

Abstract
Hearing loss has been attributed to many factors, including degeneration of sensory neurons in the auditory pathway and demyelination along the cochlear nerve. Fibroblast growth factors (FGFs), which signal through four receptors (Fgfrs), are produced by auditory neurons and play a key role in embryonic development of the cochlea and in neuroprotection against sound-induced injury. However, the role of FGF signaling in the maintenance of normal auditory function in adult and aging mice remains to be elucidated. Furthermore, the contribution of glial cells, which myelinate the cochlear nerves, is poorly understood. To address these questions, we generated transgenic mice in which Fgfr1 and Fgfr2 were specifically inactivated in Schwann cells and oligodendrocytes but not in neurons. Adult mutant mice exhibited late onset of hearing impairment, which progressed markedly with age. The hearing impairment was accompanied by significant loss of myelinated spiral ganglion neurons. The pathology extended into the cochlear nucleus, without apparent loss of myelin or of the deletion-bearing glial cells themselves. This suggests that perturbation of FGF receptor-mediated glial function leads to the attenuation of glial support of neurons, leading to their loss and impairment of auditory functions. Thus, FGF/FGF receptor signaling provides a potentially novel mechanism of maintaining reciprocal interactions between neurons and glia in adult and aging animals. Dysfunction of glial cells and FGF receptor signaling may therefore be implicated in neurodegenerative hearing loss associated with normal aging.
Keywords: oligodendrocytes, Schwann cells, myelin, aging, hearing loss
The cochlear nerve conducts sound from sensory organs in the inner ear, through the spiral ganglion, to the cochlear nucleus in the brainstem, and finally to the brain. Age-related hearing loss, or presbycusis, affects approximately half of the population over the age of 65 years (Ohlemiller, 2004) and is linked to the degeneration of sensory neurons along the cochlear nerve (Felder and Schrott-Fischer, 1995; Adams and Schulte, 1997). Demyelination in the cochlear nerve is also linked to hearing loss, because myelination is crucial to the rapid conduction of auditory signals on which acoustic coding depends (Ochikubo et al., 1992; Naito et al., 1999; Ito et al., 2004). The cochlear nerve is myelinated by Schwann cells in the peripheral nervous system (PNS) and by oligodendrocytes in the central nervous system (CNS). In addition, cell bodies of type I but not type II spiral ganglion neurons (SGN) are also myelinated by spiral ganglion Schwann cells. The unmyelinated terminal processes of SGN, which synapse with hair cells, are ensheathed by “supporting cells” in the organ of Corti.
Fibroblast growth factors (FGFs) are a family of 22 members (Itoh and Ornitz, 2004) that signal through four transmembrane tyrosine kinase receptors (Fgfrs1–Fgfr4; Zhang et al., 2006). FGF-1 and FGF-2 are expressed by SGN and neurons in the cochlear nucleus (Luo et al., 1993; Pirvola et al., 1995; Riedel et al., 1995; Silva et al., 2005). Schwann cells and oligodendrocytes express Fgfr1, Fgfr2, and Fgfr3 (Bansal et al., 1996, 2003; Grothe and Nikkah, 2001). FGF signaling is known to regulate multiple functions of oligodendrocytes and Schwann cells, including proliferation of spiral ganglion Schwann cells and forebrain oligodendrocytes, differentiation and process elongation of oligodendrocytes, regeneration of peripheral nerves following injury, and recovery following demyelination of the corpus callosum in animal models (Davis and Stroobant, 1990; Grothe and Nikkhah, 2001; Hansen et al., 2001; Bansal, 2002; Armstrong et al., 2002; Oh et al., 2003; Fortin et al., 2005; Murtie et al., 2005). We have recently shown that Fgfr1 and Fgfr2 signaling in nonmyelinating Schwann cells plays a key role in maintaining the structure and function of peripheral sensory c-fibers (Furusho et al., 2009). Furthermore, gene knockout studies have demonstrated a critical role of FGFs and their receptors in the morphogenesis of the inner ear. For example, conditional inactivation of Fgfr1 in the inner ear epithelium results in reduced sensory epithelial precursor cell proliferation and widespread auditory hair cell death (Pirvola et al., 2002); targeted deletion of the Fgfr2(IIIb) isoform results in the failure of morphological development in the inner ear, including the spiral ganglion (Pirvola et al., 2000; De Moerlooze et al., 2000), and Fgfr3-null mice show deafness as a result of defects in pillar cell differentiation and organ of Corti formation (Colvin et al., 1996). Moreover, analysis of FGF-2-overexpressing mice suggests that FGF-2 may contribute to synaptic reorganization after noise damage in the adult mouse ear (D’sa et al., 2007). Although these studies indicate a crucial role of FGF signaling in the development and pathology of the auditory system, its role in maintaining normal auditory functions in adult and aging animals, particularly in glial cells, is poorly understood. Therefore, to elucidate the in vivo role of glial-mediated FGF signaling in the auditory pathway, we generated conditional knockout mice with disrupted Fgfr1 and Fgfr2 signaling in glial cells of the PNS and CNS, namely, Schwann cells and oligodendrocytes.
We found that the lack of Fgfr1 and Fgfr2 signaling in glial cells resulted in significant loss of type I SGN, accompanied by late onset of hearing impairment, progressing with age, without an apparent loss of glial cells or myelin. These findings emphasize the importance of glial cells in providing trophic support to auditory sensory neurons/axons through a mechanism that is regulated in part by FGF signaling in glial cells.
MATERIALS AND METHODS
Generation and Characterization of Conditional Fgfr1/Fgfr2 Double Knockout Mice
Conditional Fgfr1/Fgfr2 double knockout mice using the Cre-loxP system, in which Cre was under the control of the CNP promoter (2′,3′-cyclic nucleotide 3′-phosphodiesterase), have been described previously. CNP is expressed primarily in Schwann cells, oligodendrocytes, and myelin that is produced by these cells (for review see Gravel al., 1998; Braun et al., 2004). The generation of floxed Fgfr1flox/+ and Fgfr2flox/+ (129SV/CD1 strains) and CNPcre/+ mice (C57Bl/6 strain) has been previously described (Pirvola et al., 2002; Lappe-Siefke et al., 2003; Yu et al., 2003). Briefly, loxP sites in the Fgfr1flox/+ mice flank regions that encode the transmembrane and tyrosine kinase domains and, in the Fgfr2flox/+ mice, the ligand binding IgIII domain and the transmembrane domain of the receptor. Deletion of these regions renders these receptors inactive (Pirvola et al., 2002; Yu et al., 2003; Kaga et al., 2006; Furusho et al., 2009). Suitable mating of Fgfr1flox/+; Fgfr2flox/+ transgenic mice with CNPcre/+ mice produced progeny in which CNP promoter-driven cre activity recombines loxP sites in the gene for Fgfr1 and Fgfr2, rendering null mutations of these receptors in CNP-expressing Schwann cells and oligodendrocytes, giving the final genotype Fgfr1flox/flox; Fgfr2flox/flox; CNPcre/+. Littermates were used as controls in most cases. Assessment of the specificity and efficiency of Fgfr1 and Fgfr2 recombination has been described previously (Kaga et al., 2006; Furusho et al., 2009).
