

Abstract
Muscular dystrophies comprise a genetically heterogeneous group of degenerative muscle disorders characterized by progressive muscle wasting and weakness. Two forms of limb-girdle muscular dystrophy, 2A and 2B, are caused by mutations in calpain 3 (CAPN3) and dysferlin (DYSF), respectively. While CAPN3 may be involved in sarcomere remodeling, DYSF is proposed to play a role in membrane repair. The coexistence of CAPN3 and AHNAK, a protein involved in subsarcolemmal cytoarchitecture and membrane repair, in the dysferlin protein complex and the presence of proteolytic cleavage fragments of AHNAK in skeletal muscle led us to investigate whether AHNAK can act as substrate for CAPN3. We here demonstrate that AHNAK is cleaved by CAPN3 and show that AHNAK is lost in cells expressing active CAPN3. Conversely, AHNAK accumulates when calpain 3 is defective in skeletal muscle of calpainopathy patients. Moreover, we demonstrate that AHNAK fragments cleaved by CAPN3 have lost their affinity for dysferlin. Thus, our findings suggest interconnectivity between both diseases by revealing a novel physiological role for CAPN3 in regulating the dysferlin protein complex.
INTRODUCTION
Muscular dystrophies comprise a heterogeneous group of inherited degenerative muscle disorders characterized by progressive muscle wasting and weakness with variable distribution and severity. Since the discovery of dystrophin, a large number of genes either associated with, or linked to, various forms of muscular dystrophy (MD) have been identified. Limb-girdle muscular dystrophies (LGMDs) are a genetically heterogeneous group of primary myopathies showing progressive weakness and wasting of the muscles of the pelvic and shoulder girdle, and ranging from severe forms with onset in the first decade with rapid progression to milder forms with later onset and a slower course. Three different major disease mechanisms are emerging for LGMD: a structural defect in the muscle membrane, muscle membrane repair deficiency and defects in sarcomere remodeling, cytoskeleton structure and cytoskeleton-membrane interactions.
Limb-girdle muscular dystrophy type 2A (LGMD2A; OMIM# 253600) or calpainopathy is considered the most frequent form of recessive LGMD world wide (1–3). The gene responsible for LGMD2A and coding for calpain 3 (CAPN3; OMIM# 114240) was localized by linkage analysis to the chromosomal region 15q15.1–15.3 and subsequently identified by positional cloning (3). To date well over 350 distinct pathogenic mutations in the calpain 3 gene have been described according to the Leiden Muscular Dystrophy database (http://www.DMD.nl/CAPN3). Calpain 3 is a skeletal muscle-specific member of the calpain superfamily of non-lysosomal, Ca2+-dependent cysteine proteases (4). Several cytoskeleton components were identified as partners and substrates for calpain 3 linking its function to the regulation of cytoskeleton structure and cytoskeleton–membrane interactions (5,6). Deregulation of sarcomere remodeling due to lack of proteolysis of substrates by calpain 3 was therefore postulated as a mechanism of LGMD2A pathogenesis (7), which emphasizes the importance to identify calpain 3 substrates.
One of the LGMDs in which a secondary reduction of calpain 3 can be observed is LGMD2B (OMIM# 253601). Mutations in dysferlin (DYSF; OMIM# 603009) do not only cause LGMD2B, but also Miyoshi myopathy and distal anterior compartment myopathy (8–10). Dysferlin is suggested to play a key role in muscle membrane repair, defining a new pathogenic mechanism in MD (11). However, the precise biochemical function of dysferlin remains unknown. Myoferlin, a homologue of dysferlin, is also expressed at the plasma membrane, and in addition found at the nuclear envelope (12). By co-immunoprecipitation experiments, we previously demonstrated that calpain 3 is in complex with dysferlin (13). In addition, we showed that the C-terminal domain of AHNAK nucleoprotein (AHNAK; OMIM# 103390), a novel partner of the dysferlin protein complex, can interact with the C2A domain of dysferlin and myoferlin and that AHNAK redistributes to the cytoplasm with dysferlin during muscle regeneration (14). As both dysferlin and AHNAK are implicated in membrane repair and maintenance, this may suggest a previously unrecognized role of calpain 3 in muscle membrane homeostasis.
AHNAK (also called desmoyokin, MW 700 kDa), located on human chromosome 11q12–13 (15), contains three main structural regions: the N-terminal 498 amino acids, a large central region of ∼4300 amino acids with multiple repeated units, most of which are 128 amino acids in length and the C-terminal 1002 amino acids (16). A second AHNAK nucleoprotein-like protein, AHNAK nucleoprotein 2 (AHNAK2; OMIM# 608570; MW 600 kDa), located on chromosome 14q32, was recently identified by a search for homologous sequences in the human genome (17). AHNAK-deficient mice show no obvious phenotype, and it is speculated that AHNAK2 can compensate for the loss of AHNAK (17). The exact biological function of AHNAK is largely unknown. In vitro, the C-terminal region of AHNAK (amino acids 5262–5643) binds to G-actin and cosediments with F-actin suggesting a role for AHNAK in the maintenance of the structural and functional organization of the subsarcolemmal cytoarchitecture in cardiomyocytes (18).
Haase et al. (19) analyzed AHNAK expression in rat cardiac and skeletal muscle preparations using the KIS antibody against the central domain of AHNAK and the Tail antibody against a serine-rich epitope located near the C-terminus of AHNAK. On western blots using the KIS antibody they observed a high molecular mass protein of the expected size of 700 kDa and a faint 500 kDa protein. In addition, a lower molecular mass immuno-reactive fragment was also detected by the Tail-antibody. These results suggested that AHNAK is either unstable or cleaved by proteases. To date however, the identity of the enzymes involved in post-translational processing of AHNAK is unknown. Moreover, the biological relevance of this potential cleavage is not understood.
In this study we demonstrate that AHNAK can interact with the protease calpain 3 and that AHNAK serves as a direct substrate of calpain 3 in cell culture. The interaction of both proteins and the cleavage of AHNAK by calpain 3 are also supported by their colocalization in skeletal muscle. Calpain 3 cleavage causes a loss of AHNAK in cell culture, and AHNAK is increased at the sarcolemma in patients with a calpainopathy. Interestingly, calpain 3-mediated proteolysis of AHNAK prevents the interaction of AHNAK with dysferlin and myoferlin, respectively, providing new mechanistic insight for the physiological function of calpain 3 in the dysferlin protein complex in skeletal muscle.
RESULTS
AHNAK interacts with calpain 3
To demonstrate that calpain 3 can directly interact with AHNAK and to determine which domains of AHNAK are responsible for the specific binding, we used AHNAK expressing clones DY-N (amino acids 249–498), DY-M (amino acids 1070–1579) and DY-C (amino acids 4895–5890) (Fig. 1) to generate a series of recombinant GST-AHNAK fusion proteins (Fig. 2A). These clones are commonly regarded as representative for the different structural domains of AHNAK. To allow for good recombinant protein production, we separated DY-C in two: C1 (amino acid 4895–5393) and C2 (amino acid 5394–5890) (Fig. 1). The bacterially produced GST fusion proteins (Fig. 2B) were applied in a GST pull-down assay, using N-terminally T7-tagged calpain 3 as prey (Fig. 2A). Because of the rapid autolysis of calpain 3 (5), the proteolytically inactive form calpain 3 C129S was used. Western blot analysis of the pull-down fractions of total protein extracts using an anti-T7 antibody showed strong interaction between C1-, C2-AHNAK and full-length calpain 3 C129S and its degradation fragments (Fig. 2C). M-AHNAK was also able to weakly bind to 65 and 40 kDa fragments of calpain 3 C129S, respectively (Fig. 2C). No binding was observed for equivalent amounts of the GST fusion proteins N-AHNAK or for the control GST protein.
