

Abstract
Mutations in mitochondrial DNA (mtDNA) tRNA genes can be considered functionally recessive because they result in a clinical or biochemical phenotype only when the percentage of mutant molecules exceeds a critical threshold value, in the range of 70–90%. We report a novel mtDNA mutation that contradicts this rule, since it caused a severe multisystem disorder and respiratory chain (RC) deficiency even at low levels of heteroplasmy. We studied a 13-year-old boy with clinical, radiological and biochemical evidence of a mitochondrial disorder. We detected a novel heteroplasmic C>T mutation at nucleotide 5545 of mtDNA, which was present at unusually low levels (<25%) in affected tissues. The pathogenic threshold for the mutation in cybrids was between 4 and 8%, implying a dominant mechanism of action. The mutation affects the central base of the anticodon triplet of tRNATrp and it may alter the codon specificity of the affected tRNA. These findings introduce the concept of dominance in mitochondrial genetics and pose new diagnostic challenges, because such mutations may easily escape detection. Moreover, similar mutations arising stochastically and accumulating in a minority of mtDNA molecules during the aging process may severely impair RC function in cells.
INTRODUCTION
In the past two decades, more than 200 mitochondrial DNA (mtDNA) mutations have been associated with human disease. The mutations may affect all the mtDNA molecules within a cell or a tissue (homoplasmy) or may coexist with normal mitochondrial genomes (heteroplasmy) (1). In the latter case, the clinical or biochemical phenotype becomes evident only when the percentage of mutant molecules exceeds a critical threshold value. This value differs for different mutations and in different tissues, but is usually in the range of 70–90% (2). In other words, mtDNA mutations have a clear deleterious effect only when they affect the majority of mtDNA molecules within a cell. In this sense, they can be considered functionally recessive.
Here, we describe a novel mtDNA mutation (C5545T in tRNATrp) that contradicts this rule, since it caused a severe multisystemic disorder and marked respiratory chain (RC) deficiency even at low levels of heteroplasmy, thus behaving as the first reported ‘dominant’ mtDNA mutation associated with human disease.
RESULTS
We studied a 13-year-old boy with clinical features suggestive of a mitochondrial disorder.
RC defect in the patient's muscle
Histochemical analysis of a muscle biopsy obtained at 3 years of age showed diffuse reduction of cytochrome c oxidase (COX) activity, except for some rare fibers that stained intensely. Succinate dehydrogenase (SDH) staining was normal, with no evidence of excessive mitochondrial proliferation (‘ragged-blue’ fibers) (Fig. 1A). Biochemical measurement of RC enzymes showed markedly reduced complex IV activity and mildly decreased activities of complexes I, I+III and II+III. Complex II was normal and citrate synthase was mildly increased (Fig. 1C).
Figure 1.

Morphological and biochemical analyses on tissue samples taken at 3 years of age. (A) Histochemical stain for COX and SDH of the patient's muscle. (B) Histochemical stain for COX and SDH of fibroblasts from the patient and a healthy control. (C) Respiratory chain enzyme activities in patient's muscle and fibroblasts. Data are expressed as percentage of controls. ND, not determined.
RC defect in the patient's fibroblasts
Analysis of cultured skin fibroblasts showed similar results: COX histochemical stain was decreased in most cells, but there were some COX-positive fibroblasts and SDH stain was normal (Fig. 1B). Biochemical measurements of RC enzymes showed reduced activities of complexes I+III, II+III and IV, and slight elevation of citrate synthase activity (Fig. 1C).
Mutation analysis
The clinical features of our patient were strongly suggestive of a mitochondrial disorder, as biochemical and histochemical analyses documented a RC defect involving predominantly COX. Because there was no evidence of maternal inheritance and the COX deficiency was rather diffuse by histochemistry, we first excluded mutations in nuclear COX-assembly genes, including SCO1, SCO2, COX10, COX15 and SURF1. We then sequenced the 22 mtDNA tRNA genes and identified a C-to-T transition at nucleotide 5545 in the tRNATrp gene (MIM 590095) (Fig. 2A).
Figure 2.