To confirm further, specifically in the auditory system, that Fgfr1 and Fgfr2 were deleted, as expected, from oligodendrocytes and Schwann cells and not from neurons, we generated YFP reporter mice by mating CNPcre/+ mice with Rosa26-YFP (yellow fluorescent protein; obtained from Dr. D. Rowe, University of Connecticut Medical School). In the peripheral region of the cochlear nerve, Schwann cells expressed YFP (hence CNP-Cre) and surrounded axons, which were YFP-negative (Fig. 1A, asterisks) and appeared as dark regions (Fig. 1A). In the spiral ganglion, Schwann cells surrounding the SGN also expressed YFP, whereas SGN cell bodies (N) remained unlabeled (Fig. 1B). In the cochlear nucleus, double labeling of YFP was done with APC and NeuN to identify oligodendrocytes and neurons, respectively. Complete overlap was observed between APC and YFP (Fig. 1C). In contrast, NeuN and YFP labeled distinct cell bodies (Fig. 1D). The results confirmed that the expression of CNP-Cre-YFP and, therefore, the deletion of floxed Fgfr1 and Fgfr2 occurred selectively in oligodendrocytes and Schwann cells but not in neurons of the spiral ganglion or cochlear nucleus.
Fig. 1.

CNP-Cre is selectively expressed in Schwann cells and oligodendrocytes but not neurons of the cochlear nucleus and spiral ganglion. Cross-sections of the peripheral segment of the cochlear nerve (A), spiral ganglion (B), and cochlear nucleus (C,D) from 2-week-old CNP-Cre-YFP-Rosa reporter mice were examined for the expression of yellow fluorescent protein (YFP). YFP was expressed by Schwann cells surrounding the cochlear nerve axons (*; A) and the SGNs (N; B) but was not detected in the axons (*) or neurons (N) themselves. Double labeling with APC (oligodendrocyte marker) and YFP showed complete overlap in the cochlear nucleus (C). In contrast, double labeling with NeuN (neuron marker) and YFP showed no overlap (D). Three mice were analyzed with similar results. Representative image is shown. Site at which sections were cut as referenced in Figure 2 are A: 2; B: 3; C,D: 5. Scale bars = 10 μm in A; 20 μm in B, insets; 100 μm in C,D.
Auditory Brainstem Response (ABR) Test
Mice were placed in a soundproof room and anesthetized with an isofluorane/oxygen mixture delivered through a nose cone. Needle electrodes were positioned at the vertex and right mastoid, using the neck as a ground. A Bose Mini Cube speaker was placed 10 cm above the mouse’s head. The stimulus was an alternating polarity click (100 μsec), generated digitally (TDT RP2 Processor) in SigGen and BioSig software (TDT). It was played at a rate of 21 Hz and was attenuated by a TDT PA2 attenuator in 5-dB steps, starting from 87 dB peak equivalent sound pressure level (peSPL). Signal acquisition was performed with an SA Instrumentation SK-4BA isolated bioelectric amplifier. The signal was bandpass filtered from 0.1 to 5,000 Hz at a gain of 100,000. Then, the signal was processed by a second TDT RP2 unit. The ABRs were sampled at 25,000 Hz over a 16-msec period. The ABR at each intensity was derived from 500 averages. The ABRs were then filtered again (300–500 Hz) in the BioSig software to measure peak-to-peak amplitudes and latencies more accurately. Threshold was determined as the lowest intensity at which any peak could be observed above the noise floor.
Acoustic Startle Response
The acoustic startle response measurements were made as part of the prepulse inhibition test, which was carried out for another, related study on our mutant and control mice. The data presented here relate just to the startle response. Testing was conducted as previously described (Conti et al., 2005). Briefly, mice were placed into a clear acrylic cylindrical chamber (3.5 cm inner diameter; 12.5 cm length), enclosed in a sound- and vibration-attenuating cabinet. The cabinet was equipped with a 5-W incandescent bulb and a fan for ventilation (San Diego Instruments). The chamber sits on a base under which a piezoelectric accelerometer detects whole body startle responses. Output signals from the accelerometer were collected as 100 sequential 1-msec measurements, starting at the onset of the startling stimulus (120 dB, 40 msec). The signals were rectified, digitized, and stored on computer by the SR-LAB program (San Diego Instruments). Chambers were calibrated each day and matched for sound intensity. Delivery of white noise acoustic stimuli through a horn tweeter (Radio Shack) was also controlled by the SR-LAB program. There was a 5-min acclimation period in the presence of the background noise (70 dB) prior to the delivery of any stimulus. All stimuli were presented on this background noise. There were 82 trials in the testing session, and on the first and last six trials the startling stimulus (white noise; 50 dB above background = 120 dB total; 40 msec) was presented alone. The remaining trials included additional startle stimulus-alone trials (used to calculate percentage prepulse inhibition) and trials in which a prepulse stimulus that was 3, 6, 12, 15, or 18 dB above background (20-msec duration; 10 trials with each pre-pulse intensity) preceded the startling stimulus by 100 msec. Additionally, there were eight trials in which no stimulus was presented, but activity within the testing chamber was assessed. These trials were presented in pseudorandom order, and the intertrial interval averaged 20 sec (15–25 sec). All testing took place between 10:00 AM and 3:00 PM.
Histology and Immunohistochemistry
Mice were perfused with 2% or 4% paraformaldehyde/PBS. The cochlea and brainstem were dissected out and post-fixed, and the cochlea was decalcified in 7% EDTA for 3 days. The cochlea and cochlear nucleus were cryoprotected in 20% sucrose, embedded in Tissue-Tek OCT mounting media, and frozen at −80°C. Cryostat cross-sections (15 μm) were pretreated with 0.1% hydrogen peroxide in PBS (30 min) to block endogenous peroxidase and, in some cases, then blocked in 10% FCS and 0.1% Triton X-100 (60 min). Sections were next incubated with primary antibodies (overnight at 4°C), washed, and incubated with secondary antibodies (1 hr) that were conjugated either to biotin (Vector, Burlingame, CA) for the ABC method or to Alexa 488, Alexa 594 (1:500; Molecular Probes, Eugene, OR), or Cy3 (1:600; Jackson Immunoresearch, West Grove, PA) for fluorescent detection. For the ABC method, the sections were incubated with avidin-biotin-peroxidase complex (Vectastain Elite ABC kit; Vector), and immune complexes were visualized by immersion of sections in 0.05% diaminobenzidine/0.015% H2O2. Nuclei were counterstained with Hoechst 33342 (0.1 μg/ml). The primary antibodies used were anti-MBP (1:1,000; E. Barbarese, University of Connecticut Medical School), Myo7a (1:200; Developmental Studies Hybridoma Bank), anti-GFP (1:1,000; NeuroMab, CA), anti-GFP (1:500; Molecular Probes), anti-NeuN (1:100; Millipore, Bilerica, MA), anti-GFAP (1:500; Chemicon, Temecula, CA), and antiadenomataus polyposos coli (APC; 1:40; Calbiochem, Darmstadt, Germany).
Frozen sections of cochlea (15 μm) were labeled with Nissl stain (Trettel and Morest, 2001) to identify SGNs. Approximately 90–95% of the SGNs are type I, which are >8–10 μm in size, whereas the type II SGNs and nonneuronal glial cells are <5–6 μm. Detection of purple-blue-stained Nissl body in the cytoplasm of neurons is a commonly used method to identify neurons in vivo. Combined with size criteria, this method is considered the most reliable and widely accepted means to identify type I SGN neurons. We have previously used TuJ1 staining and consistently obtained results similar to those with Nissl staining, confirming the identity of the “large” Nissl-stained cells as type I SGNs. In contrast, the 5–10% of SGNs that are type II are closer in size to the non-neuronal glial cells and are harder to distinguish with these criteria. The smaller cells were therefore grouped together for the analysis. In Norther Eclipse imaging software, images of the stained cochlea were collected with a Zeiss Axiophot microscope and a ×20 lens. For each mouse, the entire Rosenthal’s canal was visualized, and the total numbers of both small and large cells with cytoplasm, a clear nucleus, and nucleoli were counted in every fourth section of the apical, medial, and basal regions of the cochlea, in NIH Image J. The area and volume of the ganglion were also calculated in Image J. The numerical density of spiral ganglion cells was expressed as cells/mm3.
