [Google Scholar]
- Takamatsu S, Oguri S, Minowa MT, Yoshida A, Nakamura K, Takeuchi M, Kobata A. Unusually high expression of N-acetylglucosaminyltransferase-IVa in human choriocarcinoma cell lines: A possible enzymatic basis of the formation of abnormal biantennary sugar chain. Cancer Res. 1999;59:3949–3953. [PubMed] [Google Scholar]
- Taniguchi N. From the γ-glutamyl cycle to the glycan cycle: a road with many turns and pleasant surprises. J Biol Chem. 2009;284:34469–34478. doi: 10.1074/jbc.X109.023150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor ME, Kurt Drickamer K. Paradigms for glycan-binding receptors in cell adhesion. Curr Opin Cell Biol. 2007;19:572–577. doi: 10.1016/j.ceb.2007.09.004. [DOI] [PubMed] [Google Scholar]
- Tokugawa K, Oguri S, Takeuchi M. Large scale preparation of PA-oligosaccharides from glycoproteins using an improved extraction method. Glycoconj J. 1996;13:53–56. [PubMed] [Google Scholar]
- Tsunoda T, Takagi T. Estimating transcription factor bindability on DNA. Bioinformatics. 1999;15:622–630. doi: 10.1093/bioinformatics/15.7.622. [DOI] [PubMed] [Google Scholar]
- Wang Y, Tan J, Sutton-Smith M, Ditto D, Panico M, Campbell RM, Varki NM, Long JM, Jaeken J, Levinson SR, et al. Modeling human congenital disorder of glycosylation type IIa in the mouse: Conservation of asparagines-linked glycan-dependent functions in mammalian physiology and insights into disease pathogenesis. Glycobiology. 2001;11:1051–1070. doi: 10.1093/glycob/11.12.1051. [DOI] [PubMed] [Google Scholar]
- Wasylyk B, Hahn SL, Giovane A. The Ets family of transcription factors. Eur J Biochem. 1993;211:7–18. doi: 10.1007/978-3-642-78757-7_2. [DOI] [PubMed] [Google Scholar]
- Yang X, Tang J, Rogler CE, Stanley P. Reduced hepatocyte proliferation is the basis of retarded liver tumor progression and liver regeneration in mice lacking N-acetylglucosaminyltransferase III. Cancer Res. 2003;63:7753–7759. [PubMed] [Google Scholar]
- Yoshida A, Minowa MT, Takamatsu S, Hara T, Ikenaga H, Takeuchi M. A novel second isozyme of the human UDP-N-acetylglucosamine:alpha1,3-d-mannoside beta1,4-N-acetylglucosaminyltransferase family: cDNA cloning, expression, and chromosomal assignment. Glycoconj J. 1998;15:1115–1123. doi: 10.1023/a:1006951519522. [DOI] [PubMed] [Google Scholar]
- Yoshida A, Minowa MT, Takamatsu S, Hara T, Oguri S, Ikenaga H, Takeuchi M. Tissue specific expression and chromosomal mapping of a human UDP-N-acetylglucosamine: alpha1,3-d-mannoside beta1,4-N-acetylglucosaminyltransferase. Glycobiology. 1999;9:303–310. doi: 10.1093/glycob/9.3.303. [DOI] [PubMed] [Google Scholar]
- Zhang W, Revers L, Pierce M, Schachter H.2000Regulation of expression of the human beta-1,2-N-acetylglucosaminyltransferase II gene (Mgat2) by Ets transcription factors. Biochem. J. 347511–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou D, Dinter A, Gallego RG, Kamerling JP, Vliegenthart JFG, Berger EG, Hennet T. A beta-1,3-N-acetylglucosaminyltransferase with poly-N-acetyllactosamine synthase activity is structurally related to beta-1,3-galactosyltransferases. Proc Natl Acad Sci USA. 1999;96:406–411. doi: 10.1073/pnas.96.2.406. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.