

Abstract
Bardet-Biedl syndrome (BBS) is a pleiotropic, genetically heterogeneous disorder characterized by obesity, retinopathy, polydactyly, cognitive impairment, renal and cardiac anomalies, as well as hypertension and diabetes. Multiple genes are known to independently cause BBS. These genes do not appear to code for the same functional category of proteins; yet, mutation of each results in a similar phenotype. Gene knockdown of different BBS genes in zebrafish shows strikingly overlapping phenotypes including defective melanosome transport and disruption of the ciliated Kupffer's vesicle. Here, we demonstrate that individual knockdown of bbs1 and bbs3 results in the same prototypical phenotypes as reported previously for other BBS genes. We utilize the zebrafish system to comprehensively determine whether simultaneous pair-wise knockdown of BBS genes reveals genetic interactions between BBS genes. Using this approach, we demonstrate eight genetic interactions between a subset of BBS genes. The synergistic relationships between distinct combinations are not due to functional redundancy but indicate specific interactions within a multi-subunit BBS complex. In addition, we utilize the zebrafish model system to investigate limb development. Human polydactyly is a cardinal feature of BBS not reproduced in BBS-mouse models. We evaluated zebrafish fin bud patterning and observed altered Sonic hedgehog (shh) expression and subsequent changes to fin skeletal elements. The SHH fin bud phenotype was also used to confirm specific genetic interactions between BBS genes. This study reveals an in vivo requirement for BBS function in limb bud patterning. Our results provide important new insights into the mechanism and biological significance of BBS.
INTRODUCTION
Bardet-Biedl syndrome (BBS, OMIM 209900) is a genetically heterogeneous autosomal recessive disorder characterized by obesity, retinal degeneration, post-axial polydactyly, cognitive impairment, hypogenitalism, renal and cardiovascular anomalies and a high incidence of diabetes mellitus and hypertension (1–3). To date, 12 BBS genes have been identified (4–15). Although the precise function of the individual BBS proteins is yet to be elucidated, data support roles in cilia function and intracellular transport.
In order to gain insights into the pathophysiology of BBS, we developed BBS zebrafish models and demonstrated that knockdown of any of the seven zebrafish BBS orthologs (bbs2, 4, 5, 6, 7, 8 and 11) results in similar phenotypes. These phenotypes include disruption of a ciliated vesicle called Kupffer's vesicle (KV), predisposition to organ laterality defects and delayed intracellular retrograde transport (16). Mutation of BBS1 in humans leads to the most common form of BBS. The function of BBS1 is unknown. This protein contains a beta-propeller domain, indicating that it interacts with other proteins. We have recently shown that BBS1, in conjunction with BBS2, BBS4, BBS5, BBS7, BBS8 and BBS9, is part of a stable multi-protein complex known as the BBSome (17). BBS3, an ADP-ribosylation factor (ARF)-like protein known as ARL6 (18), was identified by positional cloning and the use of computational genomics (10). ARL proteins are proposed to have a role in intracellular vesicle and membrane trafficking and in microtubule assembly (18,19). Using zebrafish, we test whether bbs1 and bbs3 loss-of-function recapitulates prototypical BBS knockdown phenotypes including KV defect and intracellular transport delay. We also evaluate pair-wise genetic interaction between eight zebrafish BBS genes (bbs1–bbs8). The zebrafish model system allows for simultaneous pair-wise knockdown of multiple combinations of BBS genes, including BBS genes that code for proteins that are part of the BBSome complex, as well as proteins that are not part of the complex. Pair-wise combinations of anti-sense oligonucleotides demonstrate eight significant synergistic interactions between specific BBS genes. Of note is a central role for bbs1 in five of these genetic interactions. The in vivo genetic interactions identified in this study reveals that BBS proteins that are part of the complex can result in a synergistic genetic interaction, as can proteins that are not part of the BBSome complex.
Human polydactyly is one of the cardinal features of BBS not reproduced in BBS knockout mouse models (20–23). Given that several zebrafish BBS genes are expressed in the developing fin bud (16), we evaluated fin bud patterning in BBS morphants. We focused on the zone of polarizing activity (ZPA) owing to its critical and evolutionarily conserved role in the patterning of the tetrapod limb/fin bud. We demonstrate that individual knockdown of BBS genes in zebrafish, as well as pair-wise knockdown of BBS genes, results in the expansion of Sonic hedgehog (shh) expression, a molecular component of the ZPA, as well as changes to subsequent skeletal elements.
RESULTS
Zebrafish bbs1 and bbs3 knockdowns display BBS prototypical phenotypes
Previously, we demonstrated that individual knockdown of bbs2, bbs4–bbs8 and bbs11 (9,16) generates prototypical phenotypes in zebrafish. In order to determine whether knockdowns of bbs1 and bbs3 result in similar phenotypes, we used antisense morpholinos to knock down these genes. Antisense morpholino oligonucleitides (MOs) were designed to target bbs1 located on chromosome 21 and bbs3 located on chromosome 1 in the zebrafish genome (Wellcome Trust Sanger Institute, http://www.ensembl.org). Fluorescein-tagged MOs targeting the initiator methionine (aug-MO) or the splice site of exon2 (exon2-MO) were microinjected into embryos. The MOs were uniformly distributed at 12–14 h post-fertilization (h.p.f.) and morphant embryos did not show any gross changes in general body size and morphology compared with controls. Bbs1 and bbs3 morphants were next evaluated for prototypical alterations including reduction or loss of a ciliated vesicle, known as Kupffer's vesicle (KV), predisposition to laterality defects and delay in melanosome retrograde transport (9,16). The KV progenitors originate in the dorsal midline and form a ciliated vesicle in the tail-bud region during somite stages (24–30). In wild-type and control MO-injected embryos at the 10–13 somite stage, the sphere-shaped KV has an average size of 50 µm (Fig. 1A and B). KV defects (reduced size to less than one-third of normal or total KV absence) were observed in a dose-dependent manner in bbs1 MO and bbs3 MO-injected embryos (Fig. 1; Table 1). Consistent with the proposed function of the KV in left–right axis asymmetry (24–30), zebrafish knockdown of bbs3 showed organ laterality defects. Bbs3 MO-injected embryos show significant jogging alterations at 22–24 h.p.f. (reversed or no jog) and looping defects at 36 h.p.f. (31), whereas low-dose bbs3 MO or control MO-injected embryos do not show significant KV or laterality alterations (Fig. 1E; Table 1).
Figure 1.