(A) Sequence of the mitochondrial tRNATrp gene in DNA isolated from the patient's muscle. The arrow indicates the C5545T mutation. (B) Alignment of sequences of the anticodon loop of tRNATrp from different eukaryotic species. (C) PCR–RFLP analysis of the mutation in different tissues of the patient and of his mother. Radiolabeled fragments, after digestion with DraI, were separated on a gel, visualized and quantitated using a BioRad phosphor imager. The wild-type yields a single fragment of 286 bp, the mutant two fragments of 197 and 89 bp. In red patient tissues. M, skeletal muscle; BL, blood leukocytes; F, hair follicles; US, urinary sediment; HF, hair follicles; BS, buccal smear; C, healthy control; U, uncut fragment. (D) Schematic representation of the structure of wild-type and mutant tRNATrp and the possible consequences of the mutation in the anticodon on codon recognition.
The C5545T mutation affects the anticodon of mitochondrial tRNATrp and is present at low levels in affected tissues
The affected nucleotide encodes the central base of the anticodon triplet (Fig. 2B and D) and is conserved in all published vertebrate tRNATrp sequences (Fig. 2B). As the mutation creates a novel DraI site, we could confirm this change by PCR–RFLP analysis. The mutation was present at unusually low levels in all available tissues from our patient: 25% in muscle, 13% in leucocytes and 17% in cultured skin fibroblasts, while it was absent in all available tissues from the mother (Fig. 2C) and in 100 ethnically matched controls.
We sequenced the entire mitochondrial genome to exclude the concurrence of another known pathogenic mtDNA mutation and found none. The only other previously unreported sequence variation was a T-to-C transition at nucleotide 7797 in the COXII gene, causing an Ile>Thr substitution at amino acid 71. This change, however, was homoplasmic in all tissues from the patient and his mother (data not shown). Southern blot analysis excluded gross mtDNA rearrangements.
The presence of the mutation correlates with COX deficiency in individual muscle fibers
To assess pathogenicity, we studied the distribution of the mutation in individual muscle fibers by single-fiber PCR (3). The mutation was significantly more abundant in COX-negative fibers (31 ± 7%, n = 11) than in the rare COX-positive fibers, where it was virtually absent (3 ± 4%, n = 13) (P < 0.01) (Fig. 3A).
Figure 3.

(A) Distribution of the C5545T mutation in COX-positive and COX-negative muscle fibers. Quantification of mutant mtDNA was performed by single fiber muscle PCR–RFLP analysis. (B) Correlation between mutational load and COX activity in cybrid cell lines, compared with parental 143B and rho° cells. Measurments were performed in triplicates.
The presence of the mutation correlates with COX deficiency in cybrid cell lines
To exclude the possibility that the C5545T mutation was simply a modifier of a nuclear mutation and to assess a potential pathogenic role of the T7797C mutation, we generated cybrid cell lines by repopulating mtDNA-depleted (ρ0) 143B206 cells with mitochondria from the patient's enucleated fibroblasts (4). After selection, repopulation was assessed by Southern blot analysis, and the presence of each mutation was assayed in 50 clones by PCR–RFLP analyses. As expected, the T7797C mutation was homoplasmic in all clones, whereas the C5545T mutation showed low levels of heteroplasmy (0–28%) in different cell lines. Interestingly, we could not detect clones harboring mutation loads higher than 28%, suggesting that the selection medium, which contained the non-fermentable sugar galactose and was devoid of uridine, did not allow cells with high mutation loads to proliferate (4). Biochemical analysis showed normal COX activity (compared with parental 143B cells) in cells lacking the mutation, and progressively lower COX activities in cell lines with increasing mutation loads (Fig. 3B). Taken together, these data confirm the pathogenicity of the C5545T mutation and exclude causative roles both for unidentified nuclear genes and for the T7797C substitution, which we deem to be a neutral polymorphism.