Figure 1.

Schematic representation of full length AHNAK and the different constructs used in these studies. On the left, the ability to bind and/or act as a substrate for calpain 3 is indicated for each AHNAK fragment.
Figure 2.

Identification of the interaction sites between AHNAK and inactive calpain 3 C129S. (A) Scheme of vectors for expressing AHNAK fusion proteins and calpain 3 C129S fusion protein. (B) Induced GST (lane 1) or GST bound to the affinity beads (lane 2); lanes 3–10 represent induced and affinity beads-bound GST-N-AHNAK (lanes 3–4), GST-M-AHNAK (lanes 5–6), GST-C1-AHNAK (lanes 7–8) and GST-C2-AHNAK (lanes 9–10), respectively. Equal amounts of GST fusion proteins representing different domains of AHNAK or GST were used in pull-down assays with T7-tagged calpain 3 C129S fusion protein as described in Materials and Methods. Bound proteins were separated by SDS–PAGE and analyzed by immunoblotting with anti-T7 HRP. (C) Lanes 1–4 represent uninduced-, induced-, soluble-, and precleared T7-tagged calpain 3 C129S, respectively. Lanes 5–8 represent GST-N-AHNAK, GST-M-AHNAK, GST-C1-AHNAK and GST-C2-AHNAK pull-down fractions while lane 9 shows the result with GST as negative control. As shown, GST-C1-AHNAK and GST-C2-AHNAK pulled down T7-tagged full length calpain 3 C129S fusion protein and its degradation products (arrowheads). GST-N-AHNAK, GST-M-AHNAK and GST did not interact with calpain 3 C129S fusion proteins. A molecular mass marker is indicated on the left.
Cleavage of AHNAK by calpain 3 downregulates AHNAK in cell culture
To investigate whether the interaction between calpain 3 and AHNAK is functional, we cloned active calpain 3 and inactive calpain 3 C129S into pEGFP-N2 expression vector. These constructs were transiently transfected into COS-1 (Fig. 3A and B) and 3T3 (Fig. 3A' and B') cells, which express high levels of endogenous AHNAK and no calpain 3. We immunocytochemically examined wt calpain 3 and calpain 3 C129S expressing cells using the KIS antibody to detect AHNAK. We observed an absence of endogenous AHNAK in wt calpain 3 overexpressing COS-1 and 3T3 cells (Fig. 3A and A'), compared with non-transfected cells. In contrast, normal AHNAK immunoreactivity was observed in calpain 3 C129S expressing cells (Fig. 3B and B'). Taken together, these results suggest that expression of calpain 3 results in a loss of AHNAK, possibly due to its proteolytic activity.
Figure 3.

Cleavage of AHNAK by calpain 3 downregulates AHNAK in cell culture. To evaluate the function of AHNAK cleavage by calpain 3 in COS-1 (A and B) and 3T3 cells (A' and B'), we immunocytochemically examined wt calpain 3 and calpain 3 C129S expressing cells using KIS antibody to detect AHNAK. Absence of endogenous AHNAK was observed in wt calpain 3 overexpressing cells (A and A'), compared with non-transfected cells. In contrast, normal AHNAK signals were observed in calpain 3 C129S expressing cells (B and B'). Bars represent 20 µm.
Calpain 3 cleaves endogenous AHNAK
To confirm the immunofluorescence data, we lysed COS-1 cells transiently transfected with calpain 3 after 48 h and evaluated proteolysis of AHNAK by a possible change in molecular mass of AHNAK on western blots, with a cocktail of the affinity purified antisera CQL, which recognizes the C-terminus, and KIS, which recognizes the repeats in AHNAK (Fig. 4). Bands at 700 and 300 kDa are detected in all lanes. However, only upon expression of active calpain 3 a 120 kDa immunoreactive fragment was observed. Further analysis with immediate lysis in sample buffer to prevent aspecific cleavage showed this signal to be recognized by the CQL antibody, and therefore to represent a C-terminal fragment of AHNAK (data not shown). No such cleavage fragment was detected in non-transfected control cells or cells transfected with inactive calpain 3 C129S.
Figure 4.

Cleavage of endogenous AHNAK by recombinant calpain 3. Cos cells were transfected with either mock (lane 1), inactive calpain 3-GFP (C129S mutant) (lane 2) or active calpain 3-GFP (lane 3). Forty-eight hours after transfection cells were harvested and analyzed on western blot with a KIS/CQL antibody mixture. The arrows point out full length AHNAK and a 120 kDa band which arises upon active calpain 3 expression.
Calpain 3 cleaves N-, C1, C2-ahnak, but not M-AHNAK
In Figure 2, we showed that AHNAK interacts with calpain 3 in a GST pull-down assay. The most C-terminal domain of AHNAK showed a clear interaction but other domains also pulled down T7-tagged inactive calpain 3, indicating that calpain 3 can bind AHNAK at other sites as well. As the CQL antibody is a limited tool to study cleavage of full-length AHNAK, we constructed four fusion proteins representing the N-terminus (N), repeated domain (M) and C-terminus (C1, and C2) of AHNAK, respectively, to identify specific domains that can be cleaved by calpain 3. Fusion proteins were cloned into a pSG8-VSV-HA eukaryotic expression vector fusing the AHNAK fragments N-terminally to a VSV tag and C-terminally to a HA tag. These fusion constructs were individually transfected and cotransfected into COS-1 cells with various forms of calpain 3 (active GFP-calpain 3, GFP-calpain 3 C129S) and GFP as control. After 48 h cells were harvested and cell lysates were analyzed on western blot to detect possible proteolytic cleavage products using antibodies recognizing the N- and C-terminal tag. As shown in Figure 5A, respective antibody detection revealed that N-AHNAK was cleaved from 35 kDa to 17 and 18 kDa protein fragments. VSV antibody detection demonstrated that C1-AHNAK was cleaved from 65 to 30 kDa (Fig. 5C) but HA antibody did not detect cleaved C-terminal fragments. VSV antibody detection also showed that C2-AHNAK was cleaved from 60 kDa to 36 and 22 kDa fragments and the HA antibody detected complementary fragments of 24 and 38 kDa (Fig. 5D), respectively. These cleavage products were observed only when normal calpain 3 was transfected, but neither in calpain 3 C129S nor GFP transfected cells. No cleavage was detected for M-AHNAK (Fig. 5B). Thus, calpain 3 can cleave the N-terminus of AHNAK at two sites and the C-terminus at three, of which the N-terminal cleavage sites are at close distance.
Figure 5.