RESULTS
Generation and characterization of conditional knockout mice with deletion of Fgfr1 and Fgfr2 in Schwann cells and oligodendrocytes but not SGN or neurons in the cochlear nucleus are described in Materials and Methods (Fig. 1) and in our previous papers (Kaga et al., 2006; Furusho et al., 2009). In this study, we investigated the effect of Fgfr1 and Fgfr2 disruption on the structure and function of the auditory pathway. The regions of the peripheral and central auditory pathway under investigation, including the cochlear nucleus, spiral ganglion, and organ of Corti, are diagramed in Figure 2.
Fig. 2.
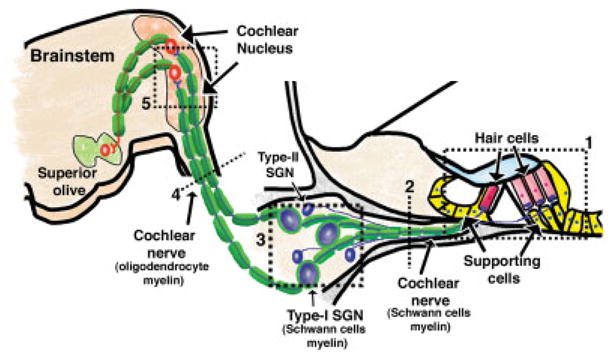
Diagramatic representation of the regions of the auditory pathway investigated in the Fgfr1−/−; Fgfr2−/− mice. Within the cochlea is the organ of Corti (1), which contains hair cells and nonneuronal support cells. Inner and outer hair cells are synaptically connected through the myelinated and unmyelinated nerve fibers of the peripheral cochlear nerve, respectively (2), to the bipolar-spiral ganglion neurons (SGNs) in the spiral ganglion (3). Approximately 90% of SGNs are type I and myelinated by Schwann cells, and 10% are type II, which are unmyelinated. From the spiral ganglion, the cochlear nerve continues (4) to the CNS via the cochlear nucleus (5) in the brainstem and is myelinated by oligodendrocytes. The numbers show the sites at which the section was cut. These numbers are referenced in each figure. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Adult Fgfr1−/−; Fgfr2−/− Mice Exhibit Progressive Hearing Loss With Age and Increased Signal Transmission Latency
Differences in ABR thresholds and latencies between control and mutant mice between 1 and 16 months of age are shown in Figure 3. Scatterplots of thresholds against age (Fig. 3A) show that the majority of the mutant mice over 4 months of age had thresholds over 60 dB peSPL, whereas the majority of the control mice had thresholds below 60 db peSPL. To examine the effect of age on hearing thresholds, we determined the percentage of mice in each group showing ABR thresholds below 60 dB as a function of age (Fig. 3B). Between 1 and 3 months of age, nearly all mice, regardless of genotype, had normal hearing (91% and 100% for mutants and controls, respectively). However, between 3 and 5 months, the percentage of mutant mice with thresholds below 60 dB peSPL decreased to 50%, compared with 85% of control mice. Between 5 and 7 months, the mutant group decreased further to 43%, whereas the control group remained nearly unchanged, and, finally, at greater than 12 months of age, just 26% of mutants retained normal hearing compared with 73% of controls. Overall, mutant mice appeared to have normal hearing up to 1–3 months of age but showed progressive impairment with age.
Fig. 3.

Fgfr1−/−; Fgfr2−/− mice exhibit progressive hearing loss with age. A: Adult mice were examined for hearing loss by auditory brainstem response (ABR) tests. Hearing sensitivity of individual control and mutant mice is shown as the thresholds of responses to a sound stimulus played at different intensities [in peak equivalent sound pressure level (peSPL), in dB], plotted as a function of age. Overall, mutant mice (solid circles) exhibited higher hearing thresholds than control mice (open circles) at all ages beyond 4 months. B: Mice that showed ABR response to stimuli below 60 dB peSPL were plotted as a percentage of total mice tested in each group. Animals with thresholds below 60 dB were considered to be within the boundary of “normal” hearing. Note the late onset of hearing impairment and progression with age in the mutant mice. Ten to fifteen mice were analyzed in each age group and genotype. Because no obvious differences were observed between males and females, data were pooled for the analysis.
In addition to hearing threshold, we also measured interpeak intervals (IPIs) in the ABR waveform. Figure 4A displays this measurement using an ABR from a control and a mutant mouse. We measured the average IPI from peak I to peaks II, III, or IV among four control and four mutant mice as they aged (Fig. 4B). Both groups at 1–3 months of age showed the same IPIs for all peaks. However, at 3–5 months, mutant mice showed significant delays in all peaks compared with controls. Similar delays continued at 5–7 and 7–10 months.
Fig. 4.
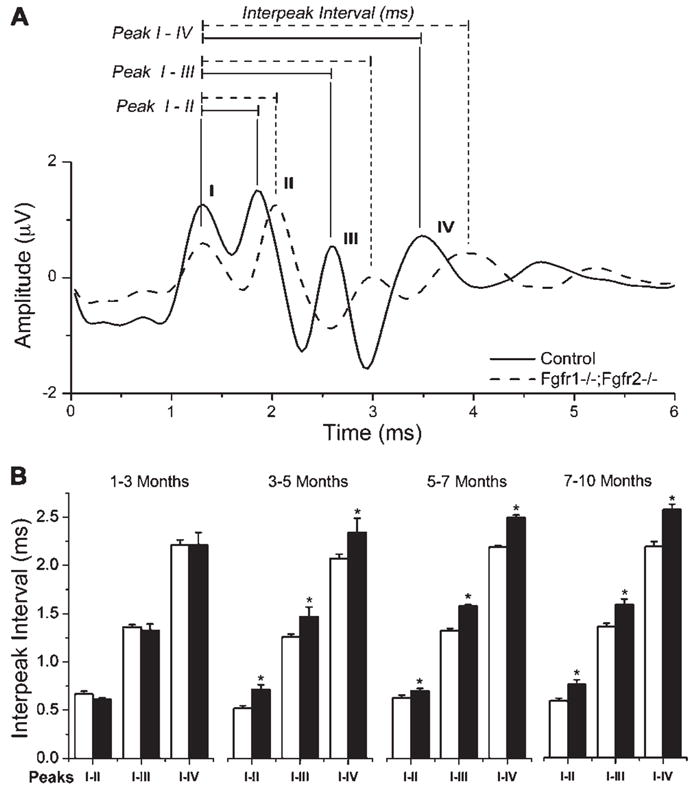
Fgfr1−/−; Fgfr2−/− mice exhibit prolonged latency between successive peaks of ABR waveforms compared with controls. A: Representative ABR waveform showing amplitude in microvolts plotted against time in milliseconds. Four ABR peaks are shown from control (solid line) and mutant (dashed line) mouse recordings. In general, a delay in the interpeak interval was observed in mutants compared with controls for all peaks. In addition, reduced peak height is also observed. B: Four control (open bars) and four mutant mice (solid bars) were analyzed at approximately 2-month interval for IPI latencies over a period of 10 months. The average interpeak intervals from 87 dB, 82 dB, and 77 dB stimuli are plotted for peaks I–II, I–III, and I–IV in each age group. Note that mutants and controls displayed similar IPIs at 1–3 months of age, but mutants showed a significant delay (*P < 0.05) at older ages. Error bars represent SEM; N = 4.
Acoustic startle response measurements were also carried out to evaluate the hearing ability of Fgfr1−/−; Fgfr2−/− mice (Fig. 5). Mice in the age groups of approximately 5 and 8 months were examined. Average startle amplitude from each of the 24 trials in which the startle stimulus sound was presented alone was calculated and plotted for both age groups. Significantly reduced acoustic startle reflex amplitudes were observed in the mutant mice compared with controls at all ages tested, suggestive of impaired hearing. We concluded that Fgfr1−/−; Fgfr2−/− mice exhibit hearing impairment that begins in adulthood and progresses with age.
