

Abstract
A characteristic feature of classic PXE, an autosomal recessive disorder caused by mutations in the ABCC6 gene, is aberrant mineralization of connective tissues, particularly the elastic fibers. Here, we report a family with PXE-like cutaneous features in association with multiple coagulation factor deficiency, an autosomal recessive disorder associated with GGCX mutations. The proband and her sister, both with severe skin findings with extensive mineralization, were compound heterozygotes for missense mutations in the GGCX gene, which were shown to result in reduced γ-glutamyl carboxylase activity and in under-carboxylation of matrix gla protein. The proband’s mother and aunt, also manifesting with PXE-like skin changes, were heterozygous carriers of a missense mutation (p.V255M) in GGCX and a null mutation (p.R1141X) in the ABCC6 gene, suggesting digenic nature of their skin findings. Thus, reduced γ-glutamyl carboxylase activity in individuals either compound heterozygous for a missense mutation in GGCX or with haploinsufficiency in GGCX in combination with heterozygosity for ABCC6 gene expression results in aberrant mineralization of skin leading to PXE-like phenotype. These findings expand the molecular basis of PXE-like phenotypes, and suggest a role for multiple genetic factors in pathologic tissue mineralization in general.
INTRODUCTION
Pseudoxanthoma elasticum (PXE; OMIM 264800, 177850) is an autosomal recessive multi-system disorder characterized by dystrophic mineralization of soft connective tissues, particularly the elastic fibers, in a number of organs, including the skin, the eyes, and the arterial blood vessels (Neldner and Struk, 2002; Ringpfeil et al., 2001; Uitto et al., 2007; Li et al., 2008). The primary cutaneous lesions are small, yellowish papules on the predilection sites at flexural areas, and these lesions progressively coalesce into larger plaques of inelastic, leathery skin with a yellowish hue. Histopathology of the affected skin shows accumulation of pleiomorphic elastotic material in upper and mid dermis, which is mineralized as visualized by special histopathologic stains. The eye manifestations consist of angioid streaks, and bleeding from the retina can result in loss of visual acuity and, rarely, blindness. The cardiovascular manifestations result from mineralization of the arterial blood vessels, and include gastrointestinal bleeding, intermittent claudication, hypertension and, occasionally, early myocardial infarcts. While the disease has considerable morbidity and mortality, the phenotypic spectrum is highly variable with both inter- and intra-familial heterogeneity. The precise incidence of this condition is undefined, but the estimates are in the range of 1 in 50,000 – 75,000.
Classic PXE is caused by mutations in the ABCC6 gene which encodes a putative transmembrane transporter protein, ABCC6 (also known as multi-drug resistance-associated protein 6 – MRP6), a member of the family of ATP-binding cassette (ABC) proteins (Bergen et al., 2000; Le Saux et al., 2000; Ringpfeil et al., 2000; Miksch et al., 2005; Pfendner et al., 2007). The ABCC6 gene is primarily expressed in the liver, to a lesser extent in the proximal tubules of kidneys, and at very low level, if at all, in tissues affected in PXE (Belinsky and Kruh, 1999; Scheffer et al., 2002; Matsuzaki et al., 2005). While the mineral deposits in the affected tissues are known to consist of calcium and phosphate, the precise mechanisms leading to aberrant mineralization remain unclear, and specifically, the substrate specificity of ABCC6 in vivo is currently unknown.
Occasionally, PXE-like phenotype have been reported in association with multiple coagulation factor deficiency (Le Corvaisier-Pieto et al., 1996; Rongioletti et al., 1989; Vanakker et al., 2007), and molecular analysis of some of these patients revealed mutations in the GGCX gene which encodes an enzyme required for γ-glutamyl carboxylation of gla-proteins (Vanakker et al., 2007). In this report we detail a family with PXE-like clinical features, and also with deficiency of vitamin K-dependent clotting factors, particularly Factor X. Strikingly, this family harbors mutations both in the GGCX and ABCC6 genes, and combinations of these mutations in two patients with PXE-like skin phenotype suggest digenic inheritance.
RESULTS
Clinical findings
The proband (III-3 in Fig. 1) is a 16-year old female who was initially evaluated in early childhood because of cardiac abnormalities, including supravalvular pulmonic stenosis and peripheral pulmonary artery stenosis. She has undergone two pulmonary valve replacement procedures. Following an episode of endocarditis, she had a stroke with vision loss in the right eye, but rigorous ophthalmologic examination with respect to angioid streaks has not been reported. She developed focal segmental glomerulosclerosis, thought to be immune complex mediated. At around age 10 years, she began developing progressively loose, sagging and redundant skin, primarily affecting the neck and trunk, and an initial diagnosis of cutis laxa was made (Fig. 1a–c). She was also found to have a coagulation disorder with Factor X deficiency (Table 1). The proband has a 19-year old sister (III-1) with similar skin changes beginning in her early teens, as well as a coagulation disorder; she has had a normal cardiac evaluation. They have a 17-year old brother (III-2) who is clinically normal. Examination of other members of the nuclear pedigree (Fig. 1g) showed that the father of the proband is clinically normal, while the mother (II-2), at the age of 40 years, has distinct folding of the skin, particularly in the axillary areas. She also has characteristic yellowish papules consistent with PXE on the sides of the neck and in the axillary areas (Fig. 1d). The mother has a fraternal twin sister (II-3) with similar cutaneous findings (Fig. 1e–f). Neither the mother nor her sister have evidence of a coagulation disorder (Table 1).
Figure 1. Cutaneous findings in a family with PXE-like phenotype.
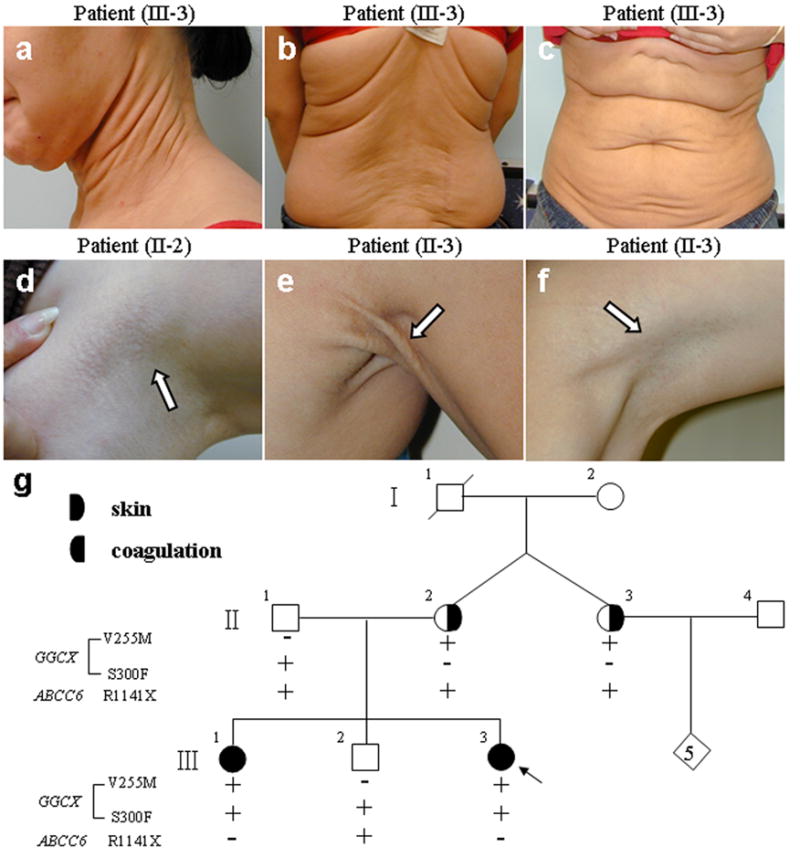
Note the loose and sagging skin in the 16 year-old proband (III-3, a–c). The proband’s mother (II-2, d) and aunt (II-3, e & f), both 40 years of age, demonstrate redundant folding of the skin in the axillary area (d, e) and popliteal fossa (f), as well as small yellowish papules characteristic of PXE (arrows). The nuclear pedigree of the family with PXE-like skin findings and coagulation factor deficiency. The mutations in the GGCX and ABCC6 genes identified in this family are indicated on the left of panel g. The proband is identified by an arrow.