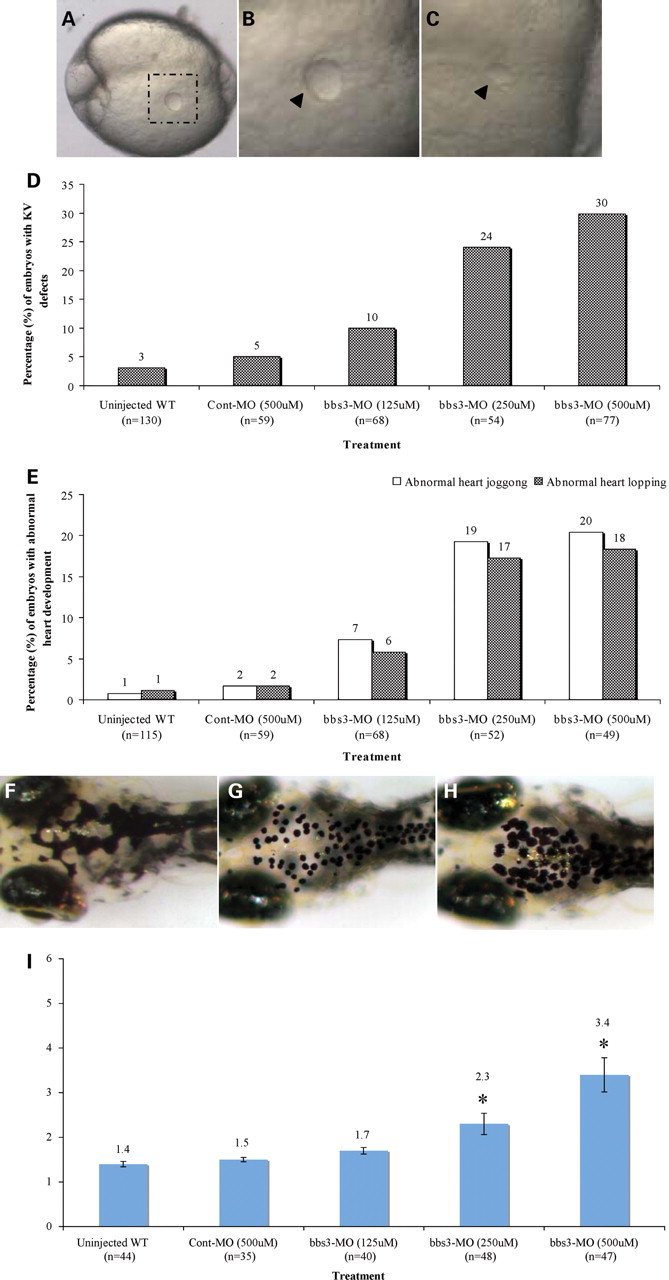
bbs3 knockdown phenotypes in zebrafish. (A–C) Photographs of live zebrafish embryos at the 10–13 somite stage. (A) KV (dashed box) is a ciliated vesicle located in the tail bud region in a wild-type embryo. (B) Higher magnification of control KV (arrowhead). (C) bbs3 MO-injected embryo with reduced KV (arrowhead). Magnification: (A) 5×; (B and C) 10×. (D) The percentage of zebrafish KV defects (reduced or absent) generated in MO and control sets. (E) Percentage of heart laterality defects observed in bbs3 morphants and controls. (F–H) Epinephrine-induced melanosome retrograde transport, dorsal anterior view of 5-day-old larvae. (F) Wild-type larvae prior to epinephrine treatment and (G) at the endpoint of 1.5 min after epinephrine treatment. (H) bbs3 morphant larvae showing a delayed response after 3.0 min of epinephrine treatment. (I) Graphical representation of epinephrine-induced melanosome retrograde transport times demonstrating a dose-dependent delay in bbs3 knockdown when compared with wild-type and control-injected embryos. Treatment and sample size noted on the x-axis and percentage noted at the top of the bar. *P < 0.001.
Table 1.
bbs1 and bbs3 knockdown phenotypes include disruption of KV formation, heart laterality defects (jogging and looping) and a significant delay in melanosome retrograde transport
| MO treatment | Percentage of KV defects | Percentage of heart jogging defects | Percentage of heart looping defects | Melanosome retrograde transport (min) |
|---|---|---|---|---|
| Wild-type | 3 (130) | 1 (115) | 1 (115) | 1.4 (44) |
| Control MO (500 μm) | 5 (59) | 2 (59) | 2 (59) | 1.5 (35) |
| bbs1 MO (125 μm) | 9 (44) | ND | ND | 1.5 (31) |
| bbs1 MO (250 μm) | 32 (78) | 22 (27) | ND | 3.5* (32) |
| bbs3 MO (125 μm) | 10 (68) | 7 (68) | 6 (68) | 1.7 (40) |
| bbs3 MO (250 μm) | 24 (54) | 19 (52) | 17 (52) | 2.3* (48) |
| bbs3 MO (500 μm) | 30 (77) | 20 (49) | 18 (49) | 3.4* (47) |
Number of embryos in each category is given in parentheses; ND, not determined.
*P < 0.001.
We previously demonstrated involvement of BBS function in general intracellular transport. Knockdown of zebrafish BBS genes (bbs2, bbs4–bbs8 and bbs11) resulted in a statistically significant delay in retrograde transport (9,16). Zebrafish shuttle melanosomes within melanophores in response to visual cues and hormonal stimuli (32–35). In dark-adapted zebrafish, the melanosomes are dispersed (Fig. 1F). Melanosome aggregation (retrograde transport) is stimulated upon treatment with epinephrine (Fig. 1G) (16,36). Wild-type and control-injected embryos rapidly retract melanosomes (Fig. 1I; Table 1). In contrast, high-dose bbs3 MO-injected embryos have significantly delayed melanosome retraction (Fig. 1H) (P < 0.001), whereas low-dose bbs3 MO-injected embryos show a similar rate as control embryos (P > 0.05) (Table 1). Similar findings are observed with bbs1 MO-injected embryos. Our data show that zebrafish bbs1 and bbs3 knockdowns result in prototypical phenotypes similar to other bbs gene knockdowns, including KV defects, predisposition to laterality defects and retrograde intracellular transport delay.
Genetic interaction between zebrafish BBS orthologs
Mutation of any of the 12 identified human BBS genes causes similar phenotypes, yet protein domain prediction analyses suggest that the BBS proteins do not belong to any one functional category (Supplementary Material, Fig. S1). It has been proposed that phenotypic heterogeneity among BBS patients may be due to genetic interactions between BBS genes (37). We therefore tested pair-wise knockdown of bbs1–bbs8 in zebrafish to evaluate synergy between these genes in vivo. We first identified a subphenotypic dose for each the eight BBS MOs. A subphenotypic dose was defined as the highest MO dose that does not generate a significant phenotype when individually injected into zebrafish embryos (indicated as '−', Fig. 2A; Table 2). We next co-injected the 28 possible pair-wise combinations of subphenotypic dose bbs1–bbs8 MOs. Interactions were considered significant (indicated as '+', Fig. 2A) if a statistically significant alteration in KV defects and an increase in melanosome retrograde transport were observed compared with treatment with a single low-dose MO. Eight of the 28 possible pair-wise injections resulted in synergistic interactions. All of these interactions involved either bbs1 or bbs2 as one of the components of the interaction. We identified five bbs1 synergistic interactions (with bbs2, bbs3, bbs4, bbs6 and bbs7 MOs) and four bbs2 synergistic interactions (with bbs1, bbs3, bbs4 and bbs6 MOs). Bbs3, bbs4 and bbs6 interacted with both bbs1 and bbs2. Bbs7 interacted only with bbs1. Bbs5 and bbs8 showed no genetic interactions with any other BBS gene. Of the eight synergistic combinations, the bbs1 with bbs7 pair-wise combination showed the most severe KV defect (Fig. 2B) and retrograde transport delay (Fig. 2C; Table 2). The other seven synergistic combinations, in order of increasing severity, are bbs2 with bbs4, bbs2 with bbs3, bbs1 with bbs2, bbs1 with bbs4, bbs1 with bbs6, bbs1 with bbs3 and bbs2 with bbs6 (Table 2). To confirm synergistic interactions, we co-injected embryos with pair-wise combinations of high-dose bbs1 MO with bbs7 MO. We also evaluated high-dose combination of bbs3 MO with bbs7 MO, a non-synergistic pair. The combined high-dose bbs1 MO with bbs7 MO generated dramatic defects, whereas high-dose pair-wise combination of bbs3 MO with bbs7 MO resulted in phenotypes similar to those generated by bbs3 MO high-dose injection alone and bbs7 high-dose injection alone (Fig. 3A and B). The synergy data provide in vivo evidence for genetic interactions between specific BBS genes. These genetic interactions could indicate functional redundancy between interacting genes and/or a required stoichiometry within a complex important for the proper function of the complex.