Fig. 5.
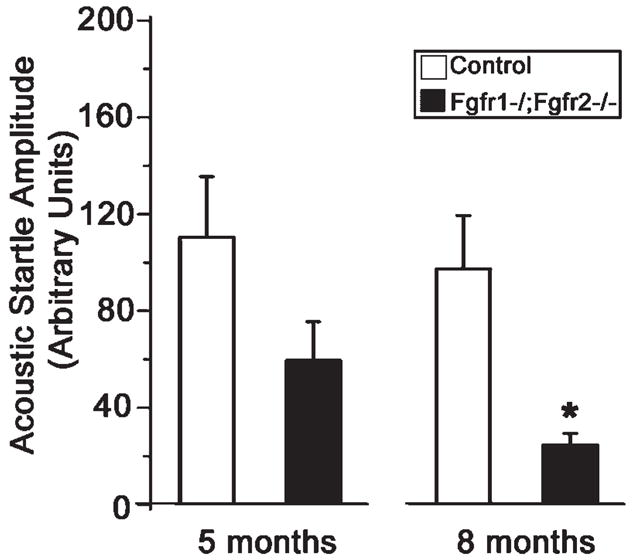
Fgfr1−/−; Fgfr2−/− mice show a lower acoustic startle response than the control mice. Mice at approximately 5 and 8 months of age were examined for hearing loss by acoustic startle reflex measurements. Average startle amplitude from each of the 24 trials in which the startle stimulus was presented alone was calculated. Mean value of startle amplitude from both age group of mutant and control mice is shown. ANOVA revealed a significant effect of genotype, F(1,30) = 26.3, *P < .001. There was no interaction between these factors. A Tukey test revealed that mutant mice showed a smaller acoustic startle response than the control mice at both ages. Error bars represent SEM; N = 3–9.
Fgfr1−/−; Fgfr2−/− Mice Exhibit Significant Loss of Type I Spiral Ganglion Neurons and Reactive Gliosis in the Cochlear Nucleus
Age-related hearing loss has been attributed to degeneration of SGNs (Felder and Schrott-Fischer, 1995; Adams and Schulte, 1997). Therefore, we examined the spiral ganglion of mice to determine whether SGNs were affected in the mutants. Sections of cochlea from control and mutant mice were Nissl stained (Fig. 6A), and numbers of type I (large; arrowheads) and type II SGNs and glial cells (small; arrows) were counted on the basis of size in the apical, medial, and basal turns of the cochlea. The mutant mice showed a significant loss of type I SGNs. Specifically, compared with control, mutants had 62%, 43%, and 40% fewer type I SGNs at the apical, medial, and basal levels of the cochlea, respectively (Fig. 6B). In contrast, type II SGNs and glial cells were not affected (data not shown).
Fig. 6.

Fgfr1−/−; Fgfr2−/− mice exhibit significant loss of type I spiral ganglion neurons and reactive gliosis in the cochlear nucleus. A: Nissl-stained sections of medial (a,b) and basal (c,d) turns of the cochlea, showing SGNs of 7-month-old control and Fgfr1−/−;Fgfr2−/− mice. A substantial loss of type I SGNs (large size, arrowhead) was observed, but not of type II SGNs (small size, arrow). Insets show magnified images of the cells denoted by arrows. B: Quantification of the numbers of type I SGNs in control (open bars) and mutant (solid bars) mice. Controls were set at baseline of 100%. Note the significant reduction (*P < 0.05) in the numbers of SGNs in the mutant apical, medial, and basal turns. Error bars represent SEM; N = 3 for all except apical and basal for controls, where N = 2. C: Transverse sections of the ventral cochlear nucleus from control (a) and mutant (b) mice were immunolabeled for glial fibrillary acidic protein (GFAP). Intense GFAP staining of thick astrocytic processes (arrowhead) and cell bodies (arrow), indicative of pathological gliosis, was observed in the mutants compared with controls. A representative experiment out of three is shown. Site at which sections were cut as referenced in Figure 2, A: 3; C: 5. Scale bars = 100 μm in A; 50 μm in inset; 20 μm in C.
We next asked whether the pathology resulting from loss of type I SGNs had extended farther into the central auditory pathway in the mutant mice. Therefore, we examined sections of the cochlear nerve in the ventral cochlear nucleus by immunostaining with antiglial fibrillary acidic protein (GFAP; Fig. 6C). Expression of GFAP in the cochlear nucleus was found to be more pronounced in the mutants compared with controls, and the astrocytic cell bodies and processes appeared enlarged. These features are suggestive of pathological gliosis in this region, perhaps in response to axonal damage. Consistent with this hypothesis, our preliminary observations have shown signs of axonal degeneration in the cochlear nerve and the ventral cochlear nucleus (data not shown). We concluded that significant loss of type I SGNs and the pathology of the central auditory pathway are consistent with hearing impairment in the Fgfr−/−; Fgfr2−/− mice.
No Obvious Loss of Peripheral or Central Myelin/Glia Was Observed in the Auditory Pathway of Fgfr1−/−; Fgfr2−/− Mice
Because we have conditionally disrupted Fgfr1 and Fgfr2 signaling in glial cells, namely, Schwann cells and oligodendrocytes, we asked whether there was a direct effect of the disruption on these cells (Figs. 7, 8). We also examined the “support cells” in the organ of Corti. On phase-contrast microscopy (Fig. 7C,D), we did not observe any gross loss of support cells. Similarly, immunolabeling with Myo7a, a marker of hair cells, showed no loss of either inner or outer hair cells (Fig. 7A,B). Myelin basic protein (MBP), a marker protein that labels Schwann cells and myelin, indicated no obvious loss of peripheral myelin wrapped around either the SGNs in the spiral ganglion (SG) or cochlear nerve (Fig. 7E–H). Similarly, in the central auditory pathway, examination of PLP mRNA+ oligodendrocyte cell bodies in the cochlear nucleus and MBP-labeled myelin in the central segment of cochlear nerve showed no obvious differences between the control and the mutant mice (Fig. 8). Overall, the present level of analysis did not identify any obvious loss of glial cells or myelin in either the peripheral or the central auditory pathways of the Fgfr1−/−; Fgfr2−/− mice.
Fig. 7.