Abnormal mitochondrial translation products in the patient's fibroblasts
To investigate the consequences of the mutation on mitochondrial protein synthesis we labeled mitochondrial translation products with 35S-methionine and 35S-cysteine in the presence of emetine, which inhibits cytoplasmic protein synthesis. As seen in Figure 4A, the levels of COX subunits I and III were the most severely reduced, whereas other polypeptides, such as ND6 and ND4L, were affected only minimally. Interestingly, ND3 levels were apparently even higher than in the controls. Moreover the proteins migrating in the COXII and COXIII region exhibited an apparently higher molecular weight compared with controls. Two other faint aberrant bands were visible in the patient, one migrating between COXIII and ATPase 6, the other with a very low molecular weight (lighter than ATPase 8). Immunoblot analysis of unlabeled proteins from the patient cells, separated under the same conditions, also revealed a slight increase in the apparent molecular weight of COXII (Fig. 4B). To exclude an artifact due to abnormal migration in the gel, we also probed the membrane with an antibody against Bcl-2, a protein with a molecular weight similar to that of COXII (26.2 versus 25.5 kDa); in this case no shift was observed.
Figure 4.
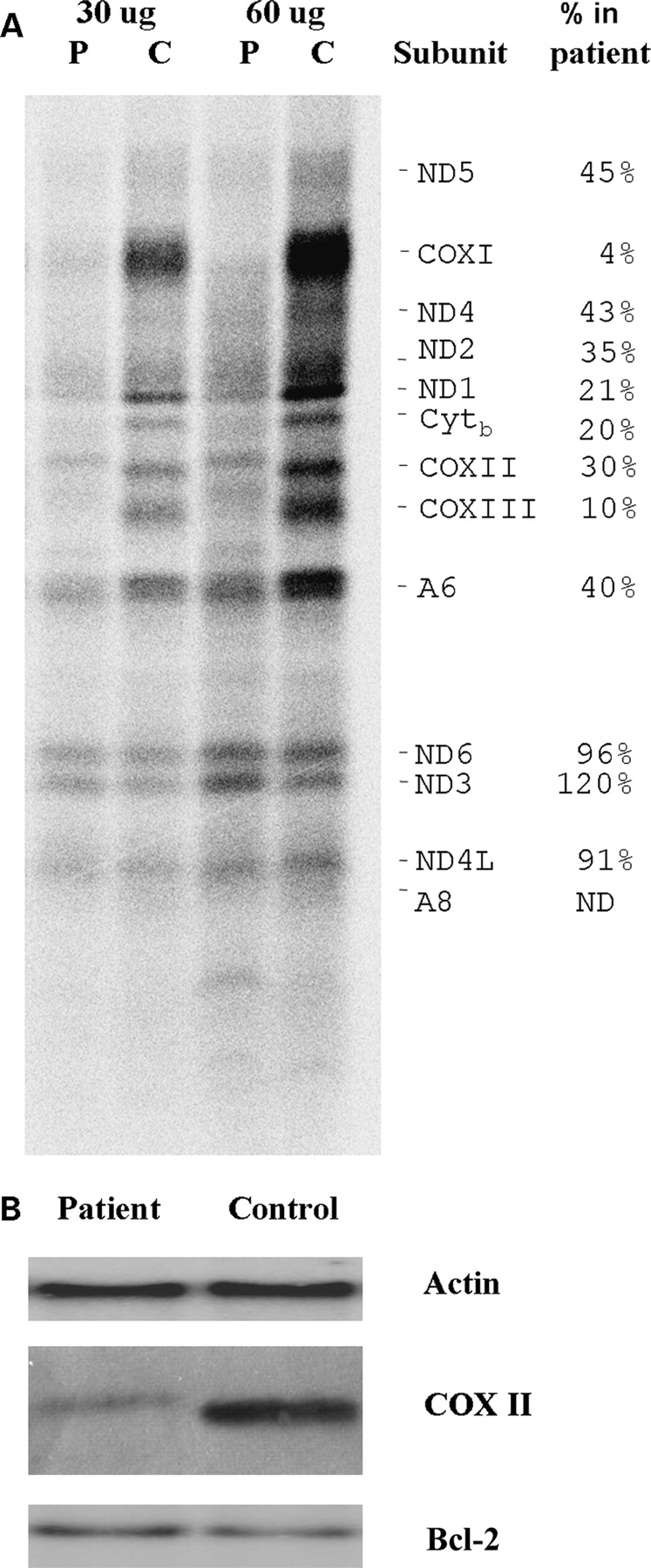
(A) In vivo 35S-labeling mitochondrial proteins of patient's fibroblast after blocking cytoplasmic protein synthesis with emetine (see Materials and Methods section in the text for details). Radiolabeled proteins were separated on a SDS–tris–glycine gel and visualized using a BioRad phosphor imager. The two sets of lanes represent two different amounts of protein from the same samples. P, patient, C, healthy control. (B) Equal amounts of unlabeled cellular extracts from patient and control fibroblasts were separated on a similar gel under the same conditions. After blotting, the membrane was probed with anti-COXII, anti-actin and anti-Bcl-2 antibodies.