Specific domains of AHNAK are cleaved by calpain 3 in cell culture. Four AHNAK fusion proteins representing N- (A), M- (B), C1- (C) and C2- of AHNAK (D), respectively, were expressed in the presence of active or inactive calpain 3 C129S to assess whether AHNAK can act as a substrate for calpain 3. The AHNAK fusion proteins were N-terminally tagged with a VSV tag and C-terminally tagged with a HA tag to detect N-terminal and C-terminal AHNAK cleavage products. Actin detection was used for equal loading control (a). Antibodies recognizing the N-terminal VSV tag (b) and C-terminal HA tag (c) were used to detect proteolytic cleavage products on western blots. In all panels, lane 1–5 represent lysates of non-transfected COS-1 cells, co-transfection of AHNAK fusion construct with calpain 3, co-transfection of AHNAK fusion construct with calpain 3 C129S, co-transfection of AHNAK fusion construct with GFP and single transfection of AHNAK fusion construct, respectively. As shown in (A), HA antibody and VSV antibody detection revealed that N AHNAK was cleaved from 35 kDa to 17 and/or 18 kDa fragments and complementary fragments were observed with the HA antibody; VSV antibody detection revealed that C1-AHNAK was cleaved from 65 to 30 kDa (C) but HA antibody could not detect cleaved C-terminal fragments. VSV antibody detection also showed that C2-AHNAK was cleaved from 60 kDa to 36 and 22 kDa fragments and the HA antibody detected complementary fragments of 24 and 38 kDa (D), respectively. These cleavage products were observed only when calpain 3 was transfected, but not in calpain 3 C129S, GFP or AHNAK fusion construct transfected cells. No cleavage was detected for M-AHNAK (C). Boxes represent cleaved fragments detected by VSV antibody and circles represent cleaved fragments detected by HA antibody. Non-marked fragments represent unspecific degradation products of the AHNAK fusion fragments. Below each panel a schematic representation of each AHNAK fusion construct with its calpain 3 cleavage sites is presented.
AHNAK colocalizes with calpain 3 in skeletal muscle fibers
Many studies highlight the diverse localization of AHNAK in a variety of tissues (20,21). We were particularly interested in the localization of AHNAK in skeletal muscle fibers. Due to the direct interaction of calpain 3 and AHNAK and the cleavage of AHNAK by calpain 3, we expected calpain 3 to colocalize with AHNAK. Previous studies have already shown that calpain 3 colocalizes with its substrates at the myotendinous junctions (MTJ) and the A-I junction region of the titin molecule. To investigate whether AHNAK also localizes in these areas, we performed double immunostaining experiments using the KIS antibody and antibody T11, staining the I-band near the A-I junction region of the titin molecule, an antibody for vinculin staining the MTJ, and an anti α-actinin antibody, staining the Z-line, respectively. In parallel, double staining was performed for calpain 3 for comparison. We observed that (i) calpain 3 and AHNAK colocalize at the I-band near the A-I junction (Fig. 6A); (ii) calpain 3 and AHNAK are enriched in most MTJ (Fig. 6B) and (iii) fluorescence signals of α-actinin alternate with doublet bands of calpain 3 and AHNAK (Fig. 6C).
Figure 6.
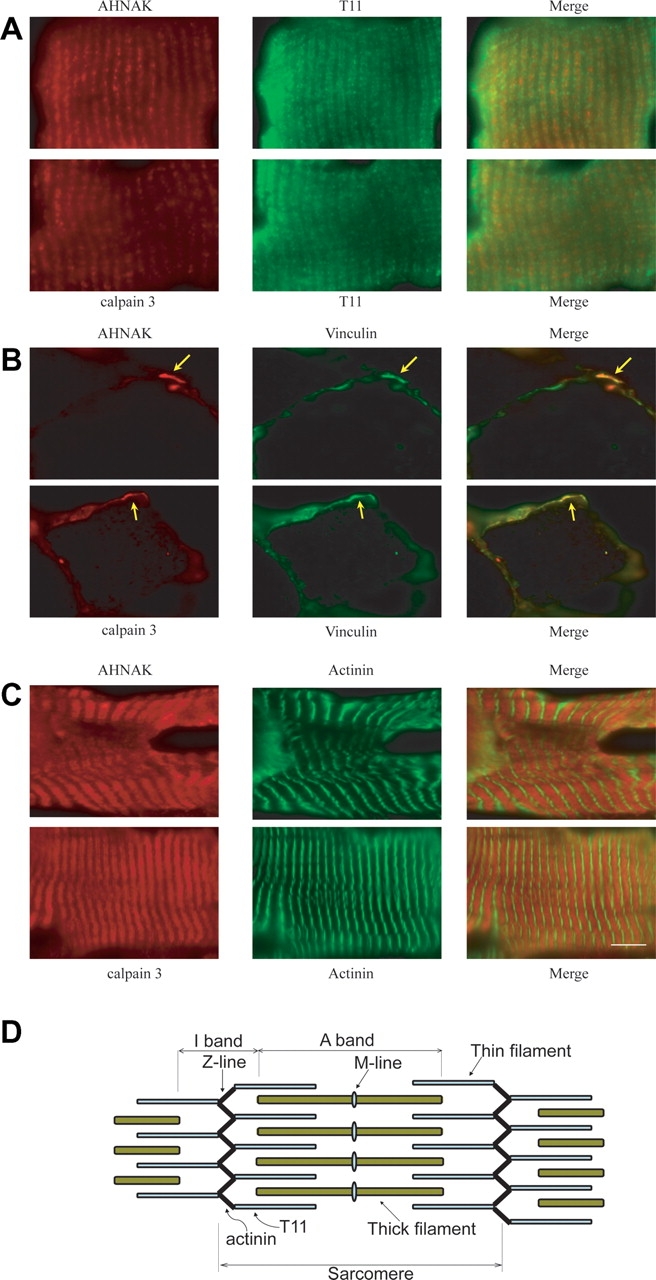
Subcellular localization of calpain 3 and AHNAK in human longitudinal skeletal muscle sections. (A) The anti-titin T11 antibody was used to specifically recognize I band near the A-I junction region of the titin molecule. The immunofluorescence images showed colocalization of calpain 3 and AHNAK with the A-I junction region of the titin. (B) MTJ was detected by the anti-vinculin antibody. Colocalization of calpain 3 and AHNAK at the MTJ is indicated by yellow arrows. (C) The anti-α actinin antibody was used to recognize the Z-lines. The immunofluorescence images showed alternating signals of α actinin and doublet bands of calpain 3 and AHNAK. (D) A scheme representing a muscle sarcomere. Bar represents 10 µm.
AHNAK is upregulated in LGMD2A patients
To investigate the expression of AHNAK in patients with a primary calpainopathy, we performed combined immunofluorescence analysis of AHNAK and dystrophin on four patients and two controls including one non-LGMD2A disease control. All four patients have different mutations, leading to mutant calpain 3 (see Materials and Methods). Dystrophin was used as a membrane staining control. We observed increased reactivity for AHNAK at the sarcolemma and in blood vessels in all LGMD2A patients, compared with the non-related MD (FSHD) and normal control (Fig. 7A). Subsequent western blot analysis on two LGMD2A patients and one control for which muscle protein lysates were available confirmed that the amount of AHNAK is increased in these patients (Fig. 7B). To further substantiate the evidence for AHNAK upregulation in skeletal muscle of calpainopathy patients, we analyzed one additional genetically confirmed calpain 3-deficient patient, and three control muscles [one healthy, one oculopharyngeal MD (OPMD) patient, and one malignant hyperthermia (MH) patient]. The results from Figure 7 were confirmed by immunohistochemistry (Supplementary Material, Fig. S1A) and western blot (Supplementary Material, Fig. S1B).
Figure 7.