Figure 2.

Genetic interaction between eight BBS genes (bbs1–bbs8). (A) Twenty-eight subphenotypic pair-wise combinations of BBS MO demonstrated only eight synergistic interactions, noted as ‘+’ sign. Genetic interactions result in (B) KV disruption and (C) epinephrine-induced melanosome retrograde transport delay when compared with control and low-dose BBS MO-injected embryos. Specific RNA can rescue KV disruption and melanosome retrograde transport delay. Control MOs were added when necessary to maintain uniform final concentration of MO in the injection mix. *P < 0.001; Cont, control.
Table 2.
Summary of genetic interaction evaluated by BBS gene knockdown
| Combined low-dose MO (n) | Percentage of KV defect | Melanosome retrograde transport (min) |
|---|---|---|
| Control + Control (61) | 5 | 1.4 |
| bbs1 + Control (44) | 9 | 1.5 |
| bbs3 + Control (68) | 8 | 1.7 |
| bbs7 + Control (85) | 6 | 1.4 |
| bbs1 + bbs7 (156) | 35 | 6.1* |
| bbs2 + bbs4 (78) | 30 | 5.0* |
| bbs2 + bbs3 (65) | 26 | 5.0* |
| bbs1 + bbs2 (58) | 24 | 3.8* |
| bbs1 + bbs4 (65) | 23 | 3.5* |
| bbs1 + bbs6 (58) | 23 | 3.0* |
| bbs1 + bbs3 (47) | 22 | 2.9* |
| bbs2 + bbs6 (43) | 21 | 2.5* |
Subphenotypic paired combinations of MOs targeting specific BBS genes demonstrated phenotypes similar to individual BBS knockdown, including KV defect and melanosome retrograde transport delay; n, number of embryos.
*P< 0.001.
Figure 3.

Medium-dose knockdown of genetically interacting BBS genes results in an increased severity and penetrance of BBS knockdown phenotypes, including (A) KV disruption and (B) epinephrine-induced melanosome retrograde transport when compared with wild-type, control injected and embryos injected with MO combinations of bbs genes that do not genetically interact. Treatment and numbers of embryos noted on the x axis and values on the top of the bars. *P < 0.001; Cont, control.
Specificity of bbs knockdown
Rescue of the MO-generated phenotypes was performed to confirm the specificity of gene knockdown. We focused this component of the study on bbs1 and bbs7 for two reasons. First, the most significant genetic interaction combination was observed with bbs1 and bbs7. Secondly, the bbs1 and bbs7 proteins show significant homology to each other. Co-injection of in vitro-transcribed bbs1 RNA with high-dose bbs1 MO, as well as co-injection of bbs7 RNA with high-dose bbs7 MO, results in rescue of both KV and retrograde transport phenotypes (Fig. 2B and C; Table 3). Thus, RNA rescue supports specificity of bbs1 and bbs7 MO-induced phenotypes. To investigate whether genetic interaction between BBS genes is due to functional redundancy, we co-injected high-dose bbs7 MO with bbs1 RNA. The high-dose bbs7 MO-induced phenotype cannot be rescued with bbs1 RNA. Similarly, high-dose bbs1 MO and high-dose bbs3 MO phenotypes cannot be rescued with bbs7 RNA (Fig. 3A and B). These RNA rescue and pair-wise combination data provide in vivo evidence for specific genetic interactions between BBS genes and suggest that these genetic interactions are not due to functional redundancy. In contrast, to high-dose MO injections which lead to phenotypes, the individual low-dose MO injections of BBS MOs do not generate phenotypes, unless combined with a specific second low-dose BBS MO. Furthermore, RNA rescue of synergistic interactions was evaluated. Phenotypes generated from combined low-dose injection of bbs1 MO with bbs7 MO were rescued with bbs7 RNA alone, as well as with bbs1 RNA alone (Table 3). The ability of bbs1 RNA or bbs7 RNA to rescue the combined low-dose knockdown indicates that partial knockdown of either bbs1 or bbs7 alone does not result in complete loss of function of the BBSome complex.
Table 3.
BBS gene knockdown rescue and test for redundant function
| MO treatment (n) | RNA | Percentage of KV defects | Melanosome retrograde transport (min) |
|---|---|---|---|
| Control (500 μm) (302) | — | 3 | 1.5 |
| Control (500 μm) (41) | bbs1 | 7 | 1.8 |
| Control (500 μm) (49) | bbs7 | 4 | 1.5 |
| bbs1 MO (250 μm) (78) | — | 32 | 3.5* |
| bbs1 MO (250 μm) (36) | bbs1 | 11 | 1.9 |
| bbs7 MO (500 μm) (95) | — | 29 | 6.0* |
| bbs7 MO (500 μm) (72) | bbs7 | 8 | 1.8 |
| bbs1 MO (250 μm) (98) | bbs7 | 32 | 3.4* |
| bbs7 MO (500 μm) (48) | bbs1 | 27 | 6.1* |
| bbs7 MO (125 μm) + bbs1 MO (125 μm) (51) | bbs1 | 10 | 1.9 |
| bbs7 MO (125 μm)+bbs1 MO (125 μm) (43) | bbs7 | 9 | 1.9 |
| bbs3 MO (500 μm) (38) | bbs7 | 34 | 3.0* |
BBS genes do not display functional redundancy. bbs7 knockdown phenotypes can be rescued only by specific bbs7 mRNA. Subphenotypic paired combination of bbs1 with bbs7 phenotypes can be rescued with either bbs1 or bbs7 mRNA. Overexpression of bbs1 or bbs7 mRNA has no significant phenotypes; n, number of embryos.
*P < 0.001.
Altered patterning in the zebrafish pectoral fin bud
Since polydactyly is one of the cardinal features of human BBS, we explored the possibility of fin bud patterning defects in BBS morphants. Indeed, BBS genes are broadly expressed, yet by 36 h.p.f. bbs7 shows anterior enrichment, with notable expression in the developing fin bud (16). In BBS morphants, overall fin size was indistinguishable from wild-type, suggesting relatively normal fin growth and viability. Zebrafish pectoral fins and tetrapod forelimbs have evolved from a common ancestral appendage and share similar morphology and similar underlying molecular mechanisms (38–43), including the ZPA (44,45) which is localized at the posterior mesenchyme of the fin/limb bud (Fig. 4A). The ZPA signals positional information important for the specification of the digits. In chick limb bud, grafting an additional ZPA anterior to the original ZPA resulted in formation of extra digits resembling human polydactyly (44). Sonic hedgehog (Shh) is expressed in the posterior vertebrate limb bud mesenchyme and mediates the functions of the ZPA, in a concentration dependent manner, in both zebrafish fin buds and tetrapod limb buds (46). Since increased ZPA function or shh activity may lead to polydactyly, we evaluated shh expression. In wild-type embryos, shh is expressed in a crescent-shaped domain in the posterior region of the pectoral fin bud (Fig. 4B). This expression domain is unchanged in control and low-dose MO-injected embryos (bbs1, bbs3 and bbs7). The pectoral fin shh expression domain in high-dose bbs1 or bbs7 morphants demonstrates an anterior expansion, whereas shh expression in the dorsal midline remains unchanged (Fig. 4C). Consistent with the previously identified genetic interactions, pair-wise combination of low-dose MO of bbs1 with bbs7 also demonstrated expanded shh expression (Fig. 4D), whereas pair-wise combination of low-dose MO of bbs3 with bbs7 appears the same as wild-type (data not shown). The average area of shh expression in the pectoral fin bud was measured using the ImageJ program (47) and confirmed statistically significant (P < 0.001) increases in high-dose bbs1 MO, high-dose bbs7 MO and bbs1 with bbs7 low-dose combination in comparison with wild-type embryos (Fig. 4E). Thus, in the developing fin bud, we observe an increased ZPA/SHH domain without perturbing overall limb/fin growth.