Table 1.
Activities of the coagulation factors and clinical laboratory values in patients with GGCX gene mutations1
| Laboratory measurement | Patient |
Normal values | ||
|---|---|---|---|---|
| III-1 | III-3 | II-3 | ||
| F II (%) | 43 | 64 | 95 | 60–160 |
| F VII (%) | 31 | 108 | 86 | 60–160 |
| F VIII (%) | - | 67 | - | 60–160 |
| F IX (%) | 56 | 62 | 76 | 60–160 |
| F X (%) | 18 | 33 | 90 | 60–160 |
| PTT (s) | 32.1 | 43.3 | 32.0 | 25–41 |
| PT (s) | 21.6 | 18.1 | 10.9 | 12–15 |
| INR | 1.9 | 1.2 | 1.1 | 0.8–1.2 |
F, factor; PTT, partial thrombin time; PT, prothrombin time; INR, international normalized ratio.
Histopathology of the skin from the proband, her sister and the aunt revealed accumulation of basophilic elastotic material in the mid dermis, which was demonstrated to show aberrant mineralization with von Kossa and Alizarin red special stains (Fig. 2). Thus, these patients have some clinical and histopathological features of PXE.
Figure 2. Histopathology of the cutaneous lesions in the sister (III-1), the proband (III-3) and aunt (II-3).
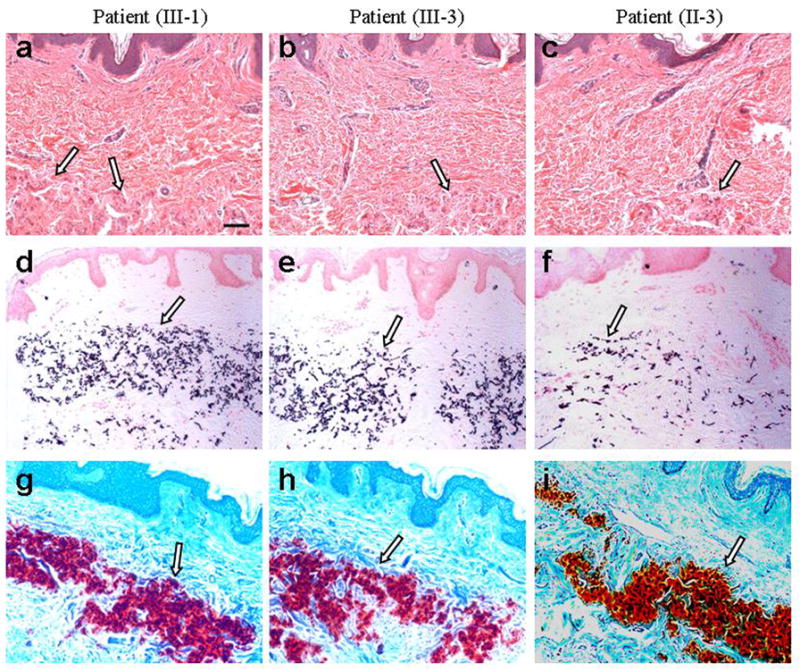
Staining with hematoxylin-eosin (a–c) demonstrates basophilic abnormal elastic structures in the mid dermis (arrows). Special stains (von Kossa, d–f; Alizarin red, g–i) reveal that these elastotic structures are mineralized (arrows). Scale bar, 0.1 mm.
Mutation analyses
Considering the presence of PXE-like cutaneous findings, we first sequenced the ABCC6 gene. The recurrent mutation, p.R1141X, which is particularly prevalent (about 30% of all detected mutations in PXE patients of the European extraction) (Pfendner et al., 2007) was found in the DNA from the father, mother, the maternal aunt and the younger brother, but not in the proband herself or her sister (Figs. 1g and 3a). The individuals were heterozygotes for this mutation, and no other ABCC6 mutation could be disclosed in this family. Sequencing of the proband’s sister’s and her aunt’s ABCC6 gene revealed the presence of eight heterozygous polymorphisms in the exons 7–19. This finding argues against large allelic deletions at the 5′ half of the gene which might not be detectable by the mutation detection strategy employed in our study (Pfendner et al., 2007).
Figure 3. Mutation detection in the family.
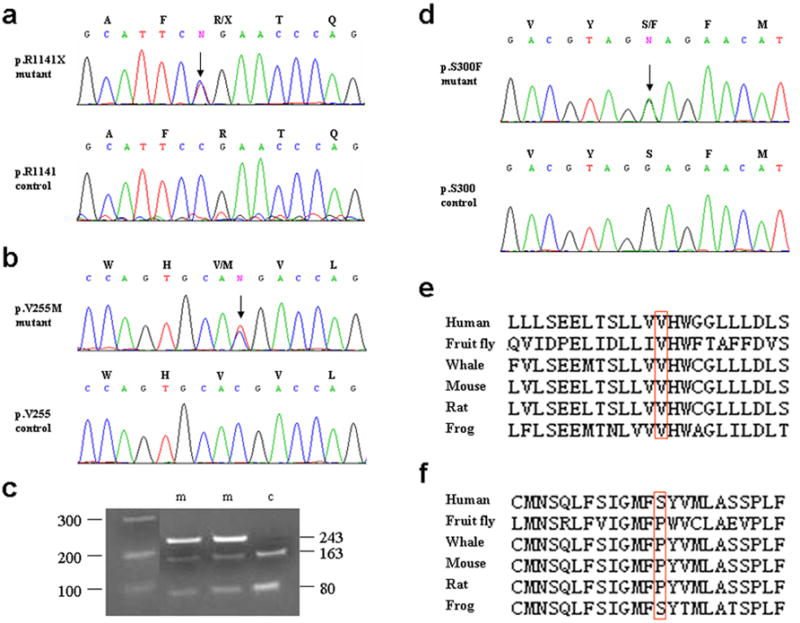
(a) Identification of the recurrent nonsense mutation p.R1141X in the ABCC6 gene. Note the heterozygous C→T transition substitution at nucleotide position 3421 (arrow). (b, d) Identification of missense mutations p.V255M and p.S300F in the GGCX gene. The sequences shown are in the complementary strand. (c) The presence of the p.V255M mutation in the control population was examined by restriction enzyme digestion with Apal I. Note that the mutation abolishes the restriction site, resulting in the 243 bp mutant allele, in addition to 163 and 80 bp fragments. In control individuals the 243 bp PCR product is digested to 163 and 80 bp fragments. (e, f) Evolutionary conservation of the amino acids V255 (panel e) and S300 panel (panel f) in the γ-glutamyl carboxylase protein.
Considering the clinical association of the PXE-like cutaneous features with coagulation disorder in this family, we also sequenced the GGCX and VKORC1 genes. The results demonstrated the presence of two missense mutations in GGCX. First, a single-base transition mutation (c.791G→A) resulting in substitution of a valine by methionine at position 255 (p.V255M) of the γ-glutamyl carboxylase enzyme was detected (Fig. 3b). This mutation was not present in 100 control alleles by restriction enzyme digestion and/or by direct nucleotide sequencing (Fig. 3c). Secondly, a single nucleotide substitution (c.927C→T) resulting in substitution of a serine by phenylalanine in position 300 (p.S300F) was detected (Fig. 3d). Direct sequencing of 150 control alleles detected the S300 codon only, suggesting that this missense substitution is not a frequent polymorphism. Determination of the evolutionary sequence conservation of GGCX in different species revealed that the amino acid corresponding to V255 is fully conserved (Fig. 3e). In contrast, the serine at position 300 was less well conserved, several species depicting proline in this position, although the surrounding amino acid sequences were highly conserved. For example, the sequence homology between the human and mouse 23-amino acid sequence segment surrounding the S300 was 95.7 percent (Fig. 3f). Sequencing of the VKORC1 gene did not disclose any pathogenic mutations.