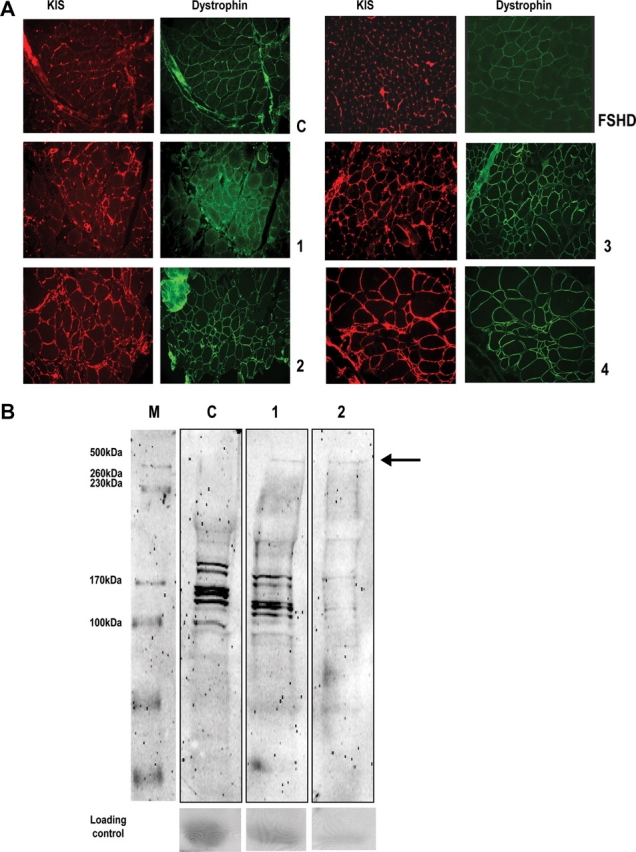
(A) AHNAK is enhanced in skeletal muscle of calpainopathy patients. Single sections of four different calpainopathy patients, one control and one non-LGMD diseased control (FSHD) were blocked for 2 h in 4% skimmed milk (Marvel) in PBS and incubated with an antiDystrophin/KIS mixture. All calpainopathy sections show increased AHNAK when compared with control. Dystrophin signal remains unchanged. (B) Single sections were dissolved in 35 µl sample buffer, loaded on 7% gel and after blotting the membrane was stained with KIS/CQL antibody mix. The arrow points out the presence of endogenous AHNAK. Ponceau S staining was used as a loading control.
Cleavage of AHNAK prevents the binding of AHNAK to dysferlin or myoferlin
Previously, we demonstrated that the C2-domain of both AHNAK proteins can interact with the C2A-domain of dysferlin and myoferlin. In order to investigate whether this interaction is perturbed after AHNAK cleavage by calpain 3, we repeated the pull-down assay in the presence of active calpain 3. After cotransfection of active calpain 3 and C2-AHNAK to COS-1 cells, cells were harvested after 48 h and total lysates were used to study the interaction between (cleaved) C2-AHNAK and the C2A-domains of dysferlin and myoferlin. Antibodies recognizing the N-terminal VSV tag (Fig. 8A) and C-terminal HA tag (Fig. 8B) were used to analyze the pull-down fractions on western blots. As shown, only full-length C2-AHNAK was pulled down by GST-C2A-dysferlin and GST-C2A-myoferlin. Cleaved fragments (22 and 38 kDa) could not be pulled down by either GST-C2A-dysferlin or GST-C2A-myoferlin. No binding was observed for equivalent amount of the control GST protein.
Figure 8.

Calpain 3-cleaved AHNAK fragments lose their binding affinity for dysferlin or myoferlin. GST-C2A-dysferlin and GST-C2A-myoferlin fusion proteins were used in pull-down assays with cell lysates which were co-transfected with active calpain 3 and C2-AHNAK as described in Materials and Methods. Bound proteins were separated by SDS–PAGE and analyzed by immunoblotting with antibodies recognizing the N-terminal VSV tag (A) and C-terminal HA tag (B). (A and B) Lanes 1–5 represent soluble fractions of cell lysates, precleared lysates, GST-C2A-dysferlin, GST-C2A-myoferlin and GST alone pull-down fractions, respectively. Only full-length C2-AHNAK (indicated by arrowheads) was pulled down by GST-C2A-dysferlin (lane 3) and GST-C2A-myoferlin (lane 4). GST did not interact with C2-AHNAK (lane 5). These results demonstrate a specific and direct interaction between full-length C2-AHNAK and C2A-dysferlin and C2A-myoferlin. Cleaved C2-AHNAK fragments (22 and 38 kD) lose their binding affinity for dysferlin and myoferlin. Boxes represent cleaved fragments (22 kD) detected by VSV antibody and circles represent cleaved fragments (38 kD) detected by HA antibody. A molecular mass marker is indicated on the left.
DISCUSSION
Mutations in the muscle-specific, non-lysosomal cysteine protease calpain 3 cause LGMD2A. The function of calpain 3 and its role in LGMD2A pathogenesis are largely unknown but recent evidence points towards an important function in cytoskeleton remodeling and cytoskeleton–membrane interactions (7). Our previous studies demonstrated that calpain 3 and AHNAK are in complex with dysferlin (14). Moreover, calpain 3 showed the ability to cleave several cytoskeletal components and actin binding proteins (5). As AHNAK also associates with actin (19) and is proposed to be cleaved by yet unidentified proteases (20), we hypothesized that AHNAK is a substrate for calpain 3.
The first step to test our hypothesis was to investigate a potential interaction between calpain 3 and AHNAK by GST pull-down assays. Using different recombinant AHNAK domains we showed a clear interaction between the C-terminus of AHNAK and full-length calpain 3.
In previous studies using the C-terminal Tail-antibody (not CQL), western blots of total protein lysates from rat heart and skeletal muscle revealed the presence of protein bands of 700, 500, 340, 170 kDa in addition to low molecular mass immunoreactive fragments (19). Moreover, a band of ∼120 kDa was also detected by the CQL antibody in lung and kidney tissues (20). It was suggested that this band, corresponding to a C-terminal AHNAK proteolytic product, was cleaved by ubiquitous calpains, possibly μ-calpain or m-calpain, but to date the identity of the proteases that cleave AHNAK is unknown. We analyzed COS-1 cells transiently transfected with active and inactive calpain 3 using the CQL antibody and revealed a 120 kDa fragment by western blot only in cells expressing active calpain 3. Thus, these results indicate that calpain 3 can cleave endogenous AHNAK in a cell culture system.
The CQL antiserum that identified the AHNAK cleavage fragment was raised against the COOH-terminus of AHNAK. It is therefore not a suitable tool to investigate cleavage if calpain 3 cleavage of AHNAK would occur at multiple sites. The possible interaction between calpain 3 and several T7-AHNAK fusion protein fragments in our GST pull-down assay seems to suggest that calpain 3 can bind AHNAK also at locations other than the C2-domain. To investigate this, four domains of AHNAK were fused to an N-terminal VSV and C-terminal HA tag and analyzed in COS-1 cells in the presence of active or inactive calpain 3. N-, C1- and C2-AHNAK fusion proteins were clearly cleaved in cells expressing calpain 3. In contrast, specific cleavage products of AHNAK were not observed in cells transfected with a mutated, inactive calpain 3 or GFP. No cleavage was observed for the repeat domain of AHNAK (M-AHNAK).