Figure 4.

Zone of polarizing activity (ZPA) of the pectoral fin bud and Sonic hedgehog (SHH). (A) Schematic representation of zebrafish fin bud prior to 48 h.p.f. ZPA is localized posteriorly and can be labeled with shh expression. Whole-mount in situ hybridization using shh as a probe in the pectoral fin bud of 42 h.p.f. (B) wild-type, (C) bbs7 morphant and (D) low-dose bbs1 MO with bbs7 MO combination. Arrowhead denotes shh expression in the limb bud on one side, and the area of expression is outlined on the other side. (E) Graphical representation of the measured area (µm2) of shh expression domain in the pectoral fin bud of BBS morphants and wild-type. *P < 0.001.
Since genetic mutants for shh demonstrate loss or reduction of the skeletal elements of the pectoral fin (46), we next determined whether the expanded shh-signaling in the fin bud of BBS morphants induces expansion of pectoral fin structures. In the pectoral fin of 6–7-day-old larvae, the most proximal skeletal element is the cleithrum (Cl), which extends dorsoventrally (Fig. 5A). Flanking the Cl is the scapulocoracoid (Sc), which extends along the base of the fin, and the most distal element is the fin. Alcian Blue staining in wild-type identifies the rod-like Cl (Fig. 5B, arrow) and adjacent Sc (Fig. 5B, arrowhead). BBS morphant embryos demonstrate an enlarged Sc (Fig. 5C, arrowhead), where the distal posterior end of the Sc appears to have disorganized and/or extra cell layers compared with wild-type (Fig. 5B). Consistent with the genetic interaction data, bbs1 with bbs3 and bbs1 with bbs7 subphenotypic dose combinations also demonstrate thickened Sc (Fig. 5D, arrowhead), whereas bbs3 with bbs7, which has no synergistic effect on KV or retrograde transport, demonstrates Sc structure similar to wild-type (data not shown). Thus, compromised BBS function by MO injection results in expanded shh expression and subsequently patterning changes in the skeletal elements of the pectoral fin. Although, zebrafish do not form digits, these results indicate that limb patterning defects observed in BBS patients result from SHH expression abnormalities.
Figure 5.

Zebrafish pectoral fin cartilage staining. (A) Schematic representation of the fin structures: Cl as a dorsoventral line (arrow); Sc distal to Cl and proximal to fin. (B–D) Alcian Blue staining of the pectoral fin shows prototypical cartilage structure in 6-day-old (B) wild-type larvae, whereas (C) bbs7 morphant larvae and (D) low-dose combination of bbs1 MO with bbs7 MO show enlarged Sc structure with two to three cell lines at the proximal posterior region of Sc (arrowhead).
DISCUSSION
Twelve genes have been shown to independently cause BBS in humans. Human BBS patients display similar features independent of the primary mutated gene. We have previously shown that individual knockdown of several zebrafish BBS genes (bbs2, bbs4–bbs8 and bbs11) results in common zebrafish phenotypes including abnormal KV formation, cilia abnormalities and melanosome transport defects (9,16). In this study, we demonstrate that the individual loss-of-function of bbs1 or bbs3 in zebrafish results in the same phenotypes as knockdown of other zebrafish BBS genes. In addition, we have now used the zebrafish model system to identify genetic interactions between BBS genes by simultaneously knocking down pair-wise combinations of genes. For this in vivo analysis of genetic interactions, the 28 possible pair-wise combinations of BBS genes (bbs1–bbs8) were tested at doses that do not produce a phenotype when an individual morpholino is used (subphenotypic dose). At the subphenotypic doses used in this study, eight pair-wise synergistic interactions were identified. Prior to this study, no animal model for BBS polydactyly has been generated. Using the zebrafish model system, we identified altered patterning of the fin bud and subsequent changes to fin skeletal elements. Since the zebrafish fin bud is orthologous to human limb bud, this study suggests a link between post-axial polydactyly observed in BBS patients and altered shh expression in the developing limb/fin bud.
BBS3 function required for cilia function and intracellular vesicular transport
BBS3 (ARL6) is an ARF-like protein (10). Data support diverse functional roles for ARL proteins, including regulation of intracellular vesicle and membrane trafficking and microtubule assembly (18,19). Our loss-of-function data demonstrates that bbs3 results in the same zebrafish phenotypes as other BBS knockdowns, including KV defects and intracellular retrograde transport delay. Cilia found in the KV appear to be initially formed in bbs3 knockdown zebrafish, but the cilia are prematurely lost and do not function in forming a normal KV. It has recently been shown that seven BBS proteins (BBS1, 2, 4, 5, 7, 8 and 9) form a complex known as the BBSome (17). The BBSome appears to play a role in ciliogenesis, at least in part, by interacting with the Rab8 GTP/GDP exchange factor (Rabin8). Rab8GTP enters primary cilia and promotes extension of the ciliary membrane. Preventing Rab8GTP production blocks ciliation and yields characteristic BBS phenotypes in zebrafish (17). Although not a part of the BBSome, the data presented here demonstrating that bbs3 knockdown results in defects in KV and nodal cilia, as well as in melanosome (lysosome related organelles) transport, support a role for BBS3 and other BBS proteins in vesicular transport to the cilium (9,16).
Genetic interaction between BBS genes
Human BBS patients show a wide range of phenotypic variability. Variation is seen among patients even within the same family (and hence with the same primary BBS mutations), indicating genetic and/or environmental modifiers of the phenotype. The phenotypic variability and the extensive genetic heterogeneity of BBS raise the possibility of complex interactions among BBS genes (37). In this study, we identified eight genetic interactions from the total of 28 possible pair-wise genetic interactions tested. Of note, each of the eight synergistic interactions resulted in KV defects and delayed melanosome transport. No synergistic interactions were observed that affected only a subset of the phenotypes. In each case, increasing the dose of both morpholinos above the subphenotypic dose resulted in increased severity of the BBS knockdown phenotypes beyond that observed for individual high-dose knockdown, supporting the hypothesis that BBS genes can modify each other's phenotypes.
BBS complex interactions could be due to redundant roles, parallel pathway function or a stoichometric requirement within a protein complex such as the BBSome, to name a few possibilities. In this study, we demonstrated that individual high-dose MO-induced defects can be rescued by co-injection of a wild-type form of the gene-specific mRNA (i.e. bbs7 mRNA rescues the high-dose bbs7 MO phenotype), but not by increased levels of other BBS proteins. For example, bbs1 mRNA does not rescue high-dose bbs7 MO-induced defects and vice versa, arguing against functional redundancy. In contrast, an individual mRNA is sufficient to rescue the subphenotypic combined knockdown MOs (i.e. bbs7 mRNA can rescue the low-dose combined bbs7 with bbs1 MO-induced defects), suggesting a required stoichiometry within a complex. A recent study by Nachury et al. (17) has identified a BBS complex (the BBSome) and reported that components of the complex are present in stoichiometric amounts.