Genotype/phenotype correlations
A correlation of the clinical findings with the genotypes revealed that the proband and her sister were compound heterozygotes for the two GGCX missense mutations, potentially explaining their hematologic findings (Brenner et al., 1998; Rost et al., 2004, 2006; Spronk et al., 2000). In contrast, the proband’s father, brother, her mother, and the mother’s twin sister were heterozygous for one of the GGCX mutations only, designating them as carriers without clinical hematologic findings (Fig. 1g). The latter individuals were also carriers of the ABCC6 nonsense mutation p.R1141X. Specifically, the mother and her twin sister were heterozygous for the GGCX missense mutation p.V255M and the ABCC6 nonsense mutation p.R1141X, suggesting digenic inheritance of their cutaneous findings. However, the proband’s younger brother and father were heterozygous carriers of the p.S300F mutation in the GGCX gene while they also carried the p.R1141X mutation in the ABCC6 gene; they did not display any signs of cutaneous findings or hematologic disorder.
Assay of γ-glutamyl carboxylase activity
Previous studies have clearly demonstrated that the p.R1141X mutation in the ABCC6 gene in heterozygous carriers does not cause PXE (Pfendner et al., 2007; Wegman et al., 2005). Similarly, combined coagulation factor deficiency is an autosomal recessive disease. The proband and her sister showed milder hematological defects than previously-reported in patients with combined deficiency of vitamin K-dependent coagulation factors, which we postulated might be due to the type of missense mutations. We ascertained, therefore, a hypothesis that the missense mutations disclosed in the GGCX gene in this family may not result in complete loss of γ-carboxylase activity, therefore explaining the milder hematologic findings in these compound heterozygous individuals. Consequently, we measured the γ-glutamyl carboxylase activity of recombinant protein expressed from the wild-type as well as from mutant alleles harboring either the p.V255M or the p.S300F mutation.
The γ-glutamyl carboxylase activity was measured by using a small peptide containing the Phe-Leu-Glu-Glu-Leu sequence as substrate, in the absence of Factor X propeptide. The assay, which utilizes incorporation of radioactive [14C]-CO2 to the substrate, revealed clearly measurable activity with the wild-type (WT) enzyme (Table 2). In contrast, the mutant protein with the p.V255M mutation showed activity which was only about 5% of that noted with the WT protein, and even more strikingly, essentially no activity (<1%) was detected with the protein harboring the p.S300F mutation (Table 2). To further examine the consequences of the mutations on the γ-glutamyl carboxylase activity, additional measurements were performed with the same peptide substrate but also with a Factor X propeptide present in reaction. This propeptide serves as the primary binding site for the carboxylase in Factor X and is conserved in vitamin K-dependent coagulation factors (Berkner, 2005). In the presence of the propeptide, the activity of the WT enzyme increased by about 3-fold as compared to that in the absence of propeptide, while the protein with p.S300F mutation continued to show low, ~3% of the WT, activity (Table 2). In contrast, the activity of the protein harboring the p.V255M mutation showed little difference (~90%) from the WT enzyme (Table 2). These observations clearly suggest that the mutations detected in the GGCX gene in this family are associated with reduced γ-carboxylase activity, but the reduced activity may result from different mechanisms with respect to enzyme-substrate interactions.
Table 2.
γ-Glutamyl carboxylase activities of wild-type and mutant proteins in vitro1
| Variant | Carboxylase activity |
|||
|---|---|---|---|---|
| Without Propeptide | With Propeptide | |||
| pmol/hr | Percent2 | pmol/hr | Percent2 | |
| Wild-type | 3,649 | 100 | 10,379 | 100 |
| S300F | 28 | <1 | 283 | 3 |
| S300F | 32 | <1 | 301 | 3 |
| V255M | 211 | 5 | 9,427 | 91 |
| V255M | 156 | 4 | 8,994 | 87 |
| Mock | 16 | 0 | 19 | 0 |
Solubilized microsomes from insect cells mock-infected or infected with baculovirus containing GGCX cDNA variants were assayed for expressed enzyme activity in the presence or absence of Factor X propeptide. Two independent viruses of each mutant were analyzed. The microsomes were also assayed by a quantitative Western analysis, and the activities shown are normalized for equivalent amounts of carboxylase protein.
The percent value was determined after subtracting the background observed in the mock-infected sample, the wild-type activity being 100 percent.
Reduced γ-glutamyl carboxylation of matrix gla protein in the skin and plasma
To examine the consequences of reduced γ-glutamyl carboxylase activity as a result of GGCX mutations, we next performed immunohistochemistry of skin biopsies with antibodies which distinguish carboxylated and under-carboxylated forms of matrix gla protein (cMGP and ucMGP, respectively), a substrate for the carboxylase activity (Schurgers et al., 2005). Staining of skin from the proband (III-3), her sister (III-1) or her aunt (II-3) revealed an abundance of MGP in the area of mineralization in the mid dermis, however, this protein was predominantly in the under-carboxylated form (ucMGP; Fig. 4a–c). In contrast, specimens from normal unrelated control individuals revealed very little, if any, of ucMGP in the corresponding areas of dermis (Fig. 4d). Low levels of cMGP both in patients’ skin (Fig. 4e–g) and in control skin (Fig. 4h) were present. Thus, the ratio of uc/cMGP is markedly higher in the patients’ skin, apparently reflecting reduced γ-glutamyl carboxylase activity. Immunostaining for two other proteins, fetuin-A and osteonectin, involved in the mineralization processes (Davies et al., 2006; Jiang et al., 2007), was also performed (Fig. 4). The presence of both these proteins was noted in the patients’ skin corresponding to the general areas of mineralization (Fig. 4; i–k, m–o). In contrast, no staining was observed in normal skin (Fig. 4; l, p).
Figure 4. Immunohistochemistry of skin in patients with PXE-like cutaneous lesions with mineralization (Patients III-1, III-3 and II-3) as well as in normal skin (control).
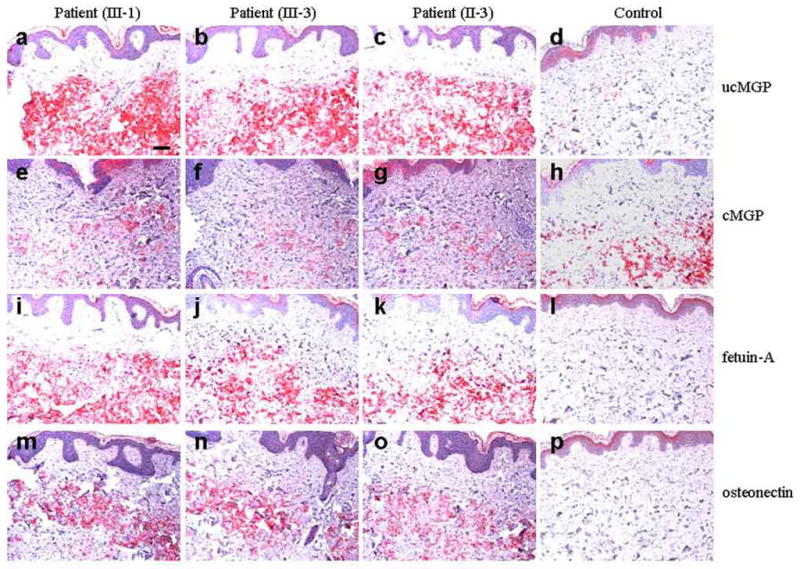
Monoclonal antibodies recognizing the under-carboxylated (ucMGP) and carboxylated (cMGP) forms of matrix gla protein, as well as polyclonal antibodies for fetuin-A and osteonectin were utilized. The secondary antibodies, biotin-conjugated anti-IgG, were recognized by avidin-alkaline phosphatase conjugates and visualized by incubation with an alkaline phosphatase substrate yielding red color. Scale bar, 0.1 mm.