DISCUSSION
Mutations in mitochondrial tRNA genes usually cause generalized impairment of mitochondrial protein synthesis. Individual mutations may affect tRNA function by different mechanisms, including inhibition of aminoacylation (5), impairment of tRNA stability (6), altered processing of the tRNA (7), modification of wobble-bases (8,9), reduced association of tRNA with the ribosome (10) or a combination of these mechanisms. In general, these mutations cause a ‘loss-of-function’ of the affected tRNA and are considered functionally recessive, as they affect most mtDNA molecules in a cell (70–90%) to cause a biochemical phenotype (2).
The C5545T mutation fulfills the canonical criteria of pathogenicity for an mtDNA mutation. First, it was absent in more than 100 controls. Second, it was heteroplasmic. Third, its relative abundance correlated with the biochemical phenotype in single muscle fibers and in cybrid cell lines. However, contrary to other mtDNA mutations, its pathogenic threshold in cybrids was between 4 and 8%. None of the aforementioned mechanisms can explain its deleterious effects, and the extremely low threshold suggests either a dominant negative effect, or, more likely, a gain-of-function of the mutated tRNA. We have considered several scenarios to explain this phenomenon.
The C5545T mutation affects the central base of the anticodon triplet of tRNATrp (tRNA position 35), which is invariably a C in all species in which UGA and UGG encode for tryptophan (11). This base interacts with the central base (G) of the codon triplet in the mRNA and is essential in determining tRNA specificity. A mutation with similar properties was described in a cell-culture model by El Meziane et al. (12), who reported that a substitution in tRNALeu(CUN) at np 12300 could suppress the RC defect in a cybrid cell line harboring high levels (>95%) of the A3243G tRNALeu(UUR) mutation. Like C5545T, the G12300A mutation also affected the anticodon triplet of tRNALeu(CUN), but at tRNA position 36, thus enabling it to recognize UUR codons. Just 12% of mutant tRNALeu(CUN) sufficed to rescue the RC defect in the 100% A3243G cybrids. In that case, the mutation produced a gain-of-function in the affected molecules, and we hypothesize a similar mechanism of action for the C5545T mutation. While tRNATrp usually recognizes UGA and UGG codons, we hypothesize that the mutation, which converts ACU (recognizing UGA and UGG) to AUU, allows the mutated tRNA to decode UAA or UAG codons (Fig. 2D).
As UAA and UAG encode termination signals, the mutated tRNATrp may cause a read-through of the mRNA (adding one Trp residue at the position of the stop codon), followed by the incorporation of a tail of lysine residues specified by the poly-A sequence at the end of the transcript. In this scenario, the mutant tRNA would act like a bacterial ochre suppressor (13). Moreover, considering that in the mitochondrial genome the third base of the codon is usually not essential to determine tRNA specificity (14), the mutated tRNA could also recognize UGC and UGU codons, thus incorporating tryptophan in the place of tyrosine (Fig. 2D). These two mechanisms probably coexist.
Not all RC complexes appeared to be equally affected by the mutation. COX was the most severely affected complex in biochemical assays, and in 35S-labeling experiments subunits COXI and COXIII had the lowest levels compared with controls. COXII and COXIII were also qualitatively abnormal, displaying an apparently higher molecular weight, a finding which is compatible with the read-through hypothesis. Two other bands migrated in regions of the gel that normally do not correspond to any mtDNA-encoded proteins; we believe these last two polypeptides may represent degradation products. However, these data must be interpreted with caution, since aberrant translation products are also associated with other mtDNA mutations, such as tRNALys A8344G (15).