This apparent discrepancy for N-AHNAK for which we found evidence of cleavage without observing a direct interaction may be explained by the different experimental design. Due to its autocatalytic activity calpain 3 is very unstable and only inactive calpain 3 can be heterologously expressed for interaction assays. Therefore, all GST pull-down assays were performed with inactive calpain 3 C129S. As the conversion from inactive to active calpain 3 involves a major conformational change including excision of insertion sequences (22), it is possible that only active, but not inactive calpain 3 C129S binds the N-terminus of AHNAK. Alternatively, the interaction between the N-terminus of AHNAK and calpain 3 may be more transient compared with the C-terminus, similar to what has been shown for the Annexin 2 binding sequence (23).
The influence of calpain 3 on the expression of endogenous AHNAK was also investigated in cell culture by immunostaining. This showed that AHNAK was absent in cells expressing active calpain 3 while normal or even increased signals were observed in cells expressing inactive calpain 3 C129S. In agreement with this observation, AHNAK immunoreactivity was increased along the sarcolemma of muscle fibers of calpainopathy patients in addition to an intense signal in blood vessels and extracellular matrix compared with normal control muscle and muscle of an unrelated MD patient. Dystrophin showed constant immunoreactivity at sarcolemma in all muscle biopsies. Moreover, semiquantitative western blot analysis confirmed the increase of AHNAK in skeletal muscle of patients with a calpainopathy. Taken together, these findings suggest that in skeletal muscle AHNAK protein turnover is regulated by calpain 3 activity and that this turnover is perturbed in patients with a calpainopathy.
Although the exact biological function of AHNAK is unknown, based on its interaction with other proteins, AHNAK has been implicated in several essential biological functions. First, in resting neuronal PC12–27 cells, AHNAK is localized within the lumen of specific vesicles called ‘enlargeosomes’, and is redistributed to the external surface of the plasma membrane in response to large increases in Ca2+ (24). These enlargeosomes have been proposed to participate in cell membrane enlargement, differentiation and repair. Second, in cardiomyocytes, AHNAK interacts specifically with the β2 subunit of cardiac L-type calcium channels at the plasma membrane (18) where it modulates channel function (25), and is suggested to play a role in the protein kinase A (PKA)-mediated signal transduction pathway (26). Third, in vitro, the C-terminal AHNAK region (amino acids 5262–5643) binds to G-actin and cosediments with F-actin (18). Finally, a C-terminal 72 kDa AHNAK fragment was found in heart tissue to stabilize muscle contraction (19). These observations suggest a role for AHNAK in the maintenance of the structural and functional organization of the subsarcolemmal cytoarchitecture. The finding that calpain 3 regulates AHNAK, combined with the observation that both proteins are found in the dysferlin protein complex raises the interesting possibility that they may function in muscle membrane repair. For m and µ calpain it has been shown that they are essential for Ca2+ induced cell membrane repair (27). Moreover, stabilization of m calpain by fetuin A (OMIM# 38680) increases the membrane resealing potential of fibroblasts (28). Fetuin A can also bind calpain 3 (28). It is therefore an interesting possibility that calpain 3 may exert the same function in muscle by regulating AHNAK levels. Based on the proteolysis of AHNAK by calpain 3, we suggest that in healthy muscle calpain 3 works as a regulatory enzyme modulating AHNAK function. Such a regulatory role would be similar to the well-documented calpain 3-dependent accumulation of IκBα in the cytoplasm and nucleus (29) and the calpain 3-mediated interaction between FLNC and γ- and δ-sarcoglycan (6).
The loss of calpain 3 activity as the cause of LGMD2A indicates that calpain 3-dependent proteolysis is required for the normal biological function and homeostasis of skeletal muscle (29). Recently, increased ubiquitination, correlated with an increase of calpain 3 expression, was observed during muscle reloading in wild-type mice, which was absent in Capn3 −/− mice (30). It was suggested that calpain 3 acts upstream of the ubiquitination–proteasome pathway to release myofibrillar proteins and provide them for proteasomal breakdown. In the present study, we provide evidence that calpain 3 activity promotes turnover of AHNAK in cell culture and in skeletal muscle. Thus, in the future, it is essential to establish whether AHNAK is ubiquitinated after calpain 3 cleavage in skeletal muscle.
To date, three major pathological mechanisms have been suggested for MD. Membrane disruption, caused by mutations in proteins of the dystrophin–glycoprotein complex, is a well established pathogenic mechanism in a large group of MD (31). Besides this membrane fragility, defects in membrane repair caused by mutations in dysferlin have been identified as second pathological mechanism (11). Recently, a new pathogenic mechanism, deregulation of sarcomere remodeling due to the mutations in calpain 3, was suggested as the cause of LGMD2A (7). Thus, the regulatory role of calpain 3 in the dysferlin protein complex may implicate an intimate relationship between muscle membrane repair and remodeling of sarcomere and subsarcolemmal cytoskeleton architecture, which thus far have been considered independent pathological mechanisms for LGMDs.
MATERIALS AND METHODS
Antibodies
The following antibodies were used in this study. The monoclonal anti-dystrophin antibody NCL-DYS2 (Novocastra, Newcastle, UK) was used in a dilution of 1:10 for immunofluorescence microscopy. The mouse monoclonal anti-calpain 3 antibody NCL-12A2 (Novocastra) was used in a dilution of 1:10 for western blot. Affinity-purified mouse anti-VSV (P5D4) (gift from Dr J. Fransen, Nijmegen, the Netherlands) was used in a dilution of 1:1000 for western blot analysis and in a dilution of 1:250 for immunofluorescent microscopy. The monoclonal anti-HA tag antibody (Upstate Cell signaling solution, VA, USA) was used in a dilution of 1:2000 for western blot analysis. The rabbit anti-calpain 3 antibody RP1Calpain3 (Triple Point Biologics, OR, USA), mouse monoclonal anti-alpha actinin antibody (clone EA-53) (Sigma, MO, USA) and mouse anti-titin T11 antibody (Sigma) were used in dilutions of 1:200, 1:2500 and 1:100 for immunofluorescence microscopy, respectively. Secondary antibodies goat anti-mousealexa488 (Molecular Probes, Eugene, OR, USA), goat anti-rabbitalexa594 (Molecular Probes) and rabbit anti-mouseHRP (DakoCytomation, Glostrup, Denmark) were diluted 1:250, 1:1000 and 1:1000, respectively. Goat anti-RabbitIRDye800 (Westburg) was used at 1:5000. KIS and CQL anti-AHNAK polyclonal antibodies were obtained from Dr Jacques Baudier (Grenoble, France) and have been described previously (20). Mouse anti-T7HRP (Novagen) was diluted 1:10 000 for western blot analysis.
Patient mutation analysis
The unrelated MD case was a patient with genetically confirmed FSHD as indicated by the presence of two D4Z4 repeat units on chromosome 4. LGMD2A patient 1 carries a X02:c.327_328delCC deletion and a X11:c.1477C>T (p.Arg493Trp) substitution. LGMD2A patient 2 carries a X05:c.759_761delGAA deletion and a X11:c.1468C>T (p.Arg490Trp) substitution. LGMD2A patient 3 carries a X05:c.633G>C (p.Lys211Asn) and a X10:c.1319G>A (p.Arg440Gln) substitution. LGMD2A patient 4 carries a X01:c.145C>T (p.Arg49Cys) and a X11:c.1468C>T (p.Arg490Trp) substitution. The MH, OPMD and calpainopathy patients used in the supplemental figure were genetically confirmed.