In addition to tissue analysis, we utilized a competitive ELISA to quantitatively determine the levels of total ucMGP in plasma, based on an antibody that recognizes both the phosphorylated and unphosphorylated forms of ucMGP. The results revealed that the father (II-1) and brother (III-2) of the proband in this family had values that were at the lower end of those in five unrelated controls (mean ± S.D., 3,902 ± 612 pM) (Fig. 5a). All the other members of the family showed reduced values, apparently reflecting deposition of ucMGP in the mineralized tissues. Assay of non-phosphorylated forms of ser-ucMGP, which display free serine residues recognized by a specific antibody, revealed that the proband (III-3) and her sister (III-1) had values far exceeding those of the control group (432.0 pM; n=100, CV 6%), while the values in plasma from other members of the family were low (Fig. 5b). These findings, together with similar assays of ser-cMGP in the patients’ plasma, showed a markedly increased ratio of ser-ucMGP/ser-cMGP in the proband and her sister (2.0 and 1.9, respectively), while the values of the rest of the family were within the normal range (mean 0.32) (Fig. 5c). Thus, the proband and her sister clearly show an abundance of undercarboxylated and non-phosphorylated species of MGP. Since these forms of MGP are inactive, unable to prevent aberrant mineralization, these findings explain the tissue mineralization shown in the skin by specific histopathologic stains (see above).
Figure 5. Quantitation of different forms of MGP in plasma by ELISA.

(a) Total, both phosphorylated and non-phosphorylated forms of under-carboxylated MGP (total ucMGP). (b) Assay of non-phosphorylated, under-carboxylated forms of MGP (ser-ucMGP). (c) The ratio of ser-ucMGP/ser-cMGP. The dashed lines in (a) and (b) represent the mean of control specimens and the coefficient of variation (CV) is indicated by the stippled areas (CV=15.7% in a, n=5; CV=6.0% in b, n=100).
DISCUSSION
In this study, we have detailed a family with PXE-like cutaneous features, in association of combined coagulation factor disorder. Specifically, the proband’s mother and her maternal aunt presented with skin manifestations typical of PXE, such as yellowish papules and/or plaques with dot-like depressions characterized by mineralization of elastic fibers in the reticular and mid-dermis as viewed by light microscopy. The proband herself and her sister were more severely affected and they have significant sagging of the skin with reduced elasticity and resilience and loss of recoil. Histopathology of the skin from the proband and her sister revealed distinct mineralization of elastotic structures in the mid dermis, as visualized with von Kossa and Alizarin red stains. These clinical and histopathologic findings in the skin are consistent with PXE (Neldner and Struk, 2002; Ringpfeil et al., 2001; Uitto et al., 2007).
A second phenotypic feature in this family is deficiency of vitamin K-dependent coagulation factors, particularly Factor X, which are synthesized in the liver and depend on vitamin K for their function (Berkner, 2005). Combined deficiency of the vitamin K-dependent factors (VKCFD) is an autosomal recessive disorder, caused either by mutations in the GGCX gene (type I: VKCFD1 – OMIM no. 277450) on chromosome 2p12, or in the VKORC1 gene (type II: VKCFD2 – OMIM no. 607473) on chromosome 16p11.2. These genes encode a vitamin K-dependent carboxylase and vitamin K 2,3 epoxide reductase, respectively (Li et al., 2004; Rost et al., 2004a, b; Wu et al., 1991). Both enzymes are essential for post-translational γ-glutamyl carboxylation of clotting factors, enabling them to attach to the phospholipid bilayer of membranes as an essential prerequisite for blood coagulation (Zhang and Ginsburg, 2004).
In order to elucidate the molecular pathogenesis of these phenotypes in this family, we initially sequenced the ABCC6 gene in consideration of the presence of PXE-like cutaneous findings. The recurrent mutation p.R1141X was found in the DNA from the father, mother, the maternal aunt and the younger brother, but not in the proband herself or her sister. In addition, no other ABCC6 mutations could be disclosed in this family. Considering the clinical finding of coagulation disorder of the proband and her sister, we also sequenced the GGCX and VKORC1 genes, in which mutations cause the type I and type II vitamin K-dependent coagulation factor deficiency disorders, respectively. Sequencing of the GGCX gene revealed two distinct missense mutations in this family, and specifically, mutations p.V255M in exon 7 and p.S300F in exon 8 of the GGCX gene were identified. The proband and her sister were compound heterozygotes for the two GGCX missense mutations, while the proband’s father, brother, her mother and maternal aunt were heterozygous for one of the GGCX mutations only (Fig. 1g). Sequencing of the whole coding region of VKORC1 gene did not reveal any pathogenic mutation.
Considering the genotype/phenotype correlations, the proband and her sister were compound heterozygotes for both GGCX missense mutations, potentially explaining their hematologic findings. In addition, their skin findings resemble those recently reported by Vanakker et al., 2007, in which 6 patients were demonstrated to have both coagulation factor deficiency and PXE-like skin changes due to mutations in the GGCX gene. In contrast, the proband’s father, brother, her mother and maternal aunt were heterozygous for one of the GGCX mutations only, designating them as carriers without clinical hematologic findings. The latter individuals were also carriers of the ABCC6 nonsense mutation p.R1141X. It should be noted that the mother and her twin sister were heterozygous for one of the GGCX missense mutation p.V255M and one ABCC6 nonsense mutation p.R1141X, suggesting digenic inheritance of their cutaneous findings. The occurrence of digenic inheritance, although rare, is well established (see e.g., Gao et al., 2007; Gropman and Adams, 2007; Jin et al., 2008; Nozu et al., 2008). The chance of a combination of mutations in the ABCC6 and GGCX genes is difficult to calculate, since the precise carrier frequency of the mutations in these genes is not known. It should be noted, however, that in a cohort of >300 families with PXE analyzed in our laboratory for mutations in the ABCC6 gene, five individuals were heterozygous carriers of a mutation (p.R1141X) in one ABCC6 allele only, yet none of them carried a mutation in the GGCX gene (unpublished). Thus, the combination of mutations reported in this study in two different genes, yet resulting in PXE-like phenotype, is rare. Finally, a recent study has suggested that heterozygosity for a single mutation in the ABCC6 gene may closely mimic PXE, particularly displaying ophthalmologic findings (Martin et al., 2008).
In order to investigate the functional consequences of the carboxylase mutants, WT and mutant proteins were expressed in insect cells which do not contain endogenous carboxylase activity but synthesize active enzyme from exogenously introduced cDNA. We tested for carboxylase activity in an assay system that measures [14C]-CO2 incorporation into a Glu-containing small peptide substrate in the presence or absence of the Factor X propeptide. This propeptide regulates the activity of carboxylases by tethering the vitamin K-dependent proteins to the enzyme and by activating the carboxylase (Berkner, 2005). In the absence of the propeptide, both mutant carboxylases showed markedly reduced enzyme activity, less than 5% of the WT activity. However, addition of the propeptide to the assay system restored the activity of the p.V255M mutant to the WT level, while the activity of the p.S300F remained low, ~3 percent of the WT. These in vitro findings clearly suggest that the mutant enzymes may be functionally deficient in vivo, but the mechanisms of inhibition may be distinct, and may involve differential propeptide as well as substrate binding.
Until now, 11 families comprising a total of 20 patients with PXE-like skin changes and/or vitamin K-dependent coagulation factor deficiency, with 16 distinct mutations in the GGCX gene, have been reported (Table 3). The functions of the residues that are mutated are for the most part unknown as only two mutants (p.L394R and p.W501S) have been previously characterized to sufficiently reveal function. Those analyses showed that Leu394 and Trp501 are part of the carboxylase and propeptide-binding sites, respectively (Lin et al., 2002; Mutucumarana et al., 2000, 2003). Both mutations therefore influence the affinity of γ-glutamyl carboxylase for the vitamin K-dependent protein substrate. To carry out the carboxylation, the enzyme must also bind three other co-substrates, which are vitamin K, CO2 and O2, but functional residues that interact with these substrates are unknown (Berkner, 2005). Thus, the carboxylation reaction is complex and only a few functional residues have been identified. It will be of interest to determine the precise mechanisms by which the p.V255M and p.S300F mutants analyzed in the present study impair carboxylation. Nevertheless, these mutants are the first ones specific to PXE-like cutaneous phenotype to have been tested for in vitro carboxylase activity. The reduced activity in p.V255M and p.S300F indicate the importance of Val255 and Ser300 to carboxylation and can explain the coagulation disorder and PXE-like cutaneous phenotype detected in the proband and her sister.