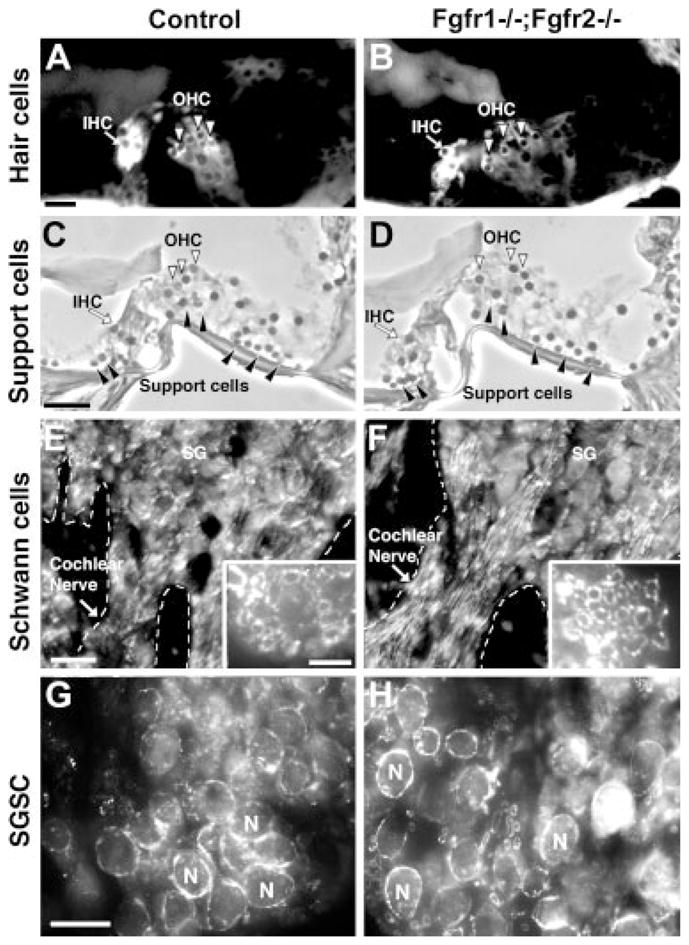
Peripheral glial cells and peripheral myelin appear similar in Fgfr1−/−; Fgfr2−/− and control mice. Sections of cochlea, immunolabeled with Myo7a (a marker of hair cells), showed that inner and outer hair cells (IHC and OHC) were present in the organ of Corti in both control and Fgfr1−/−; Fgfr2−/− mice (A,B). Similarly, phase-contrast microscopy showed that support cells were also present in the control and mutants (C,D, solid arrowheads). Expression of MBP (a marker for myelin and myelinating Schwann cells) showed a similar pattern of immunolabeling of the spiral ganglion (SG) and peripheral segment of cochlear nerve in mutant and control mice (E,F), further confirmed in cross-sections of cochlear nerves showing axons surrounded by MBP+ myelinating Schwann cells (insets in e,f). Cross-sections of spiral ganglions (G,H) immunostained with MBP show that spiral ganglion Schwann cells (SGSCs), ensheathing the spiral ganglion neurons (N) with myelin, are present in both the control and the Fgfr1−/−; Fgfr2−/− mice. Similar results were obtained when three or four mice of each genotype, ranging between 4 and 10 months of age were analyzed. Representative images are shown. Site at which sections were cut as referenced in Figure 2, A–D: 1; E–H: 3. Scale bars = 20 μm in a (applies to A,B); 20 μm in e (applies to E,F); 20 μm in in g (applies to g,h); 50 μm in c (applies to D,D); 5 μm in insets.
Fig. 8.
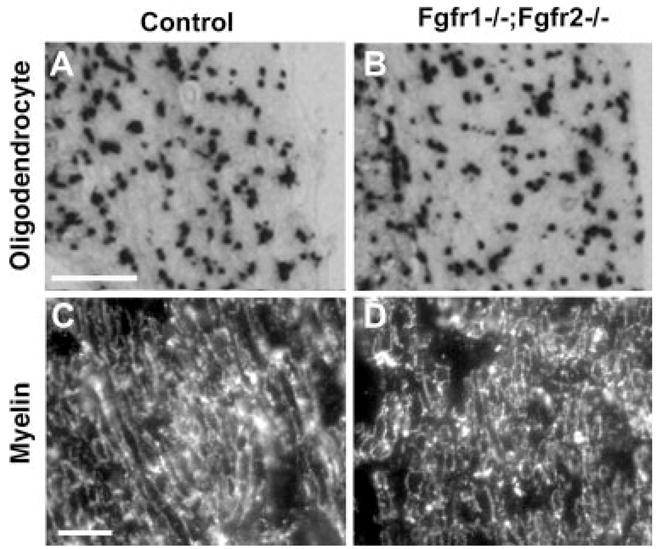
Oligodendrocytes and central myelin appear to be similar in Fgfr1−/−; Fgfr2−/− and control mice. Transverse sections of the ventral cochlear nucleus (A,B) or central segment of cochlear nerve (C,D) from control and mutant mice were analyzed by in situ hybridization for the oligodendrocyte marker PLP mRNA (A,B) or by immunohistochemistry for myelin marker MBP (C,D). Expression patterns of PLP mRNA and MBP were similar in control and Fgfr1−/−; Fgfr2−/− mice. Analysis of three or four mice of each genotype, ranging between 4 and 8 months of age, gave similar results. Representative images are shown. Site at which sections were cut as referenced in Figure 2, A,B: 5; C,D: 4. Scale bars = 200 μm in a (applies to A,B); 10 μm in c (applies to C,D).
DISCUSSION
The present study suggests that FGF/FGF receptors are an important part of the signaling network through which glial cells support neuronal functions in the auditory pathway. This conclusion is drawn from our observation that significant loss of myelinated SGNs (type I), accompanied by functional impairment of auditory function, occurred in mice in which Fgfr1 and Fgfr2 were deleted specifically from myelinating glial cells and not neurons. We hypothesize that the perturbation of glial function resulting from loss of Fgfr1 and Fgfr2 results in attenuation of trophic support of neurons by glial cells, leading to significant neuronal loss and pathology of the auditory pathway. Because neurons and nerve fibers, but not myelinating glial cells, produce FGF-1 and FGF-2 in the spiral ganglion and cochlear nucleus (Luo et al., 1993; Pirvola et al., 1995; Riedel et al., 1995; Silva et al., 2005), FGF/FGF receptors provide an efficient means of reciprocal signaling between neurons and glia.
How does disruption of Fgfr1 and Fgfr2 signaling in Schwann cells and oligodendrocytes result in hearing impairment? The cochlear nerve is myelinated by Schwann cells and oligodendrocytes in the peripheral and central auditory pathways, respectively. In addition, Schwann cells wrap a thin sheath of myelin around the somata of the bipolar type I SGNs in the spiral ganglion. Thus, any abnormality of myelin structure in the mutant mice, such as hypomyelination of the cochlear nerve, could reduce conduction velocity and thus hearing ability. Immunostaining of both peripheral and central nerve fibers and SGN cell bodies with MBP showed very similar patterns of staining in the mutants and controls, suggesting no obvious loss of myelin. Nevertheless, the possibility exists that thinner or defective myelin, not identified at the light microscopic level, could exist in the mutants. Although a detailed ultrastructural characterization of myelin, including measurement of its thickness (g-ratio) and analysis of paranodal structures, is required to resolve this issue definitively, the prolongation of ABR peak latency (indicative of slower nerve conduction) that is observed in the Fgfr1−/−; Fgfr2−/− mice points to a possible myelin defect in the mutants. Prolongation of peak latency has been reported in other PNS and CNS hypomyelinated or dysmyelinated mutants, such as TremblerJ and Po-DT-A mutants (Zhou et al., 1995), myelin-deficient (md) rat (Ito et al., 2004), myelin-deficient “bt” hamster (Naito et al., 1999), quaking mouse (Shah and Salamy, 1980), and shiverer mouse (Fujiyoshi et al., 1994). In fact, prolongation of peak latency has been used as a measure for demyelination of the cochlear nerve in patients with demyelinating diseases, such as multiple sclerosis (Starr and Achor 1975; Robinson and Rudge, 1977). Decrease in latency was also observed in normal infants and developing rodent brains that have immature myelination (Starr et al., 1977; Shah et al., 1978). Correlation of myelin thickness with prolongation of peak latency was shown elegantly in a study in which restoring partial myelin by expressing MBP in transgenic shiverer mice resulted in partial normalization of peak latency (Fujiyoshi et al., 1994).