The observed preferential susceptibility of COX subunits to the mutation, compared with other RC complexes, is not fully clear. We note that the efficiency of tRNA suppression in bacteria depends on the codon context of the suppressed stop codons (16). It is possible that also in this case the codons upstream of the termination signals (or of tyrosine codons) determine where tryptophan is inappropriately inserted in the proteins, justifying the differential involvement of individual subunits. COXI and ND6 have AGA and AGG termination signals, and therefore should not be ‘suppressed’ by the mutant tRNA. ND6 seems unaffected by the mutation, while the severe reduction of COXI (which does not display an apparent increase in molecular weight) could be due to the inappropriate insertion of tryptophan instead of tyrosine. Further experiments are required to clarify in depth all these issues. Unfortunately, to date it is still impossible to manipulate the mitochondrial genome of higher eukaryotes, therefore traditional genetic techniques cannot be employed to investigate this problem via targeted mutagenesis. We are currently trying to isolate sufficient amounts of purified COX subunits to perform direct protein sequencing in order to confirm these hypotheses.
Two other possible scenarios are that the mutant tRNA inhibits aminoacylation of wild-type tRNATrp molecules, or that it interferes with the ribosomal machinery. However, we do not favor these hypotheses because they cannot account for the apparently increased molecular weight of COXII and COXIII, and because in these cases we would expect similar effects on all mtDNA-encoded subunits. ND3 is apparently increased in the patient, and there is no clear correlation between the number of tryptophan residues in individual mitochondrial proteins and the corresponding reduction of their levels in the patient.
Finally, it should be noted that another anticodon mutation is associated with human disease, affecting tRNAPro at nt 15990 (17). However, this mutation behaved in the ‘typical’ way, displaying a high threshold that excluded a dominant effect. This discrepancy can be explained by the fact that the tRNAPro mutation impairs correct processing by inhibiting methylation of nucleotide 37 of the tRNA and causing a ‘classical’ loss-of-function of the mutated tRNA (18).
Regardless of the precise mechanism of action, our data provide a novel paradigm in mitochondrial genetics, in which a mutation affecting a very small proportion of tRNA molecules in a cell can cause severe RC deficiency via a dominant effect. In this model, a single mutant mtDNA molecule per mitochondrion is sufficient to cause RC deficiency. The low threshold values are consistent with those found for the tRNALeu(CUN) at np 12 300 (12). In that case the fact that 12% mutant mtDNA was enough to suppress the effect of the A3243G mutation, and recover mitochondrial protein synthesis, indicates that, at this percentage of heteroplasmy, most mitochondria harbor at least one molecule with the G12300A mutation. In any case, the pathogenic threshold may be different in different tissues, and may be influenced by several factors, including mitochondrial morphology and dynamics. Also, the situation may be different in replicating cells compared with that in non-dividing muscle fibers.
The abrupt decrease in COX activity seen between 4 and 8% mutant can be explained by progressive accumulation of the mutation in mitochondria, while the relatively smaller differences in cells harboring higher mutation levels can be explained by the fact that once that most mitochondria contain, on average, at least one mutated genome, more mutant molecules do not appear to produce a significantly more severe biochemical phenotype. In this view, the C5545T behaves like a ‘true dominant’ allele like the Huntington disease mutation, or mutations in the prion protein gene, where the phenotype of homozygotes does not differ from that of heterozygotes (19). In fact, at the levels found in our cybrids, the mutated alleles coexist with a significant (>70%) proportion of wild-type tRNATrp molecules. If clones with higher mutational loads become available in the future, it will be interesting to study the effect on other RC complexes, because increasing loads of the mutant allele should reduce the percentage of functional tRNAtrp molecules available for protein synthesis. In fact, the mutated tRNA might be at such high levels that there might be insufficient numbers of wild-type tRNATrp molecules to decode the normal UGA and UGG tryptophan codons.
Taken together, these findings have important implications in the study of mitochondrial disease, because mutations at low levels of heteroplasmy may easily escape detection by standard sequencing methods. Moreover, similar mutations arising stochastically and accumulating in a minority of mtDNA molecules during the aging process may severely impair RC function in cells and lead to dysfunction in organs with high energy requirements.