Fusion protein constructs and expression in prokaryotic vector
pcDNAI eukaryotic expression vectors containing N-AHNAK (N-AHNAK) (residues 249–498), M-AHNAK (residues 1068–1577), C-AHNAK (residues 4893–5890) were a kind gift from Dr Takashi Hashimoto (Keio University School of Medicine, Tokyo, Japan). DNA fragments encoding the protein fragments N-AHNAK, M-AHNAK and C-AHNAK, respectively, were obtained by BamHI/XhoI restriction from pcDNAI and ligated in the prokaryotic expression vector pET28a-GST (modified from pET28a (Novagen, Madison, WI) with an additional GST tag). Subsequently, pET28a-GST-C-AHNAK was split in two parts by BamHI/SacI digestion (designated C1-AHNAK, from residue 4893–5392) and by SacI/XhoI digestion (designated C2-AHNAK, from residue 5393–5890) and ligated in BamHI/SacI-digested or SacI/XhoI-digested pET28c-GST, respectively. Unfused GST protein was used as control.
To generate full length calpain 3 (amino acids 1–821), digestion by SalI/NotI was performed from the plasmid containing calpain 3 in pBLUESCRIPT SK+ and ligated in the SalI/NotI-digested prokaryotic expression vector pET28c. Site-directed mutagenesis was performed by use of the QuickChange site-directed mutagenesis kit (Stratagene, La Jolla, USA). Calpain 3 C129S was obtained by replacing the cysteine at position 129 with serine. All sequences were confirmed by automated sequencing (LGTC, Leiden, the Netherlands).
The expression plasmid in pGEX 4T-1 vector (encoding the first 124 amino acids of human dysferlin) and the expression plasmid in pGEX 4T-1 vector (encoding the first 125 amino acids of human myoferlin) were a kind gift of Dr E.M. McNally (Department of Human Genetics, University of Chicago, USA).
The prokaryotic expression constructs were used for recombinant protein production in BL21 (DE3)-RIL E. coli (Stratagene) cells according to the manufacturer's instructions. Briefly, bacterial cells were grown to log phase and recombinant protein production was initiated by the addition of IPTG (Fermentas, St Leon-Rot, Germany) to a final concentration of 1 mm. After 2.5 h induction at 30°C, the cells were collected by centrifugation at 3000g, lysed in lysis buffer 1 [50 mm Tris, pH 7.4, 1 mm EDTA, 1.5 mg/ml lysozyme, 0.15 M NaCl, 1% Triton X-100, plus 1× protease inhibitor cocktail (Roche Molecular Biochemicals, Basel, Switzerland)] and sonicated on ice. For the pull-down assay, after centrifugation at 13 000g for 30 min at 4°C, the supernatant containing soluble GST fusion proteins was recovered. Fusion proteins were immobilized to Gluthione–Sepharose 4B (Amersham, Uppsala, Sweden), incubated at room temperature for 30 min and then centrifuged at 500g for 5 min. The supernatant was removed and the Glutathione–Sepharose was washed three times with binding buffer (50 mm Tris, pH 7.4, 150 mm NaCl, 0.2% Triton X-100). The GST/GST-fusion protein affinity beads were ready for pull-down assay.
Fusion protein constructs and expression in eukaryotic vector
DNA fragments encoding the protein fragments N-AHNAK, M-AHNAK, C1-AHNAK and C2-AHNAK were amplified from cDNA clones (Keio University School of Medicine) with forward primer (5'-CGCGGATCCGCGCCTGGGATAAAG-3') and reverse primer (5'-CCGCTCGAGCGGAGGTTTCTGAATAATCA-3'), forward primer (5'-CGCGGATCCGCGTCTTTGCCAGATGTT-3') and reverse primer (5'-CCGCTCGAGCGGGTGCGTCTGTATATTCA-3'), forward primer (5'-CGCGGATCCGCGCGTCTGGATTTC-3') and reverse primer (5'-CCGCTCGAGCGGTTTGGGAAGTTTAAT-3'), forward primer (5'-CGCGGATCCGCGGCTCCTGATCTAA-3') and reverse primer (5'-CCGCTCGAGCGGCTCTTTCTTTGTGGAA-3'), respectively.
A pSG8-VSV eukaryotic expression vector was modified by insertion of a Heamaglutinin (HA) tag at XhoI/BglII sites using forward primer (5'-TCGAGTATCCATATGATGTTCCAGATTATGCTTGAA-3') and reverse primer (5'-GATCTTCAAGCATAATCTGGAACATCATATGGATAC-3'). All PCR products were digested with BamHI/XhoI and ligated in BamHI/XhoI -digested pSG8-VSV-HA vector.
The full length calpain 3 cDNA was amplified from the plasmid containing calpain 3 in pBLUESCRIPT KS+ with forward primer (5'-CCGCTCGAGCGGTGCCATGCCGAC-3') and reverse primer (5'-CGCGGATCCGCGATTCGGCATACATGGT-3'). Subsequently, the fragment was digested with BamHI/XhoI and ligated in BamHI/XhoI -digested eukaryotic expression vector pEGFP N2 (Clontech, CA, USA). Calpain 3 C129S was obtained by replacing the cysteine at position 129 with serine by use of the QuickChange site-directed mutagenesis kit (Stratagene).
Pull-down assays
To determine the association of calpain 3 and AHNAK, GST-AHNAK proteins were used to pull down T7-tagged fusion proteins of calpain 3 C129S produced in E. coli. To determine the association of dysferlin and myoferlin and AHNAK after the calpain 3 cleavage, respectively, GST-C2A-dysferlin and GST-C2A-myoferlin were used to pull down COS-1 cell lysates which were co-transfected with active calpain 3 and C2-AHNAK.
Lysates were centrifuged for 15 min at 4°C and the supernatants were pre-cleared with Gluthione–Sepharose 4B (Amersham) for 1 h at 4°C. Then, 500 µl aliquots of pre-cleared supernatant were incubated with 10 µg of purified GST fusion protein affinity beads in binding buffer supplemented 1% BSA for o/n at 4°C or with unfused GST protein as control. After binding, the beads were washed three times in binding buffer without BSA and one time in 50 mm Tris–HCl pH 7.4, then eluted by boiling in 50 µl of 2×SDS–PAGE sample buffer for 5 min. Ten microliters was loaded on 12% SDS–PAGE gels to detect proteolytic fragments of calpain 3 C129S. After separation, proteins were transferred to PVDF membranes and dried o/n. Then, blots were incubated with anti T7HRP (1: 10 000) for 1.5 h at RT for detection of recombinant calpain 3 C129S protein. ECL plus (Amersham) was used for visualization.