The identification of the BBSome complex consisting of seven of the 12 known BBS proteins (BBS1, BBS2, BBS4, BBS5, BBS7, BBS8 and BBS9) (17) can be used to categorize the known BBS proteins into those that are part of the BBSome and those that are not part of this complex (BBS3, BBS6, BBS10, BBS11 and BBS12). This information, along with the genetic interaction data from the current study, allows the determination of whether BBS pair-wise genetic interactions result from impairment of two BBSome proteins, impairment of a component of the BBSome and a non-BBSome component and/or simultaneous impairment of two non-BBSome proteins. The first two types of interactions were observed. For example, the strongest interaction was seen between two BBSome components (BBS1 and BBS7). In addition, the two non-BBSome proteins studied (BBS3 and BBS6) were each shown to genetically interact with specific components of the BBSome (BBS3 with BBS1 and BBS2; BBS6 with BBS1 and BBS2), but not with other BBSome components and not with each other. Furthermore, direct physical interactions within the BBSome complex do not predict genetic interactions. For example, BBS7 physically interacts directly with BBS2 and not with BBS1 (17). However, BBS7 genetically interacts with BBS1, but not with BBS2.
On the basis of finding of a heterozygous variant in MGC1203 in a few BBS patients, Badano et al. (48) utilized zebrafish to evaluate epistatic effect of mgc1203 on BBS genes and reported that modest suppression of mgc1203 exerts an epistatic effect on the developmental phenotype of BBS morphants. Similarly, our subphenotypic knockdowns of specific zebrafish BBS genes suggest that some BBS genes have an epistatic effect on the phenotype of other BBS genes. It is noteworthy that two independent studies utilized similar subphenotypic approach in zebrafish to evaluate genetic interactions of bbs10 and bbs12 genes (12,15). It should be noted that the authors reported gastrulation defects as a BBS MO-induced phenotype (12,15,48) and utilized this phenotype to identify potential interactions between BBS genes. The morphant embryos in our study do not show any gross changes in general body size and morphology. The cilia defect and transport delay phenotypes observed in the current study are biologically relevant to the human disorder.
Of clinical relevance, our data provide insight into non-Mendelian modes of BBS inheritance. Triallelic inheritance has been reported in a small subset of BBS cases (49–54). Our zebrafish data indicate that high-dose knockdown of individual BBS genes results in KV cilia and vesicle transport phenotypes (9,16). These data support mouse knockout data that complete loss of function of a single BBS protein is sufficient to induce BBS phenotypes (20–22). The genetic interaction data presented here suggest that functionally hypomorphic alleles of two specific BBS proteins could combine to result in digenic inheritance of BBS. On the basis of the zebrafish data demonstrating relevant phenotypes induced by simultaneous knockdown of two BBS genes, it is tempting to hypothesize that heterozygosity in two BBS genes (double heterozygosity) could be sufficient to manifest some BBS phenotypes. Such an occurrence has not been reported. The availability of multiple BBS mouse models will facilitate testing of this hypothesis.
A link between SSH and BBS limb defects
BBS pathophysiology has been linked to defects in cilia function and intracellular transport. Yet, polydactyly is not consistently reported in ciliary diseases (55–58). Therefore, mechanistic links between cilia function and polydactyly remain unclear. Since the zebrafish paired fins and tetrapod limbs are phylogenetically related, morphologically similar (42,43,59) and display similar gene expression patterns in the developing limb/fin buds, we can use this system to analyze the molecular mechanisms that underlie their early development (38–43). BBS loss-of-function demonstrates expanded shh expression in the pectoral fin bud without affecting shh neural tube expression. Furthermore, shh is required for fin/limb endoskeletal development (43), and the zebrafish shh mutant (syu) showed loss of pectoral girdle skeletal elements (46,60), while ectopic expression of chicken Shh in limb bud micromass culture induced novel cartilage nodules (61). Analysis of the skeletal elements in BBS knockdown larvae revealed a thickened scapulocoracoid of the pectoral fin consistent with increased shh expression in the pectoral fin bud. Although cilia function and IFT have been reported to play a role in SHH signaling (56,62,63), our current study uses shh as markers for the ZPA. In addition, shh expression in the midline is unperturbed. Further study is needed to directly address the role of cilia or intracellular transport in shh regulation in the BBS morhpants. However, our data provide in vivo evidence for a role of BBS function in limb/fin bud patterning resulting in increased shh signaling.
MATERIALS AND METHODS
MO antisense
Antisense MOs were designed and purchased from Gene Tools, LLC. MOs were microinjected in approximately 3 nanoliter volumes into one to eight cell embryos at concentrations ranging from 125 to 500 mm. Zebrafish MO sequences are bbs1_aug (GGGAACAGATGACATGGTTGTTTTG); bbs1_ exon2 (TGAAACTCACCAATGCATGAGGAGA); control MO bbs1_5mis (TAAGaGTGAtGATGGcCACTtGCAT); bbs3_aug (AGCTTGTCAAAAAGCCCCA TTTGCT); bbs3_exon2 (TACATCTTTTATTTTCACCTTGAGG) and control MO bbs3_5mis (AGgTTcTCAAAAAaCCCgATTTcCT).
Pair-wise combination of bbs morpholinos
Two different BBS MOs targeting bbs1–bbs8 were combined at subphenotypic low doses (125 µm each) and microinjected into one to eight cell embryos. Controls for these experiments were combinations of low dose of each BBS MO and a control MO (125 µm each). The final concentration of MOs injected into any embryo in this experiment was always constant (250 µm).
Analysis of the KV
Vesicles in embryos with a diameter less than one-third that of wild-type were considered reduced and embryos in which vesicles could not be morphologically identified were scored as absent. Live embryos were photographed with a Zeiss Axiocam on a stereoscope.
Melanosome transport assay
Day 5 larvae were exposed to epinephrine (50 mg/ml, Sigma, E4375) added to embryo medium (64) for a final concentration of 500 µg/ml. Melanosome retraction was continuously monitored under the microscope and the endpoint was scored when all melanosomes in the head and the trunk were perinuclear.
Rescue of knockdown phenotype
Morpholino-resistant BBS RNA was subcloned into pCS2+ expression vector (65). Synthetic RNA was made with mMessage mMachine (Ambion). mRNA (50 ng/µl bbs1 and 100 ng/µl bbs7) was co-injected with appropriate MO.
Whole-mount in situ hybridization of shh in the pectoral fin buds
Embryos, treated with pigment inhibitor at 12 h.p.f., were cultured to 42 h.p.f. and fixed with 4% paraformaldehyde/phosphate-buffered saline (PBS). shh digoxigenin-UTP RNA probes (Roche) were synthesized from the linearized templates, antisense (SpeI, T7 RNA polymerase) and sense (NotI, SP6 RNA polymerase). Whole-mount in situ hybridization was performed as described by Yen et al. (16). The area of shh expression was measured using ImageJ 1.37v (47). Student's t-test assuming unequal variance was used to evaluate statistically significant differences in the average area of shh expression (P < 0.001). Embryos were photographed at 50× magnification.
Cartilage staining
Cartilage staining was performed as described in Taylor and Van Dyke (66). Briefly, larvae at 6 day post-fertilization (d.p.f.) were fixed overnight with 4% paraformaldehyde/PBS, dehydrated gradually with absolute ethanol and stained overnight with Alcian Blue (30 mg in 8:2 ethanol:glacial acetic acid). Larvae were then bleached with 1% potassium hydroxide (1 M)/3% hydrogen peroxide, cleared with 0.5% trypsine/saturated sodium tetraborate, pH 9.1, for 2 h and stored in 7:3 glycerol:(1X) PBS. Cartilage mounts were photographed at 50× magnification.