PATIENT DATA AND METHODS
Case report
The patient, a 13-year-old boy born of non-consanguineous parents, was diagnosed at 5 months of age with hypertrophic cardiomyopathy without evidence of outflow obstruction. He also had truncal hypotonia and lactic acidosis. He progressively lost acquired developmental milestones, developed seizures, and, at 3 years of age showed slowly progressive chorea. Brain MRI showed mild diffuse brain atrophy and bilateral putaminal hyperintensities. At 13 years of age, head circumference was at the third percentile, and weight and height were below the fifth percentile. He was hypotonic but hyperreflexic, with proximal weakness but with normal extraocular movements. He was ataxic and dysmetric, and had low-amplitude choreic movements of the face, trunk and limbs. He was able to stand and walk only with support. The cardiologic situation remained stable. All studies were performed with the informed consent of the patient's parents on the samples obtained at 3 years of age.
Biochemical and histochemcal studies
Muscle histochemistry and biochemistry were performed as described elsewhere (20), (21). Fibroblast and cybrids were analyzed as described previously (22).
Molecular studies
Sequencing of SCO1, SCO2, COX10, COX15 and SURF1 and of the mitochondrial genome was carried out as described (21).
The C5545T mutation was assayed using primers 5454F (5′-ctcatcgcccttaccacgct-3′) and 5739R (5′-gcgggagaagtagattgaag-3′) PCR conditions were 94°C for 3 min 30 cycles of 94°C 30 s, 55°C 30 s and 72°C 30 s, and a final extension steps of 72°C for 7 min. A last PCR cycle was performed with the addition of 0.1 µCi of α-32P dCTP. Samples were digested overnight with 10 units of DraI, separated on a 12% polyacrylamide gel and visualized and quantitated using a BioRad phosphorimager. The ‘last cycle hot’ avoids detection of heteroduplexes between mutant and wild-type strands, which would cause underestimation of the mutant population after restriction nuclease digestion (23).
The T7797C variant was analyzed with a similar procedure using for amplification primers 7773mis F (5′-ccgtctgaactatcctgcccgGc-3′) (in capital the mismatched nucleotide) and 7938R (5′-aagattagtccgccgtagtc-3′). The mismatch creates a SfaI site in wild-type allele, and a BanI site in the mutant. PCR fragments were radiolabeled, digested separately with each enzyme and visualized as above.
Southern blot analysis of mtDNA was performed as described (24).
Single-fiber PCR analysis
Single-fiber PCR analysis of the C5545T mutation was performed as described (3). In brief, 30 µm-thick muscle were stained for COX. COX-positive and COX-negative fibers were isolated and lysed by boiling. The lysate was used as template for subsequent PCR–RFLP quantitation of the mutation employing the same procedure described above.
Generation of cybrids
Cybrids were generated by fusing enucleated patient's fibroblasts with mtDNA-depleted 143B206 cells as described (4). Twenty-four hours after fusion, cybrids were plated in DMEM containing 0.9 mg/ml galactose instead of glucose, and supplemented with 100 µg of 5-bromodeoxyuridine (BrDU) per ml, and 5% dialyzed fetal bovine serum (FBS) (selective medium). BrDU is included in the medium because it kills contaminating fibroblasts (or nucleated fibroblasts fused with 143B206 cells). The 143B206 cells lacking thymidine kinase are unaffected by BrDU. Clones were isolated by trypsinization in glass cloning cylinders 13 to 18 days after fusion. Cells were subsequently cultured in DMEM supplemented with 5% FBS.
Labeling of mitochondrial translation products
Patients’ fibroblasts growing at 70% confluency in 60 mm Petri dishes were labeled with [35S]Translabel (1000 Ci/mmol, 100–200 mCi/ml; Dupont NEN) for 2 h in 2 ml of methionine- and cysteine-free DMEM supplemented with 100 mg/ml of emetine (which blocks cytoplasmic protein synthesis, but does not affect mitochondria) and 5% dialyzed FBS as described previously by Chomyn et al. (25). Cells were then trypsinized and total protein concentration in samples was determined using the Total Protein Kit (Sigma). Equal amounts of total cellular protein were then separated on SDS–tricine gels as described (26) and visualized with a Phosphorimager.
The assignment of mtDNA-encoded translation products was determined by their similarity to those described in Chomyn et al. (25), and in MITOMAP (Mitochondrial DNA Polypeptide Assignments http://www.mitomap.org/cgi-bin/tbl2gen.pl), and by their corresponding absence in a parallel experiment performed with 143B206 cells lacking mtDNA.