Cell culture and transfection
COS-1 monkey kidney cells and 3T3 fibroblast cells were maintained in Dulbecco's modified Eagle's medium (Gibco-BRL, Grand Island, NY) supplemented with 10% fetal bovine serum (Gibco-BRL), 1% Pen/Strep (100IU/100UG/ML, Gibco-BRL). For plasmid transfections, cells were harvested, plated at 50% subconfluence (200 000 cells per well in six-well microtiter plates), and allowed to grow for 24 h. One microgram of each construct and 6 µl of FuGENE 6 transfection reagent (Roche Applied Science) were used to transfect cells. Two days after transfection, cells were lysed in buffer 2 (50 mm Tris, pH 7.4, 0.15 M NaCl, 0.2% Triton X-100, plus 1× protease inhibitor cocktail (Roche Molecular) for immunoblotting and GST pull-down assay. For immunoblotting, protein concentrations were determined using a BCA protein assay kit (Pierce Science, the Netherlands) according to manufacturer's instructions and equal amounts of lysates were loaded per well of the SDS–PAGE gel. After separation, proteins were transferred to PVDF membranes and dried o/n. Then, blots were incubated with mouse anti-VSV antibody (1:1000), mouse anti HA antibody (1:2000) and rabbit anti-actin antibody (1:200) for 1.5 h at RT. Rabbit anti mouseHRP and swine anti rabbitHRP were used for detection of cleaved AHNAK fusion protein fragments and actin, respectively. ECL plus (Amersham) was used for visualization.
Immunostaining
For immunohistochemical examinations, transverse muscle cryosections of 5 µm thickness were fixed in 3.7% formaldehyde in phosphate-buffer saline (PBS) containing 0.1% triton X for 30 min, following by pre-incubation with 4% skimmed milk (Marvel) in PBS at room temperature for 2 h. The sections were next incubated with primary antibody fragments o/n at 4°C, and subsequently by incubation of fluorescein-labeled secondary antibody for 40 min at RT. Background staining was performed by omitting the primary antibody from the first step. The sections were washed with PBS, dehydrated with 70, 90, 100% ethanol and mounted in a DAPI (50 ng/µl)/ Vectashield™ mounting medium (Burlingame, CA). Final preparations were analyzed with a Leica Aristoplan fluorescence microscope and images were obtained using Cytovision (Applied imaging) digital system.
For immunocytochemical examinations, 2 days after transfection, cells were fixed in 3.7% formaldehyde containing 0.1% triton X for 10 min and then blocked in 1% BSA for 30 min. Primary polyclonal rabbit KIS antibody for detection of endogenous AHNAK was incubated for 2 h at a dilution of 1:100 and secondary goat anti-rabbitalexa594 antibody (Molecular probes) was incubated for 1 h at a dilution of 1: 1000.
Oddyssey western blot analysis
For both transient transfections and single sections total cell lysates were prepared and run on a 7% gel and blotted with a Criterion plate electrode blotting device o/n at 30 V on nitrocellulose membrane. Membrane was stained with Ponceau S and washed several times with water. Next the membrane was blocked with 4% skimmed milk (Marvel) in PBS for 2 h. The membrane was then incubated with a pool of KIS and CQL in blocking buffer for 2 h. After three 5 min wash steps the membrane was probed with antiRabbitIRDye800 (Westburg, Leusden, The Netherlands) at 1:5000 for 1 h in blocking buffer. Next the membrane was washed as before and dried o/n. Next day an odyssey scanner (Licor, Lincoln, Nebraska, USA) was used for visualization.
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by grants from SenterNovem (IOP-Genomics IGE01019), the National Institutes of Health (NIH-NIAMS R21-AR48327-01) and the Prinses Beatrix Fonds.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to Dr Takashi Hashimoto, Keio University School of Medicine, for providing us with cDNA clones of human AHNAK. We also thank Prof. E.M. McNally, University of Chicago for providing us with GST-C2A-dysferlin and GST-C2A-myoferlin cDNA, and Dr I. Ginjaar, Leiden University Medical Center and Dr R. Charlton, Freeman Hospital, Newcastle-Upon-Tyne for providing muscle cryosections. We thank Dr J. Baudier for providing us rabbit polyclonal anti-AHNAK KIS and CQL antibodies.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Dincer P., Leturcq F., Richard I., Piccolo F., Yalnizoglu D., deToma C., Akcoren Z., Broux O., Deburgrave N., Brenguier L., et al. A biochemical, genetic, and clinical survey of autosomal recessive limb girdle muscular dystrophies in Turkey. Ann. Neurol. 1997;42:222–229. doi: 10.1002/ana.410420214. [DOI] [PubMed] [Google Scholar]
- 2.Piluso G., Politano L., Aurino S., Fanin M., Ricci E., Ventriglia V.M., Belsito A., Totaro A., Saccone V., Topaloglu H., et al. Extensive scanning of the calpain-3 gene broadens the spectrum of LGMD2A phenotypes. J. Med. Genet. 2005;42:686–693. doi: 10.1136/jmg.2004.028738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Richard I., Broux O., Allamand V., Fougerousse F., Chiannilkulchai N., Bourg N., Brenguier L., Devaud C., Pasturaud P., Roudaut C., et al. Mutations in the proteolytic-enzyme calpain-3 cause limb-girdle muscular-dystrophy type-2A. Cell. 1995;81:27–40. doi: 10.1016/0092-8674(95)90368-2. [DOI] [PubMed] [Google Scholar]
- 4.Goll D.E., Thompson V.F., Li H., Wei W., Cong J. The calpain system. Physiol. Rev. 2003;83:731–801. doi: 10.1152/physrev.00029.2002. [DOI] [PubMed] [Google Scholar]
- 5.Taveau M., Bourg N., Sillon G., Roudaut C., Bartoli M., Richard I. Calpain 3 is activated through autolysis within the active site and lyses sarcomeric and sarcolemmal components. Mol. Cell Biol. 2003;23:9127–9135. doi: 10.1128/MCB.23.24.9127-9135.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guyon J.R., Kudryashova E., Potts A., Dalkilic I., Brosius M.A., Thompson T.G., Beckmann J.S., Kunkel L.M., Spencer M.J. Calpain 3 cleaves filamin C and regulates its ability to interact with gamma- and delta-sarcoglycans. Muscle Nerve. 2003;28:472–483. doi: 10.1002/mus.10465. [DOI] [PubMed] [Google Scholar]
- 7.Duguez S., Bartoli M., Richard I. Calpain 3: a key regulator of the sarcomere? FEBS J. 2006;273:3427–3436. doi: 10.1111/j.1742-4658.2006.05351.x. [DOI] [PubMed] [Google Scholar]
- 8.Bashir R., Britton S., Strachan T., Keers S., Vafiadaki E., Lako M., Richard I., Marchand S., Bourg N., Argov Z., et al. A gene related to Caenorhabditis elegans spermatogenesis factor fer-1 is mutated in limb-girdle muscular dystrophy type 2B. Nat. Genet. 1998;20:37–42. doi: 10.1038/1689. [DOI] [PubMed] [Google Scholar]