FUNDING
This research work was supported by the National Institutes of Health (EY011298 and EY017168 to V.C.S. and CA112369 to D.C.S.). V.C.S. is an investigator of the Howard Hughes Medical Institute.
SUPPLEMENTARY MATERIAL
ACKNOWLEDGEMENTS
We thank the members of V.C.S. and D.C.S. laboratories for their insightful discussions.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Green J.S., Parfrey P.S., Harnett J.D., Farid N.R., Cramer B.C., Johnson G., Heath O., McManamon P.J., O'Leary E., Pryse-Phillips W. The cardinal manifestations of Bardet-Biedl syndrome, a form of Laurence–Moon–Biedl syndrome. N Engl. J. Med. 1989;321:1002–1009. doi: 10.1056/NEJM198910123211503. [DOI] [PubMed] [Google Scholar]
- 2.Harnett J.D., Green J.S., Cramer B.C., Johnson G., Chafe L., McManamon P., Farid N.R., Pryse-Phillips W., Parfrey P.S. The spectrum of renal disease in Laurence–Moon–Biedl syndrome. N. Engl. J. Med. 1988;319:615–618. doi: 10.1056/NEJM198809083191005. [DOI] [PubMed] [Google Scholar]
- 3.Elbedour K., Zucker N., Zalzstein E., Barki Y., Carmi R. Cardiac abnormalities in the Bardet-Biedl syndrome: echocardiographic studies of 22 patients. Am. J. Med. Genet. 1994;52:164–169. doi: 10.1002/ajmg.1320520208. [DOI] [PubMed] [Google Scholar]
- 4.Slavotinek A.M., Stone E.M., Mykytyn K., Heckenlively J.R., Green J.S., Heon E., Musarella M.A., Parfrey P.S., Sheffield V.C., Biesecker L.G. Mutations in MKKS cause Bardet-Biedl syndrome. Nat. Genet. 2000;26:15–16. doi: 10.1038/79116. [DOI] [PubMed] [Google Scholar]
- 5.Mykytyn K., Braun T., Carmi R., Haider N.B., Searby C.C., Shastri M., Beck G., Wright A.F., Iannaccone A., Elbedour K., et al. Identification of the gene that, when mutated, causes the human obesity syndrome BBS4. Nat. Genet. 2001;28:188–191. doi: 10.1038/88925. [DOI] [PubMed] [Google Scholar]
- 6.Nishimura D.Y., Searby C.C., Carmi R., Elbedour K., Van Maldergem L., Fulton A.B., Lam B.L., Powell B.R., Swiderski R.E., Bugge K.E., et al. Positional cloning of a novel gene on chromosome 16q causing Bardet-Biedl syndrome (BBS2) Hum. Mol. Genet. 2001;10:865–874. doi: 10.1093/hmg/10.8.865. [DOI] [PubMed] [Google Scholar]
- 7.Nishimura D.Y., Swiderski R.E., Searby C.C., Berg E.M., Ferguson A.L., Hennekam R., Merin S., Weleber R.G., Biesecker L.G., Stone E.M., et al. Comparative genomics and gene expression analysis identifies BBS9, a new Bardet-Biedl syndrome gene. Am. J. Hum. Genet. 2005;77:1021–1033. doi: 10.1086/498323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ansley S.J., Badano J.L., Blacque O.E., Hill J., Hoskins B.E., Leitch C.C., Kim J.C., Ross A.J., Eichers E.R., Teslovich T.M., et al. Basal body dysfunction is a likely cause of pleiotropic Bardet-Biedl syndrome. Nature. 2003;425:628–633. doi: 10.1038/nature02030. [DOI] [PubMed] [Google Scholar]
- 9.Chiang A.P., Beck J.S., Yen H.J., Tayeh M.K., Scheetz T.E., Swiderski R.E., Nishimura D.Y., Braun T.A., Kim K.Y., Huang J., et al. Homozygosity mapping with SNP arrays identifies TRIM32, an E3 ubiquitin ligase, as a Bardet-Biedl syndrome gene (BBS11) Proc. Natl Acad. Sci. USA. 2006;103:6287–6292. doi: 10.1073/pnas.0600158103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chiang A.P., Nishimura D., Searby C., Elbedour K., Carmi R., Ferguson A.L., Secrist J., Braun T., Casavant T., Stone E.M., et al. Comparative genomic analysis identifies an ADP-ribosylation factor-like gene as the cause of Bardet-Biedl syndrome (BBS3) Am. J. Hum. Genet. 2004;75:475–484. doi: 10.1086/423903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li J.B., Gerdes J.M., Haycraft C.J., Fan Y., Teslovich T.M., May-Simera H., Li H., Blacque O.E., Li L., Leitch C.C., et al. Comparative genomics identifies a flagellar and basal body proteome that includes the BBS5 human disease gene. Cell. 2004;117:541–552. doi: 10.1016/s0092-8674(04)00450-7. [DOI] [PubMed] [Google Scholar]
- 12.Stoetzel C., Laurier V., Davis E.E., Muller J., Rix S., Badano J.L., Leitch C.C., Salem N., Chouery E., Corbani S., et al. BBS10 encodes a vertebrate-specific chaperonin-like protein and is a major BBS locus. Nat. Genet. 2006;38:521–524. doi: 10.1038/ng1771. [DOI] [PubMed] [Google Scholar]
- 13.Badano J.L., Ansley S.J., Leitch C.C., Lewis R.A., Lupski J.R., Katsanis N. Identification of a novel Bardet-Biedl syndrome protein, BBS7, that shares structural features with BBS1 and BBS2. Am. J. Hum. Genet. 2003;72:650–658. doi: 10.1086/368204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Katsanis N., Beales P.L., Woods M.O., Lewis R.A., Green J.S., Parfrey P.S., Ansley S.J., Davidson W.S., Lupski J.R. Mutations in MKKS cause obesity, retinal dystrophy and renal malformations associated with Bardet-Biedl syndrome. Nat. Genet. 2000;26:67–70. doi: 10.1038/79201. [DOI] [PubMed] [Google Scholar]
- 15.Stoetzel C., Muller J., Laurier V., Davis E.E., Zaghloul N.A., Vicaire S., Jacquelin C., Plewniak F., Leitch C.C., Sarda P., et al. Identification of a novel BBS gene (BBS12) highlights the major role of a vertebrate-specific branch of chaperonin-related proteins in Bardet-Biedl syndrome. Am. J. Hum. Genet. 2007;80:1–11. doi: 10.1086/510256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yen H.J., Tayeh M.K., Mullins R.F., Stone E.M., Sheffield V.C., Slusarski D.C. Bardet-Biedl syndrome genes are important in retrograde intracellular trafficking and Kupffer's vesicle cilia function. Hum. Mol. Genet. 2006;15:667–677. doi: 10.1093/hmg/ddi468. [DOI] [PubMed] [Google Scholar]
- 17.Nachury M.V., Loktev A.V., Zhang Q., Westlake C.J., Peranen J., Merdes A., Slusarski D.C., Scheller R.H., Bazan J.F., Sheffield V.C., et al. A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell. 2007;129:1201–1213. doi: 10.1016/j.cell.2007.03.053. [DOI] [PubMed] [Google Scholar]