Immunoblot analysis
Equal amounts of total cell proteins were separated on SDS–tricine gels as descried above. After blotting, membranes were probed with an anti COXII antibody (Molecular Probes), with an anti-actin antibody (Sigma), and with an anti-Bcl-2 antibody (Sigma) according to the manufacturer's protocol. Detection was performed using the ECL advanced kit (GE biosciences).
FUNDING
This work was supported by NIH Grant HD32062, a grant from the Muscular Dystrophy Association, and the Marriott Mitochondrial Disorders Clinical Research Fund (MMDCRF). L.S. has been Supported by Telethon Italy Grants 439b and GGP06256.
ACKNOWLEDGEMENTS
We are grateful to Dr Valerio Dorrello for helpful discussion.
Conflict of Interest statement. None declared.
REFERENCES
- 1.DiMauro S., Hirano M. Mitochondrial encephalomyopathies: an update. Neuromuscul. Disord. 2005;15:276–286. doi: 10.1016/j.nmd.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 2.Rossignol R., Faustin B., Rocher C., Malgat M., Mazat J.P., Letellier T. Mitochondrial threshold effects. Biochem. J. 2003;370:751–762. doi: 10.1042/BJ20021594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moraes C.T., Ricci E., Bonilla E., DiMauro S., Schon E.A. The mitochondrial tRNA(Leu(UUR)) mutation in mitochondrial encephalomyopathy, lactic acidosis, and strokelike episodes (MELAS): genetic, biochemical, and morphological correlations in skeletal muscle. Am. J. Hum Genet. 1992;50:934–949. [PMC free article] [PubMed] [Google Scholar]
- 4.King M.P., Attardi G. Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science. 1989;246:500–503. doi: 10.1126/science.2814477. [DOI] [PubMed] [Google Scholar]
- 5.Enriquez J.A., Chomyn A., Attardi G. MtDNA mutation in MERRF syndrome causes defective aminoacylation of tRNA(Lys) and premature translation termination. Nat. Genet. 1995;10:47–55. doi: 10.1038/ng0595-47. [DOI] [PubMed] [Google Scholar]
- 6.Hao H., Moraes C.T. A disease-associated G5703A mutation in human mitochondrial DNA causes a conformational change and a marked decrease in steady-state levels of mitochondrial tRNA(Asn) Mol. Cell. Biol. 1997;17:6831–6837. doi: 10.1128/mcb.17.12.6831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schon E.A., Koga Y., Davidson M., Moraes C.T., King M.P. The mitochondrial tRNA(Leu)(UUR)) mutation in MELAS: a model for pathogenesis. Biochim. Biophys. Acta. 1992;1101:206–209. [PubMed] [Google Scholar]
- 8.Yasukawa T., Suzuki T., Ueda T., Ohta S., Watanabe K. Modification defect at anticodon wobble nucleotide of mitochondrial tRNAs(Leu)(UUR) with pathogenic mutations of mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes. J. Biol. Chem. 2000;275:4251–4257. doi: 10.1074/jbc.275.6.4251. [DOI] [PubMed] [Google Scholar]
- 9.Kirino Y., Goto Y., Campos Y., Arenas J., Suzuki T. Specific correlation between the wobble modification deficiency in mutant tRNAs and the clinical features of a human mitochondrial disease. Proc. Natl. Acad. Sci. USA. 2005;102:7127–7132. doi: 10.1073/pnas.0500563102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chomyn A., Enriquez J.A., Micol V., Fernandez-Silva P., Attardi G. The mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episode syndrome-associated human mitochondrial tRNALeu(UUR) mutation causes aminoacylation deficiency and concomitant reduced association of mRNA with ribosomes. J. Biol. Chem. 2000;275:19198–19209. doi: 10.1074/jbc.M908734199. [DOI] [PubMed] [Google Scholar]