In addition to slower nerve conduction, myelin defects could also compromise the long-term integrity of the cochlear nerve and the viability of SGN. Consistently with this, TremblerJ and Po-DT-A, mutants with peripheral hypomyelination, showed elevation in ABR thresholds and loss of spiral ganglion neurons (Zhou et al., 1995). In these mutants, inner and outer hair cells appeared normal, suggesting that elevation of ABR thresholds in the mutant mice is caused by loss of SGNs and, most likely, degeneration of myelinated fibers. Our Fgfr1−/−; Fgfr2−/− mutants also display elevated ABR thresholds and reduced acoustic startle amplitudes, consistent with a loss of SGNs and most likely an accompanied degeneration of nerve fibers. As for TremblerJ mutants, no obvious loss of hair cells or support cells was noted in Fgfr1−/−; Fgfr2−/− mutants. Overall, the probability remains that subtle defects in myelin that are unidentified at this stage of analysis may play a part in the manifestation of hearing defects in the Fgfr1−/−; Fgfr2−/− mutant mice.
The attenuation of trophic support of SGN by Fgfr1- and Fgfr2-deficient glial cells may also be caused by mechanisms independent of (or in addition to) their myelination function, as has been suggested for mutants of certain myelin proteins (Nave and Trapp, 2008). Specifically, oligodendrocytes of transgenic mice with deletion of myelin proteins CNP or PLP remain viable and make compact myelin but develop late-onset axonal degeneration (Griffiths et al., 1998; Lappe-Siefke et al., 2003). Peripheral neuropathies, in which the primary defect resides in the myelinating Schwann cells (Berger et al., 2006), further provide compelling evidence for a role of glia in the maintenance of neuronal function, sometimes without loss of glial cells themselves. We have recently shown that conditional disruption of Fgfr1 and Fgfr2 signaling in nonmyelinating Schwann cells in the sciatic nerve results in neuropathy of sensory c-fibers and impairment of thermal nociception (Furusho et al., 2009). This was not accompanied by loss of these cells or their ensheathment of c-fibers in the Remak bundles, providing further evidence that glial support of neurons can be independent of their ensheathment function.
The mechanism of how FGF signaling in myelinating glial cells may support long-term maintenance and survival of SGNs remains unclear. Because FGF is known to regulate and interact with a number of other neurotrophic factors, it is likely that it operates in concert with other factors (for review see Bansal, 2002). For example, FGF-1 stimulates NGF synthesis and secretion in astrocytes and fibroblasts (Yoshida and Gage, 1991), and FGF-2 signaling up-regulates GDNF (Suter-Crazzolara and Unsicker, 1996) and NGF mRNA in cells expressing Fgfr1 and Fgfr2 (Ferhat et al., 1997). Neurotrophins are important for survival of SGNs (Davies and Wright, 1995; Hossain et al., 2002; Wei et al., 2007) and are expressed by Schwann cells in a neuroprotective role (Bandtlow et al., 1987; Hammarberg et al., 1996; Davies, 1998; Thippeswamy et al., 2005). A model can be proposed in which FGF that is produced by SGN signals to FGF receptors on Schwann cells, which regulates the synthesis and/or release of other trophic factors that in turn support survival of type I SGNs and their projections.
It is interesting to note that, when Neuregulin-erbB signaling was disrupted in transgenic mice by the expression of dominant-negative erbB receptor driven by GFAP promoter, a loss of type I SGN was also observed (Stankovic et al., 2004). The mechanism of this loss must be different from that in the Fgfr1−/−; Fgfr2−/− mutant, because it was concluded from this study that “support cells” of the organ of Corti and not myelinating Schwann cells were involved in this process. Furthermore, in contrast to the present study, no prolongation of normal neural response latency was observed in the erbB mutants, consistent with intact Schwann cell function. This suggests that, in the adult mice, maintenance of SGNs and their myelinated fibers is supported by more than one growth factor receptor signaling within different glial cells of the auditory pathway. Future studies directed at understanding the interplay between these factors is expected to elucidate the mechanism of these neuron–glial interactons.
Hearing loss and neurodegeneration in the auditory system, which progressively increase with advancing age, are highly prevalent in the elderly population (Rowe, 1978; Morton, 1991). It appears from our studies that age-related hearing loss is accelerated in mice with nonfunctional Fgfr1 and Fgfr2 in oligodendrocytes and Schwann cells. To our knowledge, the role of Fgfr signaling in aging, especially in the myelin-producing cells, is poorly understood. The present study provides new insights into this role, which opens new avenues of investigation in hearing research.
In summary, selective loss of Fgfr1 and Fgfr2 signaling in Schwann cells and oligodendrocytes in the peripheral and central auditory pathways results in significant loss of myelinated spiral ganglion neurons, accompanied by age-related hearing impairment in adulthood, without apparent loss of glia or myelin. These findings emphasize the importance of Fgfr1 and Fgfr2 signaling as potential mediators of reciprocal axon–glial interaction in the auditory pathway, primarily via influencing Schwann cell (and possibily oligodendrocyte) functions, which in turn modulates survival of sensory spiral ganglion neurons. This study has important implications for understanding the potentially protective role of FGF signaling in glia against hearing loss that is associated with normal aging.
Acknowledgments
Contract grant sponsor: National Institutes of Health; Contract grant number: NS38878 (to R.B.); Contract grant number: DC000127 (to D.K.M.); Contract grant number: DC006387 (to D.K.M.); Contract grant sponsor: National Multiple Multiple Sclerosis Society; Contract grant number: RG4087-A-3 (to R.B.); Contract grant sponsor: National Organization for Hearing Research Foundation (to C.D., D.K.M.); Contract grant sponsor: NIDCD; Contract grant number: DC002178 (to S.K.).
We thank Dr. D.M. Ornitz (Washington University School of Medicine, St. Louis, MO) for providing us with floxed Fgfr1 and Fgfr2 transgenic mice; Dr. K. Nave (Max Planck Institute of Experimental Medicine, Goettingen, Germany) for CNP-Cre mice; and Melissa Bryant, Ukti Gohel, and Tamair Holden for technical assistance. The monoclonal antibody Myo7a developed by Dana Jo Orten was obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD and maintained by the University of Iowa (Department of Biological Sciences, Iowa City, IA 52242).