Table 3.
Distinct mutations reported in the GGCX gene in patients with Pseudoxanthoma elasticum-like skin findings and/or vitamin K-dependent coagulation factor deficiency (VKCFD)1
| Family (No)2 | Age (yrs) | Mutations | Phenotype | Reference | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Allele 1 | Exon | Allele 2 | Exon | PXE-like | VKCFD | |||||
| I (4) | NB | p.L394R | c.1209T>G | 9 | p.L394R | c.1209T>G | 9 | NR | + | Brenner et al., 1998 |
| II (1) | NB | p.W501S | c.1530G>C | 11 | p.W501S | c.1530G>C | 11 | NR | + | Spronk et al., 2000 |
| III (1) | 1 | p.R485P | c.1454G>C | 11 | Loss of exon 3 | Intron 2G>T | 3 | NR | + | Rost et al., 2004 |
| IV (1) | 38 | p.R485P | c.1454G>C | 11 | p.H404P | c.1239A>C | 9 | NR | + | Rost et al., 2006 |
| V (1) | 2 | p.W157R | c.497T>C | 4 | p.T591K | c.1800C>A | 13 | NR | + | Darghouth et al., 2006 |
| VI (1) | 46 | p.W493S | c.1506G>C | 114 | p.W493S | c.1506G>C | 114 | + | + | Vanakker et al., 2007 |
| VII (1) | 67 | p.R476C | c.1454C>T | 10 | − | − | − | + | + | Vanakker et al., 2007 |
| VIII (1) | 32 | p.R476H | c.1455G>A | 10 | − | − | − | + | + | Vanakker et al., 2007 |
| IX (2) | 46, 44 | p.Q374X | c.1148C>T3 | 8 | p.G537Y | c.1339G>T5 | 12 | + | + | Vanakker et al., 2007 |
| X (1) | 40 | p.F299S | c.924T>C | 8 | p.G558R | c.1700G>A | 12 | + | + | Vanakker et al., 2007 |
| XI (2) | 16, 19 | p.V255M | c.791G>A | 7 | p.S300F | c.927C>T | 8 | + | + | Present study |
| XI (2) | 40 | p.V255M | c.791G>A | 7 | − | − | − | + | ND | Present study |
| XI (2) | 17, 39 | p.S300F | c.927C>T | 8 | − | − | − | ND | ND | Present study |
PXE, Pseudoxanthoma elasticum; NB, new-born; NR, not reported; ND, not detected.
The numbers in parenthesis indicate the number of individuals depicting the combination of mutations in the family.
This mutation was reported as c.1149C>T(Vanakker et al., 2007).
This mutation was reported to reside in exon 12 (Vanakker et al., 2007).
In the reference sequence (NM000821), this nucleotide resides in exon 10 and encodes a tryptophan.
Less apparent, and perhaps more complex, are the mechanisms leading to cutaneous PXE-like phenotypes in this family. Mutations in the ABCC6 gene have been shown to cause PXE, an autosomal recessive disorder (Ringpfeil et al., 2006), but heterozygous carriers of mutations, such as p.R1141X detected in the proband’s mother and aunt, are asymptomatic (Pfendner et al., 2007; Wegman et al., 2005). At the same time, the proband and her sister did not harbor mutations in ABCC6, yet their skin lesions clearly showed profound mineralization. These findings raise the hypothesis that reduced carboxylase activity results in under-carboxylation of not only the coagulation factors but other proteins, such as MGP, as well. MGP plays a critical role in preventing aberrant mineralization but in order to be activated, this protein has to be fully carboxylated while under-carboxylated forms of the protein are inactive and can not prevent aberrant mineralization (Price et al., 1998; Shearer, 2000). In fact, skin biopsies from the proband and her sister depicted preponderance of under-carboxylated MGP, when examined by immunohistochemistry using specific antibodies that distinguish the fully carboxylated and under-carboxylated forms of the protein (Schurgers et al., 2005). Furthermore, assay of total ucMGP in plasma, which has been recently suggested to serve as a biomarker of cardiovascular calcification (Cranenburg et al., 2008), was reduced in patients with skin findings, apparently reflecting tissue mineralization.
An intriguing observation in our family was the presence of PXE-like cutaneous features, with profound mineralization, in the proband’s mother and aunt. These two individuals were heterozygous carriers of p.R1141X mutation in ABCC6 and p.V255M in GGCX. Since heterozygous carriers of p.R1141X in ABCC6 alone do not manifest PXE and GGCX mutations with respect to coagulation disorder are recessive, these findings suggest that the skin phenotype in these two individuals may be due to digenic inheritance. In this case, haploinsufficiency of the carboxylase activity and reduced ABCC6 functions could be complementary or synergistic. The reasons for the fact that the proband’s father and her brother were heterozygous carriers of mutations in the ABCC6 gene (p.R1141X) and the GGCX gene (p.S300F) yet did not display any cutaneous findings are not clear. Specifically, while both GGCX mutations resulted in reduced enzyme activity, the reduction in case of protein harboring the p.S300F mutation was more pronounced than that of p.V255M. In this context, it should be noted that the substrate employed in the carboxylase assay is a pentapeptide, Phe-Leu-Glu-Glu-Leu, and it is possible that the activity measurements if done on full-length MGP as substrate would show differential activity with these two mutant enzymes. Finally, these two individuals have not been examined by an ophthalmologist nor has a skin biopsy been performed, due to their unwillingness to participate in further studies. Consequently, the possibility that the father and the brother of the proband may have subclinical, PXE-associated features can not be rigorously excluded.
The findings of this study have potential implications regarding the pathomechanisms of PXE, a multisystem disorder with extensive tissue mineralization. Our results suggest that under-carboxylation of MGP plays a critical role in aberrant mineralization of tissues in PXE, a proposition that has been supported by recent electron microscopic examination of skin of patients with PXE (Gheduzzi et al., 2007) and analysis of the carboxylation status of MGP in Abcc6−/− null mice which recapitulate the features of PXE (Jiang et al., 2007; Li et al., 2007). How the absence of ABCC6, a putative transmembrane transporter expressed primarily in the liver, contributes to reduced γ-glutamyl carboxylase of MGP remains to be explored, but one possibility revolves around cellular transport and redistribution of vitamin K, an obligatory co-factor of γ-glutamyl carboxylase (Berkner, 2005; Schurgers et al., 2007; Borst et al., 2008; Li et al., 2008). Such notion would have broader implications for aberrant tissue mineralization including pathologic processes, such as arteriosclerosis, beyond PXE. Finally, the ratio of ser-ucMGP/ser-cMGP was markedly elevated in the proband and her sister, individuals with most severe skin manifestations, including profound mineralization of the dermis. This ratio may reflect vitamin K status of the individual patient, and consequently, one could ascertain the possibility that treatment with appropriate vitamin K preparations could ameliorate or even reverse tissue mineralization in PXE, a currently intractable disorder.
MATERIAL AND METHODS
Patients
The patients were clinically evaluated at Washington University School of Medicine (St. Louis Children’s Hospital and the Center for Advanced Medicine) and at St. Louis University. Informed consent was obtained from all patients. Skin biopsies were evaluated with light microscopy using hematoxylin-eosin, von Kossa (calcium phosphate), and Alizarin red (calcium) stains. Coagulation assays were performed on EDTA blood samples and the activities of Factors II, VII, VIII, IX, and X were measured in one-stage clotting assays.
Molecular analysis
Genomic DNA was isolated from peripheral blood samples (QIAamp Blood Maxi Kit; Qiagen Inc., Valencia, CA). PCR was performed using 1.5 U Taq polymerase (Qiagen) mixed with 5 U Optimase Taq polymerase (Transgenomic, Gaithersburg, MD) and Q buffer (Qiagen), according to the manufacturers’ instructions. PCR reactions contained 200 ng DNA as template and 20 ng of each primer in a final volume of 50 μl.