- 11.Sprinzl M., Horn C., Brown M., Ioudovitch A., Steinberg S. Compilation of tRNA sequences and sequences of tRNA genes. Nucleic Acids Res. 1998;26:148–153. doi: 10.1093/nar/26.1.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.El Meziane A., Lehtinen S.K., Hance N., Nijtmans L.G., Dunbar D., Holt I.J., Jacobs H.T. A tRNA suppressor mutation in human mitochondria. Nat. Genet. 1998;18:350–353. doi: 10.1038/ng0498-350. [DOI] [PubMed] [Google Scholar]
- 13.Yoshimura M., Kimura M., Ohno M., Inokuchi H., Ozeki H. Identification of transfer RNA suppressors in Escherichia coli. III. Ochre suppressors of lysine tRNA. J. Mol. Biol. 1984;177:609–625. doi: 10.1016/0022-2836(84)90040-8. [DOI] [PubMed] [Google Scholar]
- 14.Jukes T.H. Mitochondrial codes and evolution. Nature. 1983;301:19–20. doi: 10.1038/301019a0. [DOI] [PubMed] [Google Scholar]
- 15.Masucci J.P., Davidson M., Koga Y., Schon E.A., King M.P. In vitro analysis of mutations causing myoclonus epilepsy with ragged-red fibers in the mitochondrial tRNA(Lys)gene: two genotypes produce similar phenotypes. Mol. Cell. Biol. 1995;15:2872–2881. doi: 10.1128/mcb.15.5.2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mottagui-Tabar S., Bjornsson A., Isaksson L.A. The second to last amino acid in the nascent peptide as a codon context determinant. Embo J. 1994;13:249–257. doi: 10.1002/j.1460-2075.1994.tb06255.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moraes C.T., Ciacci F., Bonilla E., Ionasescu V., Schon E.A., DiMauro S. A mitochondrial tRNA anticodon swap associated with a muscle disease. Nat. Genet. 1993;4:284–288. doi: 10.1038/ng0793-284. [DOI] [PubMed] [Google Scholar]
- 18.Brule H., Holmes W.M., Keith G., Giege R., Florentz C. Effect of a mutation in the anticodon of human mitochondrial tRNAPro on its post-transcriptional modification pattern. Nucleic Acids Res. 1998;26:537–543. doi: 10.1093/nar/26.2.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zlotogora J. Dominance and homozygosity. Am. J. Med. Genet. 1997;68:412–416. doi: 10.1002/(sici)1096-8628(19970211)68:4<412::aid-ajmg8>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 20.Sciacco M., Bonilla E. Cytochemistry and immunocytochemistry of mitochondria in tissue sections. Methods Enzymol. 1996;264:509–521. doi: 10.1016/s0076-6879(96)64045-2. [DOI] [PubMed] [Google Scholar]
- 21.Sacconi S., Salviati L., Sue C.M., Shanske S., Davidson M.M., Bonilla E., Naini A.B., De Vivo D.C., DiMauro S. Mutation screening in patients with isolated cytochrome c oxidase deficiency. Pediatr. Res. 2003;53:224–230. doi: 10.1203/01.PDR.0000048100.91730.6A. [DOI] [PubMed] [Google Scholar]
- 22.Salviati L., Hernandez-Rosa E., Walker W.F., Sacconi S., DiMauro S., Schon E.A., Davidson M.M. Copper supplementation restores cytochrome c oxidase activity in cultured cells from patients with SCO2 mutations. Biochem. J. 2002;363:321–327. doi: 10.1042/0264-6021:3630321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shoffner J.M., Lott M.T., Lezza A.M., Seibel P., Ballinger S.W., Wallace D.C. Myoclonic epilepsy and ragged-red fiber disease (MERRF) is associated with a mitochondrial DNA tRNA(Lys) mutation. Cell. 1990;61:931–937. doi: 10.1016/0092-8674(90)90059-n. [DOI] [PubMed] [Google Scholar]
- 24.Salviati L., Sacconi S., Mancuso M., Otaegui D., Camano P., Marina A., Rabinowitz S., Shiffman R., Thompson K., Wilson C.M., et al. Mitochondrial DNA depletion and dGK gene mutations. Ann. Neurol. 2002;52:311–317. doi: 10.1002/ana.10284. [DOI] [PubMed] [Google Scholar]
- 25.Chomyn A. In vivo labeling and analysis of human mitochondrial translation products. Methods Enzymol. 1996;264:197–211. doi: 10.1016/s0076-6879(96)64020-8. [DOI] [PubMed] [Google Scholar]
- 26.Schagger H., von Jagow G. Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal. Biochem. 1987;166:368–379. doi: 10.1016/0003-2697(87)90587-2. [DOI] [PubMed] [Google Scholar]