- 18.Pasqualato S., Renault L., Cherfils J. Arf, Arl, Arp and Sar proteins: a family of GTP-binding proteins with a structural device for ‘front-back’ communication. EMBO Rep. 2002;3:1035–1041. doi: 10.1093/embo-reports/kvf221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bhamidipati A., Lewis S.A., Cowan N.J. ADP ribosylation factor-like protein 2 (Arl2) regulates the interaction of tubulin-folding cofactor D with native tubulin. J. Cell Biol. 2000;149:1087–1096. doi: 10.1083/jcb.149.5.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fath M.A., Mullins R.F., Searby C., Nishimura D.Y., Wei J., Rahmouni K., Davis R.E., Tayeh M.K., Andrews M., Yang B., et al. Mkks-null mice have a phenotype resembling Bardet-Biedl syndrome. Hum. Mol. Genet. 2005;14:1109–1118. doi: 10.1093/hmg/ddi123. [DOI] [PubMed] [Google Scholar]
- 21.Mykytyn K., Mullins R.F., Andrews M., Chiang A.P., Swiderski R.E., Yang B., Braun T., Casavant T., Stone E.M., Sheffield V.C. Bardet-Biedl syndrome type 4 (BBS4)-null mice implicate Bbs4 in flagella formation but not global cilia assembly. Proc. Natl Acad. Sci USA. 2004;101:8664–8669. doi: 10.1073/pnas.0402354101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nishimura D.Y., Fath M., Mullins R.F., Searby C., Andrews M., Davis R., Andorf J.L., Mykytyn K., Swiderski R.E., Yang B., et al. Bbs2-null mice have neurosensory deficits, a defect in social dominance, and retinopathy associated with mislocalization of rhodopsin. Proc. Natl Acad. Sci. USA. 2004;101:16588–16593. doi: 10.1073/pnas.0405496101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Davis R.E., Swiderski R.E., Rahmouni K., Nishimura D.Y., Mullins R.F., Agassandian K., Philp A.R., Searby C.C., Andrews M.P., Thompson S., et al. A knockin mouse model of the Bardet Biedl syndrome 1 M390R mutation has cilia defects, ventriculomegaly, retinopathy, and obesity. Proc. Natl Acad. Sci. USA. 2007;104:19422–19427. doi: 10.1073/pnas.0708571104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Supp D.M., Brueckner M., Kuehn M.R., Witte D.P., Lowe L.A., McGrath J., Corrales J., Potter S.S. Targeted deletion of the ATP binding domain of left-right dynein confirms its role in specifying development of left–right asymmetries. Development. 1999;126:5495–5504. doi: 10.1242/dev.126.23.5495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Supp D.M., Witte D.P., Potter S.S., Brueckner M. Mutation of an axonemal dynein affects left–right asymmetry in inversus viscerum mice. Nature. 1997;389:963–966. doi: 10.1038/40140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Essner J.J., Amack J.D., Nyholm M.K., Harris E.B., Yost H.J. Kupffer's vesicle is a ciliated organ of asymmetry in the zebrafish embryo that initiates left–right development of the brain, heart and gut. Development. 2005;132:1247–1260. doi: 10.1242/dev.01663. [DOI] [PubMed] [Google Scholar]
- 27.Essner J.J., Vogan K.J., Wagner M.K., Tabin C.J., Yost H.J., Brueckner M. Conserved function for embryonic nodal cilia. Nature. 2002;418:37–38. doi: 10.1038/418037a. [DOI] [PubMed] [Google Scholar]
- 28.Melby A.E., Warga R.M., Kimmel C.B. Specification of cell fates at the dorsal margin of the zebrafish gastrula. Development. 1996;122:2225–2237. doi: 10.1242/dev.122.7.2225. [DOI] [PubMed] [Google Scholar]
- 29.Cooper M.S., D'Amico L.A. A cluster of noninvoluting endocytic cells at the margin of the zebrafish blastoderm marks the site of embryonic shield formation. Dev. Biol. 1996;180:184–198. doi: 10.1006/dbio.1996.0294. [DOI] [PubMed] [Google Scholar]
- 30.D'Amico L.A., Cooper M.S. Spatially distinct domains of cell behavior in the zebrafish organizer region. Biochem. Cell. Biol. 1997;75:563–577. [PubMed] [Google Scholar]
- 31.Chen C.Y., Schwartz R.J. Recruitment of the tinman homolog Nkx-2.5 by serum response factor activates cardiac alpha-actin gene transcription. Mol. Cell. Biol. 1996;16:6372–6384. doi: 10.1128/mcb.16.11.6372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Skold H.N., Aspengren S., Wallin M. The cytoskeleton in fish melanophore melanosome positioning. Microsc. Res. Tech. 2002;58:464–469. doi: 10.1002/jemt.10164. [DOI] [PubMed] [Google Scholar]
- 33.Barral D.C., Seabra M.C. The melanosome as a model to study organelle motility in mammals. Pigment Cell Res. 2004;17:111–118. doi: 10.1111/j.1600-0749.2004.00138.x. [DOI] [PubMed] [Google Scholar]
- 34.Marks M.S., Seabra M.C. The melanosome: membrane dynamics in black and white. Nat. Rev. Mol. Cell Biol. 2001;2:738–748. doi: 10.1038/35096009. [DOI] [PubMed] [Google Scholar]
- 35.Blott E.J., Griffiths G.M. Secretory lysosomes. Nat. Rev. Mol. Cell Biol. 2002;3:122–131. doi: 10.1038/nrm732. [DOI] [PubMed] [Google Scholar]
- 36.Nascimento A.A., Roland J.T., Gelfand V.I. Pigment cells: a model for the study of organelle transport. Annu. Rev. Cell. Dev. Biol. 2003;19:469–491. doi: 10.1146/annurev.cellbio.19.111401.092937. [DOI] [PubMed] [Google Scholar]
- 37.Mykytyn K., Sheffield V.C. Establishing a connection between cilia and Bardet-Biedl syndrome. Trends Mol. Med. 2004;10:106–109. doi: 10.1016/j.molmed.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 38.Krauss S., Concordet J.P., Ingham P.W. A functionally conserved homolog of the Drosophila segment polarity gene hh is expressed in tissues with polarizing activity in zebrafish embryos. Cell. 1993;75:1431–1444. doi: 10.1016/0092-8674(93)90628-4. [DOI] [PubMed] [Google Scholar]
- 39.Ekker M., Wegner J., Akimenko M.A., Westerfield M. Coordinate embryonic expression of three zebrafish engrailed genes. Development. 1992;116:1001–1010. doi: 10.1242/dev.116.4.1001. [DOI] [PubMed] [Google Scholar]
- 40.Akimenko M.A., Ekker M. Anterior duplication of the Sonic hedgehog expression pattern in the pectoral fin buds of zebrafish treated with retinoic acid. Dev. Biol. 1995;170:243–247. doi: 10.1006/dbio.1995.1211. [DOI] [PubMed] [Google Scholar]
- 41.Akimenko M.A., Johnson S.L., Westerfield M., Ekker M. Differential induction of four msx homeobox genes during fin development and regeneration in zebrafish. Development. 1995;121:347–357. doi: 10.1242/dev.121.2.347. [DOI] [PubMed] [Google Scholar]
- 42.Sordino P., van der Hoeven F., Duboule D. Hox gene expression in teleost fins and the origin of vertebrate digits. Nature. 1995;375:678–681. doi: 10.1038/375678a0. [DOI] [PubMed] [Google Scholar]