References
- Adams JC, Schulte BA. Histopathologic observations of the aging gerbil cochlea. Hear Res. 1997;104:101–111. doi: 10.1016/s0378-5955(96)00184-0. [DOI] [PubMed] [Google Scholar]
- Armstrong RC, Le TQ, Frost EE, Borke RC, Vana AC. Absence of fibroblast growth factor 2 promotes oligodendroglial repopulation of demyelinated white matter. J Neurosci. 2002;22:8574–8585. doi: 10.1523/JNEUROSCI.22-19-08574.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandtlow CE, Heumann R, Schwab ME, Thoenen H. Cellular localization of nerve growth factor synthesis by in situ hybridization. EMBO J. 1987;6:891–899. doi: 10.1002/j.1460-2075.1987.tb04835.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal R. Fibroblast growth factors and their receptors in oligodendrocyte development: implications for demyelination and remyelination. Dev Neurosci. 2002;24:35–46. doi: 10.1159/000064944. [DOI] [PubMed] [Google Scholar]
- Bansal R, Kumar M, Murray K, Morrison RS, Pfeiffer SE. Regulation of FGF receptors in the oligodendrocyte lineage. Mol Cell Neurosci. 1996;7:263–275. doi: 10.1006/mcne.1996.0020. [DOI] [PubMed] [Google Scholar]
- Bansal R, Lakhina V, Remedios R, Tole S. Expression of FGF receptors 1, 2, 3 in the embryonic and postnatal mouse brain compared with Pdgfrα, Olig2 and Plp/dm20, implications for oligodendrocyte development. Dev Neurosci. 2003;25:83–95. doi: 10.1159/000072258. [DOI] [PubMed] [Google Scholar]
- Berger P, Niemann A, Suter U. Schwann cells and the pathogenesis of inherited motor and sensory neuropathies (Charcot-Marie-Tooth disease) Glia. 2006;54:243–257. doi: 10.1002/glia.20386. [DOI] [PubMed] [Google Scholar]
- Braun PE, Lee J, Gravel M. 2′,3′-Cyclic nucleotide 3′-phosphodiesterase: structure, biology, and function. In: Lazzarini RA, editor. Myelin biology and disorders. Vol. 1. San Diego: Elsevier Academic Press; 2004. pp. 499–522. [Google Scholar]
- Colvin JS, Bohne BA, Harding GW, McEwen DG, Ornitz DM. Skeletal overgrowth and deafness in mice lacking fibroblast growth factor receptor 3. Nat Genet. 1996;12:390–397. doi: 10.1038/ng0496-390. [DOI] [PubMed] [Google Scholar]
- Conti LH, Costill JE, Flynn S, Tayler JE. Effects of a typical and an atypical antipsychotic on the disruption of prepulse inhibition caused by corticotropin-releasing factor. Behav Neusosci. 2005;119:1052–1060. doi: 10.1037/0735-7044.119.4.1052. [DOI] [PubMed] [Google Scholar]
- D’Sa C, Gross J, Francone VP, Morest DK. Plasticity of synaptic endings in the cochlear nucleus following noise-induced hearing loss is facilitated in the adult FGF2 overexpressor mouse. Eur J Neurosci. 2007;26:666–680. doi: 10.1111/j.1460-9568.2007.05695.x. [DOI] [PubMed] [Google Scholar]
- Davies AM. Neuronal survival: early dependence on Schwann cells. Curr Biol. 1998;8:R15–R18. doi: 10.1016/s0960-9822(98)70009-0. [DOI] [PubMed] [Google Scholar]
- Davies AM, Wright EM. Neurotrophic factors. Neurotrophin autocrine loops. Curr Biol. 1995;5:723–726. doi: 10.1016/s0960-9822(95)00144-8. [DOI] [PubMed] [Google Scholar]
- Davis JB, Stroobant P. Platelet-derived growth factors and fibroblast growth factors are mitogens for rat Schwann cells. J Cell Biol. 1990;110:1353–1360. doi: 10.1083/jcb.110.4.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Moerlooze L, Spencer-Dene B, Revest JM, Hajihosseini M, Rose-well I, Dickson C. An important role for the IIIb isoform of fibroblast growth factor receptor 2 (FGFR2) in mesenchymal-epithelial signalling during mouse organogenesis. Development. 2000;127:483–492. doi: 10.1242/dev.127.3.483. [DOI] [PubMed] [Google Scholar]
- Felder E, Schrott-Fischer A. Quantitative evaluation of myelinated nerve fibres and hair cells in cochleae of humans with age-related high-tone hearing loss. Hear Res. 1995;91:19–32. doi: 10.1016/0378-5955(95)00158-1. [DOI] [PubMed] [Google Scholar]
- Ferhat L, Represa A, Zouaoui-Aggoun D, Ferhat W, Ben-Ari Y, Khrest-chatisky M. FGF-2 induces nerve growth factor expression in cultured rat hippocampal neurons. Eur J Neurosci. 1997;9:1282–1289. doi: 10.1111/j.1460-9568.1997.tb01483.x. [DOI] [PubMed] [Google Scholar]
- Fortin D, Rom E, Sun H, Yayon A, Bansal R. Distinct fibroblast growth factor (FGF)/FGF receptor signaling pairs initiate diverse cellular responses in the oligodendrocyte lineage. J Neurosci. 2005;25:7470–7479. doi: 10.1523/JNEUROSCI.2120-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiyoshi T, Hood L, Yoo TJ. Restoration of brain stem auditory-evoked potentials by gene transfer in shiverer mice. Ann Otol Rhinol Laryngol. 1994;103:449–456. doi: 10.1177/000348949410300606. [DOI] [PubMed] [Google Scholar]
- Furusho M, Dupree JL, Bryant M, Bansal R. Disruption of fibroblast growth factor receptor signaling in nonmyelinating Schwann cells causes sensory axonal neuropathy and impairment of thermal pain sensitivity. J Neurosci. 2009;29:1608–1614. doi: 10.1523/JNEUROSCI.5615-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gravel M, Di Polo A, Valera PB, Braun PE. Four-kilobase sequence of the mouse CNP gene directs spatial and temporal expression of lacZ in transgenic mice. J Neurosci Res. 1998;53:393–404. doi: 10.1002/(SICI)1097-4547(19980815)53:4<393::AID-JNR1>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Griffiths I, Klugmann M, Anderson T, Yool D, Thomson C, Schwab MH, Schneider A, Zimmermann F, McCulloch M, Nadon N, Nave KA. Axonal swellings and degeneration in mice lacking the major proteolipid of myelin. Science. 1998;280:1610–1613. doi: 10.1126/science.280.5369.1610. [DOI] [PubMed] [Google Scholar]
- Grothe C, Nikkhah G. The role of basic fibroblast growth factor in peripheral nerve regeneration. Anat Embryol. 2001;204:171–177. doi: 10.1007/s004290100205. [DOI] [PubMed] [Google Scholar]
- Hammarberg H, Piehl F, Cullheim S, Fjell J, Hökfelt T, Fried K. GDNF mRNA in Schwann cells and DRG satellite cells after chronic sciatic nerve injury. Neuroreport. 1996;7:857–860. doi: 10.1097/00001756-199603220-00004. [DOI] [PubMed] [Google Scholar]
- Hansen MR, Vijapurkar U, Koland JG, Green SH. Reciprocal signaling between spiral ganglion neurons and Schwann cells involves neuregulin and neurotrophins. Hear Res. 2001;161:87–98. doi: 10.1016/s0378-5955(01)00360-4. [DOI] [PubMed] [Google Scholar]
- Hossain WA, Brumwell CL, Morest DK. Sequential interactions of fibroblast growth factor-2, brain-derived neurotrophic factor, neurotrophin-3, and their receptors define critical periods in the development of cochlear ganglion cells. Exp Neurol. 2002;175:138–151. doi: 10.1006/exnr.2002.7872. [DOI] [PubMed] [Google Scholar]
- Ito T, Tokuriki M, Shibamori Y, Saito T, Nojyo Y. Cochlear nerve demyelination causes prolongation of wave I latency in ABR of the myelin deficient (md) rat. Hear Res. 2004;191:119–124. doi: 10.1016/j.heares.2003.12.019. [DOI] [PubMed] [Google Scholar]
- Itoh N, Ornitz DM. Evolution of the Fgf and Fgfr gene families. Trends Genet. 2004;20:563–569. doi: 10.1016/j.tig.2004.08.007. [DOI] [PubMed] [Google Scholar]