- 9.Illa I., Serrano-Munuera C., Gallardo E., Lasa A., Rojas-Garcia R., Palmer J., Gallano P., Baiget M., Matsuda C., Brown R.H. Distal anterior compartment myopathy: a dysferlin mutation causing a new muscular dystrophy phenotype. Ann. Neurol. 2001;49:130–134. [PubMed] [Google Scholar]
- 10.Liu J., Aoki M., Illa I., Wu C., Fardeau M., Angelini C., Serrano C., Urtizberea J.A., Hentati F., Hamida M.B., et al. Dysferlin, a novel skeletal muscle gene, is mutated in Miyoshi myopathy and limb girdle muscular dystrophy. Nat. Genet. 1998;20:31–36. doi: 10.1038/1682. [DOI] [PubMed] [Google Scholar]
- 11.Bansal D., Miyake K., Vogel S.S., Groh S., Chen C.C., Williamson R., Mcneil P.L., Campbell K.P. Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature. 2003;423:168–172. doi: 10.1038/nature01573. [DOI] [PubMed] [Google Scholar]
- 12.Davis D.B., Delmonte A.J., Ly C.T., Mcnally E.M. Myoferlin, a candidate gene and potential modifier of muscular dystrophy. Hum. Mol. Genet. 2000;9:217–226. doi: 10.1093/hmg/9.2.217. [DOI] [PubMed] [Google Scholar]
- 13.Huang Y.C., Verheesen P., Roussis A., Frankhuizen W., Ginjaar I., Haldane F., Laval S., Anderson L.V.B., Verrips T., Frants R.R., et al. Protein studies in dysferlinopathy patients using llama-derived antibody fragments selected by phage display. Eur. J. Hum. Genet. 2005;13:721–730. doi: 10.1038/sj.ejhg.5201414. [DOI] [PubMed] [Google Scholar]
- 14.Huang Y.C., Laval S.H., van Remoortere A., Baudier J., Benaud C., Anderson L.V.B., Straub V., Deelder A., Frants R.R., den Dunnen J.T., et al. AHNAR, a novel component of the dysferlin protein complex, redistributes to the cytoplasm with dysferlin during skeletal muscle regeneration. FASEB J. 2007;21:732–742. doi: 10.1096/fj.06-6628com. [DOI] [PubMed] [Google Scholar]
- 15.Kudoh J., Wang Y., Minoshima S., Hashimoto T., Amagai M., Nishikawa T., Shtivelman E., Bishop J.M., Shimizu N. Localization of the human AHNAK/desmoyokin gene (AHNAK) to chromosome band 11q12 by somatic cell hybrid analysis and fluorescence in situ hybridization. Cytogenet. Cell Genet. 1995;70:218–220. doi: 10.1159/000134037. [DOI] [PubMed] [Google Scholar]
- 16.Shtivelman E., Cohen F.E., Bishop J.M. A human gene (AHNAK) encoding an unusually large protein with a 1.2-microns polyionic rod structure. Proc. Natl Acad. Sci. USA. 1992;89:5472–5476. doi: 10.1073/pnas.89.12.5472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Komuro A., Masuda Y., Kobayashi K., Babbitt R., Gunel M., Flavell R.A., Marchesi V.T. The AHNAKs are a class of giant propeller-like proteins that associate with calcium channel proteins of cardiomyocytes and other cells. Proc. Natl Acad. Sci. USA. 2004;101:4053–4058. doi: 10.1073/pnas.0308619101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hohaus A., Person V., Behlke J., Schaper J., Morano I., Haase H. The carboxyl-terminal region of ahnak provides a link between cardiac L-type Ca2+ channels and the actin-based cytoskeleton. FASEB J. 2002;16:1205–1216. doi: 10.1096/fj.01-0855com. [DOI] [PubMed] [Google Scholar]
- 19.Haase H., Pagel I., Khalina Y., Zacharzowsky U., Person V., Lutsch G., Petzhold D., Kott M., Schaper J., Morano I. The carboxyl-terminal ahnak domain induces actin bundling and stabilizes muscle contraction. FASEB J. 2004;18:839–841. doi: 10.1096/fj.03-0446fje. [DOI] [PubMed] [Google Scholar]
- 20.Gentil B.J., Delphin C., Benaud C., Baudier J. Expression of the giant protein AHNAK (desmoyokin) in muscle and lining epithelial cells. J. Histochem. Cytochem. 2003;51:339–348. doi: 10.1177/002215540305100309. [DOI] [PubMed] [Google Scholar]
- 21.Gentil B.J., Benaud C., Delphin C., Remy C., Berezowski V., Cecchelli R., Feraud O., Vittet D., Baudier J. Specific AHNAK expression in brain endothelial cells with barrier properties. J. Cell Physiol. 2005;203:362–371. doi: 10.1002/jcp.20232. [DOI] [PubMed] [Google Scholar]
- 22.Diaz B.G., Moldoveanu T., Kuiper M.J., Campbell R.L., Davies P.L. Insertion sequence 1 of muscle-specific calpain, p94, acts as an internal propeptide. J. Biol. Chem. 2004;279:27656–27666. doi: 10.1074/jbc.M313290200. [DOI] [PubMed] [Google Scholar]
- 23.De Seranno S., Benaud C., Assard N., Khediri S., Gerke V., Baudier J., Delphin C. Identification of an AHNAK binding motif specific for the Annexin2/S100A10 tetramer. J. Biol. Chem. 2006;281:35030–35038. doi: 10.1074/jbc.M606545200. [DOI] [PubMed] [Google Scholar]
- 24.Borgonovo B., Cocucci E., Racchetti G., Podini P., Bachi A., Meldolesi J. Regulated exocytosis: a novel, widely expressed system. Nat. Cell Biol. 2002;4:955–962. doi: 10.1038/ncb888. [DOI] [PubMed] [Google Scholar]
- 25.Haase H., Alvarez J., Petzhold D., Doller A., Behlke J., Erdmann J., Hetzer R., Regitz-Zagrosek V., Vassort G., Morano I. Ahnak is critical for cardiac Ca(V)1.2 calcium channel function and its beta-adrenergic regulation. FASEB J. 2005;19:1969–1977. doi: 10.1096/fj.05-3997com. [DOI] [PubMed] [Google Scholar]
- 26.Haase H., Podzuweit T., Lutsch G., Hohaus A., Kostka S., Lindschau C., Kott M., Kraft R., Morano I. Signaling from beta-adrenoceptor to L-type calcium channel: identification of a novel cardiac protein kinase A target possessing similarities to AHNAK. FASEB J. 1999;13:2161–2172. doi: 10.1096/fasebj.13.15.2161. [DOI] [PubMed] [Google Scholar]
- 27.Mellgren R.L., Zhang W., Miyake K., Mcneil P.L. Calpain is required for the rapid, calcium-dependent repair of wounded plasma membrane. J. Biol. Chem. 2007;282:2567–2575. doi: 10.1074/jbc.M604560200. [DOI] [PubMed] [Google Scholar]
- 28.Mellgren R.L., Huang X. Fetuin A stabilizes m-calpain and facilitates plasma membrane repair. J. Biol. Chem. 2007;282:35868–35877. doi: 10.1074/jbc.M706929200. [DOI] [PubMed] [Google Scholar]
- 29.Richard I., Roudaut C., Marchand S., Baghdiguian S., Herasse M., Stockholm D., Ono Y., Suel L., Bourg N., Sorimachi H., et al. Loss of calpain 3 proteolytic activity leads to muscular dystrophy and to apoptosis-associated I kappa B alpha/nuclear factor kappa B pathway perturbation in mice. J. Cell Biol. 2000;151:1583–1590. doi: 10.1083/jcb.151.7.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kramerova I., Kudryashova E., Venkatraman G., Spencer M.J. Calpain 3 participates in sarcomere remodeling by acting upstream of the ubiquitin-proteasome pathway. Hum. Mol. Genet. 2005;14:2125–2134. doi: 10.1093/hmg/ddi217. [DOI] [PubMed] [Google Scholar]
- 31.Lapidos K.A., Kakkar R., Mcnally E.M. The dystrophin glycoprotein complex: signaling strength and integrity for the sarcolemma. Circ. Res. 2004;94:1023–1031. doi: 10.1161/01.RES.0000126574.61061.25. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.