The GGCX gene was analyzed using primers and PCR conditions as previously described (Oldenburg et al., 2000). The ABCC6 sequences were analyzed using PCR primers as described previously (Pfendner et al., 2007). For the detection of the deletion of exons 23–29, the primers described by Le Saux et al. (2001) were used. The VKORC1 coding region was analyzed using primers described previously (Darghouth et al., 2006).
The entire coding region and intron/exon boundaries of GGCX, ABCC6 and VKORC1 were analyzed with direct sequencing using an Applied Biosystems 3730 Sequencer (Applied Biosystems, Foster City, CA). Within the exon 7 of the GGCX gene, the 791G→A substitution abolished an Apal I restriction site. Digestion of the 243-bp PCR product resulted in the case of the normal allele in 163- and 80-bp fragments, while the heterozygotes showed 243-, 163- and 80-bp bands.
For GGCX sequences the nucleotide numbers are derived from cDNA (GenBank accession no. NM000821); the +1 corresponds to the −28 with respect to the translation initiation codon. In the ABCC6 gene, the +1 correspond to the A nucleotide in the ATG translation initiation codon (GenBank accession no. AF076622). The alignments of the GGCX protein sequences in different species were generated with Clustal W program.
Construction and analysis of mutant carboxylase enzymes
Generation of carboxylase cDNAs with the individual mutations p.V255M and p.S300F was accomplished using the Quick Change XL site-directed mutagenesis kit (Stratagene, La Jolla, CA) according to the manufacturer’s instructions and using the following oligonucleotide primers: p.V255M: (forward) CTGACTAGCCTGCTGGTCATGCACTGGGGTGGGCTGCTG and (reverse) CAGCAGCCCACCCCAGTGCATGACCAGCAGGCTAGTCAG; p.S300F: (forward) TTCAGCATTGGTATGTTCTTCTACGTCATGCTGGCCAGC and (reverse) GCTGGCCAGCATGACGTAGAAGAACATACCAATGCTGAA.
Briefly, a full-length GGCX cDNA was hybridized to the indicated primers and subjected to Pfu polymerase (Stratagene) extension. After 20 cycles of amplification, parental wild-type strands were digested with the methyl-sensitive restriction enzyme DpnI, and amplified mutant strands were self-ligated and used to transform DH5α E. coli. A BamHI fragment containing the full-length cDNA was isolated and subcloned into the baculovirus expression vector pBacPak9 (Invitrogen Corp., Carlsbad, CA), and the cDNA was sequenced on both strands. Baculoviruses containing each variant were generated by cotransfecting SF21 cells with the plasmids and Bsu36I-digested viral DNA (BacPAK6, Clontech, Palo Alto, CA) as before (Rishavy et al., 2004). Virus was isolated from individual plaques and screened for carboxylase expression by infecting cells, preparing lysates and performing Western analysis using an antibody against the C-terminus of the carboxylase (Berkner and McNally, 1997; Berkner and Pudota, 1998). Two independent viral isolates of each mutant were then amplified and used in large-scale infections to generate microsomes. The microsomes were solubilized (at 4 mg/ml) in 0.5% CHAPS, 0.1% phosphatidyl choline and then assayed in a quantitative Western analysis to determine the amount of carboxylase protein. Carboxylase activity was measured by incubating the solubilized microsomes (40 μg) in 160 μl of 0.8 M ammonium sulfate, 0.16% CHAPS, 0.16% phosphatidyl choline, 2.5 mM Phe-Leu-Glu-Glu-Leu peptide as substrate, 50 mM BES (pH 6.9), 2.5 mM dithiothreitol, 1.3 mM [14C]-CO2 and 200 μM vitamin K hydroquinone. Samples were assayed in duplicate and the assays were performed either in the presence or absence of 10 μM Factor X propeptide (Berkner, 2005).
Immunohistochemistry
Immunohistochemistry was performed on tissues embedded in paraffin. Sections of 5 μm were stained with moAb-cMGP (4.5 μg/ml) (monoclonal antibody towards the carboxylated forms of human MGP), moAb-ucMGP (4.5 μg/ml) (monoclonal antibody towards the noncarboxylated forms of human MGP) (Schurgers et al., 2005), goat anti-human fetuin-A (1:40; R&D Systems, Minneapolis, MN) and rabbit anti-human osteonectin (1:1000; Calbiochem, San Diego, CA), respectively. Immunostaining was performed using either biotinylated rabbit anti-mouse IgG (DAKO, Carpinteria, CA) or biotinylated rabbit anti-goat IgG (DAKO) or biotinylated swine anti-rabbit IgG (DAKO) as secondary antibody (60 minutes at room temperature), followed by incubation with avidin-linked alkaline phosphatase complex (30 minutes at room temperature; DAKO); staining was performed using the Vector® Red Alkaline Phosphatase Substrate Kit I (Vector Laboratories, Burlingame, CA). Sections were counterstained with hematoxylin and mounted with Permount (Fisher, Fair Lawn, NJ). Controls for the immunoreactions were performed by omitting the primary antibody or by incubating the sections with nonimmune sera instead of the primary antibody.
ELISA for plasma MGP
A conformation-specific monoclonal antibody (Schurgers et al., 2005) was used to construct an ELISA for detection of total ucMGP in plasma, as described elsewhere (Cranenburg et al., 2008). Two additional sandwich ELISAs were developed at VitaK BV (Maastricht, the Netherlands) to quantitatively measure the non-phosphorylated forms of both ucMGP and cMGP in plasma. In brief, a monoclonal antibody specific for non-phosphorylated-MGP (moAb serMGP) was coupled to the microtiter plate (5 μg/mL). After incubation, non-specific sites were blocked using a solution of 2% BSA in HN (Hepes/NaCl) buffer. After stringent washing, standards, controls, or experimental samples were applied, diluted in 0.2% protifar in HN buffer. The second antibody (either directed against the uncarboxylated or carboxylated conformation of MGP) was biotinylated and diluted in 0.2% protifar HN buffer. After stringent washing, streptavidin in 0.5% BSA/HN was added and staining was carried out using the substrate TMB (30 min at room temperature). The reaction was stopped with 2 N H2SO4, and the OD was measured at 450 nm.
We have received institutional approval of experiments and adhere to the Helsinki Guidelines.
Acknowledgments
The authors thank Drs. Joel Rosenbloom and Brian Carr (Jefferson Medical College) and Dr. Andras Varadi (Hungarian Academy of Sciences) for insightful discussions, Dr. Olivier Vanakker (University of Ghent) for sharing sequence information on GGCX, and Carol Kelly for assistance. This study was support by National Institutes of Health/National Institute of Arthritis and Musculoskeletal and Skin Diseases (R01AR28450 and R01AR52627 to J.U.). Dr. Jiang is recipient of a Dermatology Foundation Research Career Development Award.
Abbreviations
- PXE
pseudoxanthoma elasticum
Footnotes
Conflict of Interest
The authors state no conflict of interest.