- Kaga Y, Shoemaker WJ, Furusho M, Bryant M, Rosenbluth J, Pfeiffer SE, Oh L, Rasband M, Lappe-Siefke C, Yu K, Ornitz DM, Nave KA, Bansal R. Mice with conditional inactivation of fibroblast growth factor receptor-2 signaling in oligodendrocytes have normal myelin but display dramatic hyperactivity when combined with Cnp1 inactivation. J Neurosci. 2006;26:12339–12350. doi: 10.1523/JNEUROSCI.3573-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappe-Siefke C, Goebbels S, Gravel M, Nicksch E, Lee J, Braun PE, Griffiths IR, Nave KA. Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nat Genet. 2003;33:366–374. doi: 10.1038/ng1095. [DOI] [PubMed] [Google Scholar]
- Luo L, Koutnouyan H, Baird A, Ryan AF. Acidic and basic FGF mRNA expression in the adult and developing rat cochlea. Hear Res. 1993;69:182–193. doi: 10.1016/0378-5955(93)90106-b. [DOI] [PubMed] [Google Scholar]
- Morton LP, Reynolds L. High frequency thresholds: variations with age and industrial noise exposure. S Afr J Commun Disord. 1991;38:13–17. [PubMed] [Google Scholar]
- Murtie JC, Zhou YX, Le TQ, Armstrong RC. In vivo analysis of oligodendrocyte lineage development in postnatal FGF2 null mice. Glia. 2005;49:542–554. doi: 10.1002/glia.20142. [DOI] [PubMed] [Google Scholar]
- Naito R, Murofushi T, Mizutani M, Kaga K. Auditory brainstem responses, electrocochleograms, and cochlear microphonics in the myelin deficient mutant hamster “bt. Hear Res. 1999;136:44–48. doi: 10.1016/s0378-5955(99)00107-0. [DOI] [PubMed] [Google Scholar]
- Nave KA, Trapp BD. Axon–glial signaling and the glial support of axon function. Annu Rev Neurosci. 2008;31:535–561. doi: 10.1146/annurev.neuro.30.051606.094309. [DOI] [PubMed] [Google Scholar]
- Ochikubo F, Gomi H, Kitamura K, Yoshikawa Y, Yamanouchi K. Auditory brainstem response in zitter rats with genetic spongiform encephalopathy. Electroencephalogr Clin Neurophysiol. 1992;82:145–151. doi: 10.1016/0013-4694(92)90158-e. [DOI] [PubMed] [Google Scholar]
- Oh LY, Denninger A, Colvin JS, Vyas A, Tole S, Ornitz DM, Bansal R. Fibroblast growth factor receptor 3 signaling regulates the onset of oligodendrocyte terminal differentiation. J Neurosci. 2003;23:883–894. doi: 10.1523/JNEUROSCI.23-03-00883.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlemiller KK. Age-related hearing loss: the status of Schuknecht’s typology. Curr Opin Otolaryngol Head Neck Surg. 2004;12:439–443. doi: 10.1097/01.moo.0000134450.99615.22. [DOI] [PubMed] [Google Scholar]
- Pirvola U, Cao Y, Oellig C, Suoqiang Z, Pettersson RF, Ylikoski J. The site of action of neuronal acidic fibroblast growth factor is the organ of Corti of the rat cochlea. Proc Natl Acad Sci USA. 1995;92:9269–9273. doi: 10.1073/pnas.92.20.9269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirvola U, Spencer-Dene B, Xing-Qun L, Kettunen P, Thesleff I, Fritzsch B, Dickson C, Ylikoski J. FGF/FGFR-2(IIIb) signaling is essential for inner ear morphogenesis. J Neurosci. 2000;20:6125–6134. doi: 10.1523/JNEUROSCI.20-16-06125.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirvola U, Ylikoski J, Trokovic R, Hébert JM, McConnell SK, Partanen J. FGFR1 is required for the development of the auditory sensory epithelium. Neuron. 2002;35:671–680. doi: 10.1016/s0896-6273(02)00824-3. [DOI] [PubMed] [Google Scholar]
- Riedel B, Friauf E, Grothe C, Unsicker K. Fibroblast growth factor-2-like immunoreactivity in auditory brainstem nuclei of the developing and adult rat: correlation with onset and loss of hearing. J Comp Neurol. 1995;354:353–360. doi: 10.1002/cne.903540305. [DOI] [PubMed] [Google Scholar]
- Robinson K, Rudge P. Abnormalities of the auditory evoked potentials in patients with multiple sclerosis. Brain. 1977;100:19–40. doi: 10.1093/brain/100.1.19. [DOI] [PubMed] [Google Scholar]
- Rowe MJ., 3rd Normal variability of the brain-stem auditory evoked response in young and old adult subjects. Electroencephalogr Clin Neurophysiol. 1978;44:459–470. doi: 10.1016/0013-4694(78)90030-5. [DOI] [PubMed] [Google Scholar]
- Shah SN, Salamy A. Auditory-evoked far-field potentials in myelin deficient mutant quaking mice. Neuroscience. 1980;5:2321–2323. doi: 10.1016/0306-4522(80)90148-7. [DOI] [PubMed] [Google Scholar]
- Shah SN, Bhargava VK, McKean CM. Maturational changes in early auditory evoked potentials and myelination of the inferior colliculus in rats. Neuroscience. 1978;3:561–563. doi: 10.1016/0306-4522(78)90020-9. [DOI] [PubMed] [Google Scholar]
- Silva VA, Gomide VC, Chadi G. Fibroblast growth factor-2 immunoreactivity is present in the central and peripheral auditory pathways of adult rats. J Morphol. 2005;265:141–151. doi: 10.1002/jmor.10345. [DOI] [PubMed] [Google Scholar]
- Stankovic K, Rio C, Xia A, Sugawara M, Adams JC, Liberman MC, Corfas G. Survival of adult spiral ganglion neurons requires erbB receptor signaling in the inner ear. J Neurosci. 2004;24:8651–8661. doi: 10.1523/JNEUROSCI.0733-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr A, Achor J. Auditory brain stem responses in neurological disease. Arch Neurol. 1975;32:761–768. doi: 10.1001/archneur.1975.00490530083009. [DOI] [PubMed] [Google Scholar]
- Starr A, Amlie RN, Martin WH, Sanders S. Development of auditory function in newborn infants revealed by auditory brainstem potentials. Pediatrics. 1977;60:831–839. [PubMed] [Google Scholar]
- Suter-Crazzolara C, Unsicker K. GDNF mRNA levels are induced by FGF-2 in rat C6 glioblastoma cells. Brain Res Mol Brain Res. 1996;41:175–182. doi: 10.1016/0169-328x(96)00089-7. [DOI] [PubMed] [Google Scholar]
- Thippeswamy T, McKay JS, Morris R, Quinn J, Wong LF, Murphy D. Glial-mediated neuroprotection: evidence for the protective role of the NO-cGMP pathway via neuron–glial communication in the peripheral nervous system. Glia. 2005;49:197–210. doi: 10.1002/glia.20105. [DOI] [PubMed] [Google Scholar]
- Trettel J, Morest DK. Cytoarchitectonic atlas of the cochlear nucleus of the mouse. In: Willott JF, editor. Handbook of mouse auditory research. Boca Raton, FL: CRC Press; 2001. pp. 279–298. [Google Scholar]
- Wei D, Jin Z, Järlebark L, Scarfone E, Ulfendahl M. Survival, synaptogenesis, and regeneration of adult mouse spiral ganglion neurons in vitro. Dev Neurobiol. 2007;67:108–122. doi: 10.1002/dneu.20336. [DOI] [PubMed] [Google Scholar]
- Yoshida K, Gage FH. Fibroblast growth factors stimulate nerve growth factor synthesis and secretion by astrocytes. Brain Res. 1991;538:118–126. doi: 10.1016/0006-8993(91)90385-9. [DOI] [PubMed] [Google Scholar]
- Yu K, Xu J, Liu Z, Sosic D, Shao J, Olson EN, Towler DA, Ornitz DM. Conditional inactivation of FGF receptor 2 reveals an essential role for FGF signaling in the regulation of osteoblast function and bone growth. Development. 2003;130:3063–3074. doi: 10.1242/dev.00491. [DOI] [PubMed] [Google Scholar]
- Zhang X, Ibrahimi OA, Olsen SK, Umemori H, Mohammadi M, Ornitz DM. Receptor specificity of the fibroblast growth factor family. The complete mammalian FGF family. J Biol Chem. 2006;281:15694–15700. doi: 10.1074/jbc.M601252200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R, Assouline JG, Abbas PJ, Messing A, Gantz BJ. Anatomical and physiological measures of auditory system in mice with peripheral myelin deficiency. Hear Res. 1995;88:87–97. doi: 10.1016/0378-5955(95)00104-c. [DOI] [PubMed] [Google Scholar]