- 43.Neumann C.J., Grandel H., Gaffield W., Schulte-Merker S., Nusslein-Volhard C. Transient establishment of anteroposterior polarity in the zebrafish pectoral fin bud in the absence of sonic hedgehog activity. Development. 1999;126:4817–4826. doi: 10.1242/dev.126.21.4817. [DOI] [PubMed] [Google Scholar]
- 44.Tickle C., Summerbell D., Wolpert L. Positional signalling and specification of digits in chick limb morphogenesis. Nature. 1975;254:199–202. doi: 10.1038/254199a0. [DOI] [PubMed] [Google Scholar]
- 45.Saunders J.W., Gasseling M.T. Ectoderm-mesenchymal interactions in the origin of wing symmetry. In: Fleischmajer R., Billingham R.E., editors. Epithelial–Mesenchymal Interactions. Baltimore, MA: Williams and Wikins; 1968. [Google Scholar]
- 46.Grandel H., Draper B.W., Schulte-Merker S. Dackel acts in the ectoderm of the zebrafish pectoral fin bud to maintain AER signaling. Development. 2000;127:4169–4178. doi: 10.1242/dev.127.19.4169. [DOI] [PubMed] [Google Scholar]
- 47.Rasband W. ImageJ 1.37v. USA: National Institute of Health; 2006. [Google Scholar]
- 48.Badano J.L., Leitch C.C., Ansley S.J., May-Simera H., Lawson S., Lewis R.A., Beales P.L., Dietz H.C., Fisher S., Katsanis N. Dissection of epistasis in oligogenic Bardet-Biedl syndrome. Nature. 2006;439:326–330. doi: 10.1038/nature04370. [DOI] [PubMed] [Google Scholar]
- 49.Katsanis N. The oligogenic properties of Bardet-Biedl syndrome. Hum. Mol. Genet. 2004;13:R65–R71. doi: 10.1093/hmg/ddh092. [DOI] [PubMed] [Google Scholar]
- 50.Katsanis N., Ansley S.J., Badano J.L., Eichers E.R., Lewis R.A., Hoskins B.E., Scambler P.J., Davidson W.S., Beales P.L., Lupski J.R. Triallelic inheritance in Bardet-Biedl syndrome, a Mendelian recessive disorder. Science. 2001;293:2256–2259. doi: 10.1126/science.1063525. [DOI] [PubMed] [Google Scholar]
- 51.Laurier V., Stoetzel C., Muller J., Thibault C., Corbani S., Jalkh N., Salem N., Chouery E., Poch O., Licaire S., et al. Pitfalls of homozygosity mapping: an extended consanguineous Bardet-Biedl syndrome family with two mutant genes (BBS2, BBS10), three mutations, but no triallelism. Eur. J. Hum. Genet. 2006;14:1195–1203. doi: 10.1038/sj.ejhg.5201688. [DOI] [PubMed] [Google Scholar]
- 52.Badano J.L., Kim J.C., Hoskins B.E., Lewis R.A., Ansley S.J., Cutler D.J., Castellan C., Beales P.L., Leroux M.R., Katsanis N. Heterozygous mutations in BBS1, BBS2 and BBS6 have a potential epistatic effect on Bardet-Biedl patients with two mutations at a second BBS locus. Hum. Mol. Genet. 2003;12:1651–1659. doi: 10.1093/hmg/ddg188. [DOI] [PubMed] [Google Scholar]
- 53.Beales P.L., Badano J.L., Ross A.J., Ansley S.J., Hoskins B.E., Kirsten B., Mein C.A., Froguel P., Scambler P.J., Lewis R.A., et al. Genetic interaction of BBS1 mutations with alleles at other BBS loci can result in non-Mendelian Bardet-Biedl syndrome. Am. J. Hum. Genet. 2003;72:1187–1199. doi: 10.1086/375178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Katsanis N., Eichers E.R., Ansley S.J., Lewis R.A., Kayserili H., Hoskins B.E., Scambler P.J., Beales P.L., Lupski J.R. BBS4 is a minor contributor to Bardet-Biedl syndrome and may also participate in triallelic inheritance. Am. J. Hum. Genet. 2002;71:22–29. doi: 10.1086/341031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ferrante M.I., Zullo A., Barra A., Bimonte S., Messaddeq N., Studer M., Dolle P., Franco B. Oral–facial–digital type I protein is required for primary cilia formation and left–right axis specification. Nat. Genet. 2006;38:112–117. doi: 10.1038/ng1684. [DOI] [PubMed] [Google Scholar]
- 56.Marshall W.F., Nonaka S. Cilia: tuning in to the cell's antenna. Curr. Biol. 2006;16:R604–R614. doi: 10.1016/j.cub.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 57.Otto E.A., Loeys B., Khanna H., Hellemans J., Sudbrak R., Fan S., Muerb U., O'Toole J.F., Helou J., Attanasio M., et al. Nephrocystin-5, a ciliary IQ domain protein, is mutated in Senior-Loken syndrome and interacts with RPGR and calmodulin. Nat. Genet. 2005;37:282–288. doi: 10.1038/ng1520. [DOI] [PubMed] [Google Scholar]
- 58.Romio L., Fry A.M., Winyard P.J., Malcolm S., Woolf A.S., Feather S.A. OFD1 is a centrosomal/basal body protein expressed during mesenchymal–epithelial transition in human nephrogenesis. J. Am. Soc. Nephrol. 2004;15:2556–2568. doi: 10.1097/01.ASN.0000140220.46477.5C. [DOI] [PubMed] [Google Scholar]
- 59.Grandel H., Schulte-Merker S. The development of the paired fins in the zebrafish (Danio rerio) Mech. Dev. 1998;79:99–120. doi: 10.1016/s0925-4773(98)00176-2. [DOI] [PubMed] [Google Scholar]
- 60.van Eeden F.J., Granato M., Schach U., Brand M., Furutani-Seiki M., Haffter P., Hammerschmidt M., Heisenberg C.P., Jiang Y.J., Kane D.A., et al. Genetic analysis of fin formation in the zebrafish, Danio rerio. Development. 1996;123:255–262. doi: 10.1242/dev.123.1.255. [DOI] [PubMed] [Google Scholar]
- 61.Stott N.S., Chuong C.M. Dual action of Sonic hedgehog on chondrocyte hypertrophy: retrovirus mediated ectopic sonic hedgehog expression in limb bud micromass culture induces novel cartilage nodules that are positive for alkaline phosphatase and type X collagen. J. Cell. Sci. 1997;110:2691–2701. doi: 10.1242/jcs.110.21.2691. [DOI] [PubMed] [Google Scholar]
- 62.Huangfu D., Anderson K.V. Cilia and Hedgehog responsiveness in the mouse. Proc. Natl Acad. Sci. USA. 2005;102:11325–11330. doi: 10.1073/pnas.0505328102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Huangfu D., Liu A., Rakeman A.S., Murcia N.S., Niswander L., Anderson K.V. Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature. 2003;426:83–87. doi: 10.1038/nature02061. [DOI] [PubMed] [Google Scholar]
- 64.Westerfield M. The Zebrafish Book. A Guide for the Laboratory Use of Zebrafish. 3rd edn. Eugene: University of Oregon Press; 1995. [Google Scholar]
- 65.Turner D.L., Weintraub H. Expression of achaete-scute homolog 3 in Xenopus embryos converts ectodermal cells to a neural fate. Genes Dev. 1994;8:1434–1447. doi: 10.1101/gad.8.12.1434. [DOI] [PubMed] [Google Scholar]
- 66.Taylor W.R., Dyke G.C.V. Revised procedures for staining and clearing small fishes and other vertebrates for bone and cartilage study. Cybium. 1985;9:107–119. [Google Scholar]