References
- Belinsky MG, Kruh GD. MOAT-E (ARA) is a full length MRP/cMOAT subfamily transporter expressed in kidney and liver. Br J Cancer. 1999;80:1342–1349. doi: 10.1038/sj.bjc.6690527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergen AA, Plomp AS, Schuurman EJ, Terry S, Breuning M, Dauwerse H, et al. Mutations in ABCC6 cause pseudoxanthoma elasticum. Nat Genet. 2000;25:228–231. doi: 10.1038/76109. [DOI] [PubMed] [Google Scholar]
- Berkner KL, McNally BA. Purification of vitamin K-dependent carboxylase from cultured cells. Methods Enzymol. 1997;282:313–333. doi: 10.1016/s0076-6879(97)82117-9. [DOI] [PubMed] [Google Scholar]
- Berkner KL, Pudota BN. Vitamin K-dependent carboxylation of the carboxylase. Proc Natl Acad Sci USA. 1998;95:466–471. doi: 10.1073/pnas.95.2.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkner KL. The vitamin K-dependent carboxylase. Ann Rev Nutr. 2005;25:127–149. doi: 10.1146/annurev.nutr.25.050304.092713. [DOI] [PubMed] [Google Scholar]
- Borst P, van de Wetering K, Schlingemann R. Does the absence of ABCC6 (Multidrug Resistance Protein 6) in patients with pseudoxanthoma elasticum prevent the liver from providing sufficient vitamin K to the periphery? Cell Cycle. 2008;7:1575–1579. doi: 10.4161/cc.7.11.6005. [DOI] [PubMed] [Google Scholar]
- Brenner B, Sánchez-Vega B, Wu SM, Lanir N, Stafford DW, Solera J. A missense mutation in gamma-glutamyl carboxylase gene causes combined deficiency of all vitamin K-dependent blood coagulation factors. Blood. 1998;92:4554–4559. [PubMed] [Google Scholar]
- Cranenburg ECM, Vermeer C, Roos R, Boumans ML, Hackeng TM, Bouwman FG, et al. The circulating inactive form of matrix Gla protein (ucMGP) as a biomarker for cardiovascular calcification. J Vasc Res. 2008;45:427–436. doi: 10.1159/000124863. [DOI] [PubMed] [Google Scholar]
- Darghouth D, Hallgren KW, Shtofman RL, Mrad A, Gharbi Y, Maherzi A, et al. Compound heterozygosity of novel missense mutations in the gamma-glutamyl-carboxylase gene causes hereditary combined vitamin K–dependent coagulation factor deficiency. Blood. 2006;108:1925–1931. doi: 10.1182/blood-2005-12-010660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies CA, Jeziorska M, Freemont AJ, Herrick AL. Expression of osteonectin and matrix Gla protein in scleroderma patients with and without calcinosis. Rheumatology. 2006;45:1349–1355. doi: 10.1093/rheumatology/kei277. [DOI] [PubMed] [Google Scholar]
- Ebberink RH, Vermeer C. Congenital deficiency of all vitamin K-dependent blood coagulation factors due to a defective vitamin K-dependent carboxylase in Devon Rex cats. Thromb Haemost. 1992;68:521–525. [PubMed] [Google Scholar]
- Gao YQ, Danciger M, Ozgul RK, Gribanova Y, Jacobson S, Farber DB. Association of the Asn306Ser variant of the SP4 transcription factor and an intronic variant in the beta-subunit of transducin with digenic disease. Mol Vis. 2007;13:287–292. [PMC free article] [PubMed] [Google Scholar]
- Gheduzzi D, Boraldi F, Annovi G, DeVincenzi CP, Schurgers LJ, Vermeer C, et al. Matrix Gla protein is involved in elastic fiber calcification in the dermis of pseudoxanthoma elasticum patients. Lab Invest. 2007;87:998–1008. doi: 10.1038/labinvest.3700667. [DOI] [PubMed] [Google Scholar]
- Gropman AL, Adams DR. Atypical patterns of inheritance. Semin Pediatr Neurol. 2007;14:34–45. doi: 10.1016/j.spen.2006.11.007. [DOI] [PubMed] [Google Scholar]
- Jiang Q, Li Q, Uitto J. Aberrant mineralization of connective tissues in a mouse model of pseudoxanthoma elasticum: Systemic and local regulatory factors. J Invest Dermatol. 2007;127:1392–1402. doi: 10.1038/sj.jid.5700729. [DOI] [PubMed] [Google Scholar]
- Jin ZB, Mandai M, Yokota T, Higuchi K, Ohmori K, Ohtsuki F, et al. Identifying pathogenic genetic background of simplex or multiplex retinitis pigmentosa patients: A large-scale mutation screening study. J Med Genet. 2008 doi: 10.1136/jmg.2007.056416. [Epub ahead of print – PMID:18310263] [DOI] [PubMed] [Google Scholar]
- Knobloch JE, Suttie JW. Vitamin K-dependent carboxylase. Control of enzyme activity by the “propeptide” region of factor X. J Biol Chem. 1987;262:15334–15337. [PubMed] [Google Scholar]
- Le Corvaisier-Pieto C, Joly P, Thomine E, Lair G, Lauret P. Generalized pseudoxanthoma elasticum combined with vitamin K dependent clotting factors deficiency. Ann Dermatol Venereol. 1996;123:555–558. [PubMed] [Google Scholar]
- Le Saux O, Urban Z, Tschuch C, Csiszar K, Bacchelli B, Quaglino D, et al. Mutations in a gene encoding an ABC transporter cause pseudoxanthoma elasticum. Nat Genet. 2000;25:223–227. doi: 10.1038/76102. [DOI] [PubMed] [Google Scholar]
- Le Saux O, Beck K, Sachsinger C, Silvestri C, Treiber C, Göring HH, et al. A spectrum of ABCC6 mutations is responsible for pseudoxanthoma elasticum. Am J Hum Genet. 2001;69:749–764. doi: 10.1086/323704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Chang CY, Jin DY, Lin PJ, Khvorova A, Stafford DW. Identification of the gene for vitamin K epoxide reductase. Nature. 2004;427:541–544. doi: 10.1038/nature02254. [DOI] [PubMed] [Google Scholar]
- Li Q, Jiang Q, Schurgers LJ, Uitto J. Pseudoxanthoma elasticum: Reduced gamma-glutamyl carboxylation of matrix gla protein in a mouse model (Abcc6−/−) Biochem Biophys Res Commun. 2007;364:208–213. doi: 10.1016/j.bbrc.2007.09.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Jiang Q, Pfendner E, Varadi A, Uitto J. Pseudoxanthoma elasticum: Clinical phenotypes, molecular genetics and putative pathomechanisms. 2008. (submitted) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin PJ, Jin DY, Tie JK, Presnell SR, Straight DL, Stafford DW. The putative vitamin K-dependent gamma-glutamyl carboxylase internal propeptide appears to be the propeptide binding site. J Biol Chem. 2002;277:28584–28591. doi: 10.1074/jbc.M202292200. [DOI] [PubMed] [Google Scholar]
- Martin L, Maître F, Bonicel P, Daudon P, Verny C, Bonneau D, et al. Heterozygosity for a single mutation in the ABCC6 gene may closely mimic PXE: consequences of this phenotype overlap for the definition of PXE. Arch Dermatol. 2008;144:301–306. doi: 10.1001/archderm.144.3.301. [DOI] [PubMed] [Google Scholar]
- Matsuzaki Y, Nakano A, Jiang QJ, Pulkkinen L, Uitto J. Tissue-specific expression of the ABCC6 gene. J Invest Dermatol. 2005;125:900–905. doi: 10.1111/j.0022-202X.2005.23897.x. [DOI] [PubMed] [Google Scholar]
- Miksch S, Lunsden A, Guenther UP, Foernzler D, Christen-Zäch S, Daugherty C, et al. Molecular genetics of pseudoxanthoma elasticum: Type and frequency of mutations in ABCC6. Hum Mutat. 2005;26:235–248. doi: 10.1002/humu.20206. [DOI] [PubMed] [Google Scholar]
- Mutucumarana VP, Stafford DW, Stanley TB, Jin DY, Solera J, Brenner B, et al. Expression and characterization of the naturally occurring mutation L394R in human γ–glutamyl carboxylase. J Biol Chem. 2000;275:32572–32577. doi: 10.1074/jbc.M006808200. [DOI] [PubMed] [Google Scholar]
- Mutucumarana VP, Acher F, Straight DL, Jin DY, Stafford DW. A conserved region of human vitamin K-dependent carboxylase between residues 393 and 404 is important for its interaction with the glutamate substrate. J Biol Chem. 2003;278:46488–46493. doi: 10.1074/jbc.M307707200. [DOI] [PubMed] [Google Scholar]
- Neldner KH, Struk B. Pseudoxanthoma elasticum. In: Royce PM, Steinmann B, editors. Connective tissue and its heritable disorders: Molecular, genetic and medical aspects. Wiley-Liss, Inc; New York: 2002. pp. 561–583. [Google Scholar]
- Nozu K, Inagaki T, Fu XJ, Nozu Y, Kaito H, Kanda K, et al. Molecular analysis of digenic inheritance in Bartter syndrome with sensorineural deafness. J Med Genet. 2008;45:182–186. doi: 10.1136/jmg.2007.052944. [DOI] [PubMed] [Google Scholar]
- Oldenburg J, von Brederlow B, Fregin A, Rost S, Wolz W, Eberl W, et al. Congenital deficiency of vitamin K dependent coagulation factors in two families presents as a genetic defect of the vitamin K-epoxidereductase- complex. Thromb Haemost. 2000;84:937–941. [PubMed] [Google Scholar]
- Pfendner EG, Vanakker O, Terry SF, Vourthis S, McAndrew P, McLain MR, et al. Mutation detection in the ABCC6 gene and genotype-phenotype analysis in a large international case series affected by pseudoxanthoma elasticum. J Med Genet. 2007;44:621–628. doi: 10.1136/jmg.2007.051094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price PA, Faus SA, Williamson MK. Warfarin causes rapid calcification of the elastic lamellae in rat arteries and heart valves. Arterioscl Throm Vasc Biol. 1998;18:1400–1407. doi: 10.1161/01.atv.18.9.1400. [DOI] [PubMed] [Google Scholar]
- Ringpfeil F, Lebwohl MG, Christiano AM, Uitto J. Pseudoxanthoma elasticum: Mutations in the MRP6 gene encoding a transmembrane ATP-binding cassette (ABC) transporter. Proc Natl Acad Sci USA. 2000;97:6001–6006. doi: 10.1073/pnas.100041297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringpfeil F, Pulkkinen L, Uitto J. Molecular genetics of pseudoxanthoma elasticum. Exp Dermatol. 2001;10:221–228. doi: 10.1034/j.1600-0625.2001.100401.x. [DOI] [PubMed] [Google Scholar]
- Ringpfeil F, McGuigan K, Fuchsel L, Kozic H, Larralde M, Lebwohl M, et al. Pseudoxanthoma elasticum is a recessive disease characterized by compound heterozygosity. J Invest Dermatol. 2006;126:782–786. doi: 10.1038/sj.jid.5700115. [DOI] [PubMed] [Google Scholar]
- Rishavy MA, Pudota BN, Hallgren KW, Qian W, Yakubenko AV, Song JH, et al. A new model for vitamin K-dependent carboxylation: the catalytic base that deprotonates vitamin K hydroquinone is not Cys but an activated amine. Proc Natl Acad Sci USA. 2004;101:13732–13737. doi: 10.1073/pnas.0404989101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rongioletti F, Bertamino R, Rebora A. Generalized pseudoxanthoma elasticum with deficiency of vitamin K-dependent clotting factors. J Am Acad Dermatol. 1989;21:1150–1152. doi: 10.1016/s0190-9622(89)70320-0. [DOI] [PubMed] [Google Scholar]
- Rost S, Fregin A, Loch D, Compes M, Müller CR, Oldenburg J. Compound heterozygous mutations in the gamma-glutamyl carboxylase gene cause combined deficiency of all vitamin K-dependent blood coagulation factors. Br J Haematol. 2004;126:546–549. doi: 10.1111/j.1365-2141.2004.05071.x. [DOI] [PubMed] [Google Scholar]
- Rost S, Fregin A, Ivaskevicius V, Conzelmann E, Hörtnagel K, Pelz HJ, et al. Mutations in VKORC1 cause warfarin resistance and multiple coagulation factor deficiency type 2. Nature. 2004b;427:537–541. doi: 10.1038/nature02214. [DOI] [PubMed] [Google Scholar]
- Rost S, Geisen C, Fregin A, Seifried E, Müller CR, Oldenburg J. Founder mutation Arg485Pro led to recurrent compound heterozygous GGCX genotypes in two German patients with VKCFD type 1. Blood Coagulation and Fibrinolysis. 2006;17:503–507. doi: 10.1097/01.mbc.0000240927.88177.d1. [DOI] [PubMed] [Google Scholar]
- Scheffer GL, Hu X, Pijnenborg AC, Wijnholds J, Bergen AA, Scheper RJ. MRP6 (ABCC6) detection in normal human tissues and tumors. Lab Invest. 2002;82:515–518. doi: 10.1038/labinvest.3780444. [DOI] [PubMed] [Google Scholar]
- Schurgers LJ, Teunissen KJ, Knapen MH, Kwaijtaal M, van Diest R, Appels A, et al. Novel conformation specific antibodies against matrix γ-carboxyglutamic acid (Gla) protein: Undercarboxylated matrix Gla protein as marker for vascular calcification. Arterioscler Thromb Vasc Biol. 2005;25:1629–1633. doi: 10.1161/01.ATV.0000173313.46222.43. [DOI] [PubMed] [Google Scholar]
- Schurgers LJ, Spronk HM, Soute BA, Schiffers PM, DeMey JG, Vermeer C. Regression of warfarin-induced medial elastocalcinosis by high intake of vitamin K in rats. Blood. 2007;109:2823–2831. doi: 10.1182/blood-2006-07-035345. [DOI] [PubMed] [Google Scholar]
- Shearer MJ. Role of vitamin K and Gla proteins in the pathophysiology of osteoporosis and vascular calcification. Curr Opin Clin Nutr Mtab Care. 2000;3:433–438. doi: 10.1097/00075197-200011000-00004. [DOI] [PubMed] [Google Scholar]
- Spronk HM, Farahm RA, Buchanan GR, Vermeer C, Soute BA. Novel mutation in the gamma-glutamyl carboxylase gene resulting in congenital combined deficiency of all vitamin K-dependent blood coagulation factors. Blood. 2000;96:3650–3652. [PubMed] [Google Scholar]
- Uitto J, Ringpfeil F. Heritable diseases affecting the elastic fibers: cutis laxa, pseudoxanthoma elasticum and related disorders. In: Rimoin DL, Connor JM, Pyeritz RE, Korf BR, editors. Principles and Practice of Genetics. 5. Churchill Livingstone; Philadelphia: 2007. pp. 3647–3670. [Google Scholar]
- Vanakker OM, Martin L, Gheduzzi D, Leroy BP, Loeys BL, Guerci VI, et al. Pseudoxanthoma elasticum-like phenotype with cutis laxa and multiple coagulation factor deficiency represents a separate genetic entity. J Invest Dermatol. 2007;27:581–587. doi: 10.1038/sj.jid.5700610. [DOI] [PubMed] [Google Scholar]
- Wegman JJ, Hu X, Tan H, Bergen AA, Trip MD, Kastelein JJ, et al. Patients with premature coronary artery disease who carry the ABCC6 R1141X mutation have no pseudoxanthoma elasticum phenotype. Int J Cardiol. 2005;100:389–393. doi: 10.1016/j.ijcard.2004.07.012. [DOI] [PubMed] [Google Scholar]
- Wu SM, Cheung WF, Frazier D, Stafford DW. Cloning and expression of the cDNA for human γ-glutamyl carboxylase. Science. 1991;54:1634–1636. doi: 10.1126/science.1749935. [DOI] [PubMed] [Google Scholar]
- Wu SM, Mutucumarana VP, Geromanos S, Stafford DW. The propeptide binding site of the bovine γ–glutamyl carboxylase. J Biol Chem. 1997;272:11718–11722. doi: 10.1074/jbc.272.18.11718. [DOI] [PubMed] [Google Scholar]
- Yamada M, Kuliopulos A, Nelson NP, Roth DA, Furie B, Furie BC, et al. Localization of the factor IX propeptide binding site on recombinant vitamin K dependent carboxylase using benzoylphenylalanine photoaffinity peptide activators. Biochemistry. 1995;34:481–489. doi: 10.1021/bi00002a012. [DOI] [PubMed] [Google Scholar]
- Zhang B, Ginsburg D. Familial multiple coagulation factor deficiencies: new biologic insight from rare genetic bleeding disorders. J Thromb Haemost. 2004;2:1564–1572. doi: 10.1111/j.1538-7836.2004.00857.x. [DOI] [PubMed] [Google Scholar]