

Abstract
Dexamethasone-treated L6 (a rat cell line) and C2C12 (a mouse cell line) myotubes are frequently used as in vitro models of muscle wasting. We compared the effects of different concentrations of dexamethasone and corticosterone (the naturally occurring glucocorticoid in rodents) on protein breakdown rates, myotube size, and atrogin-1 and MuRF1 mRNA levels in the two cell lines. In addition, the expression of the glucocorticoid receptor (GR) and its role in glucocorticoid-induced metabolic changes were determined. Treatment with dexamethasone or corticosterone resulted in dose-dependent increases in protein degradation rates in both L6 and C2C12 myotubes accompanied by 25-30% reduction of myotube diameter. The same treatments increased atrogin-1 mRNA levels in L6 and C2C12 myotubes but, surprisingly, upregulated the expression of MuRF1 in L6 myotubes only. Both cell types expressed the GR and treatment with dexamethasone or corticosterone downregulated total cellular GR levels while increasing nuclear translocation of the GR in both L6 and C2C12 myotubes. The GR antagonist RU38486 inhibited the dexamethasone- and corticosterone-induced increases in atrogin-1 and MuRF1 expression in L6 myotubes but not in C2C12 myotubes. Interestingly, RU38486 exerted agonist effects in the C2C12, but not in the L6 myotubes. The present results suggest that muscle wasting-related responses to dexamethasone and corticosterone are similar, but not identical, in L6 and C2C12 myotubes. Most notably, the regulation by glucocorticoids of MuRF1 and the role of the GR may be different in the two cell lines. These differences need to be taken into account when cultured myotubes are used in future studies to further explore mechanisms of muscle wasting.
Keywords: Muscle wasting, glucocorticoids, myotubes
INTRODUCTION
Muscle wasting, mainly reflecting increased myofibrillar protein breakdown, is commonly seen in patients with various catabolic conditions, including severe injury [Pereira et al., 2005], sepsis [Tiao et al., 1997; Klaude et al., 2007], and cancer [Williams et al., 1999; Acharyya and Guttridge, 2007]. Glucocorticoids are important mediators of muscle proteolysis and were found in previous studies to upregulate ubiquitin-proteasome-dependent protein degradation in skeletal muscle [Hasselgren, 1999; Menconi et al., 2007]. In addition, muscle wasting induced by sepsis or burn injury was prevented by treatment with the glucocorticoid receptor antagonist RU38486 [Fang et al., 1995; Tiao et al., 1996].
Glucocorticoid-treated cultured myotubes have been used in multiple previous studies as an in vitro model of muscle wasting [Hong and Forsberg, 1995; Wang et al., 1998; Thompson et al., 1999; Du et al., 2000; Sacheck et al., 2004; Stitt et al., 2004; Evenson et al., 2005; Yang et al., 2005a; Marinovic et al., 2006]. In most of those studies, rat L6 or mouse C2C12 myotubes were treated with dexamethasone, resulting in increased protein degradation and upregulated expression of several genes in the ubiquitin-proteasome proteolytic pathway [Wang et al., 1998; Thompson et al., 1999; Evenson et al., 2005; Du et al., 2000; Sacheck et al., 2004; Stitt et al., 2004; Latres et al., 2005; Marinovic et al., 2006; Sultan et al., 2006]. Because these and other metabolic changes induced by dexamethasone resemble changes seen in atrophying muscle in experimental animals [Tiao et al., 1994, 1996; Wray et al., 2003] and patients [Tiao et al., 1997; Williams et al., 1999; Acharyya and Guttridge, 2007; Klaude et al., 2007], dexamethasone-treated myotubes have been used to define mechanisms of muscle wasting.
Despite the frequent use of dexamethasone-treated myotubes in muscle wasting research, there are a number of important questions that have not been addressed regarding the validity of this experimental model. First, it is not known if the effects are specific for the artificial glucocorticoid dexamethasone or whether similar changes can be induced by corticosterone, the naturally occurring glucocorticoid in rodents. Second, the concentrations of dexamethasone used in previous studies varied substantially (10,000-fold) from 10 nM to 100 μM [Du et al., 2000; Stitt et al., 2004; Latres et al., 2005; Marinovic et al., 2006]. Considering the approximately 25-fold higher potency of dexamethasone compared with corticosterone [Orth et al., 1992], the dexamethasone concentrations used in many previous studies were supraphysiologic and it is not known if L6 and C2C12 myotubes respond similarly to different concentrations of dexamethasone, including concentrations reflecting physiological glucocorticoid concentrations. Finally, it is not known if similar mechanisms of muscle wasting are activated by glucocorticoids in L6 and C2C12 myotubes. This is particularly important because recent studies suggest that the expression of the glucocorticoid receptor (GR) may be different in L6 and C2C12 myotubes [Sultan et al., 2006] and different responses of gene regulation were reported in the two cell lines after treatment with dexamethasone [Tortorella and Pilch, 2002].
In the present study, we compared the effects of different concentrations of dexamethasone and corticosterone on protein degradation and the expression of the muscle wasting-associated ubiquitin ligases atrogin-1 and MuRF1 in L6 and C2C12 mytotubes. Results suggest that both dexamethasone and corticosterone induce similar changes in protein degradation and atrogin-1 mRNA levels in the two cell lines. In contrast, the regulation of MuRF1 expression by the glucocorticoids as well as the role of the glucocorticoid receptor (GR) in dexamethasone- and corticosterone-induced changes were different in L6 and C2C12 myotubes.
MATERIALS AND METHODS
Materials
Corticosterone, dexamethasone, mifepristone (RU38486), and fetal bovine serum (FBS), were from Sigma-Aldrich (St. Louis, MO). L-[ring-3,5-3H]-tyrosine (40-60 Ci/mmol), Western Lightning™ Chemiluminescence Reagent Kit for enhanced chemiluminescence detection, and Solvable™ tissue solubilizer were purchased from PerkinElmer Life Sciences (Boston, MA). The NE-PER® Nuclear and Cytoplasmic Extraction Kit was purchased from Pierce Biotechnology (Rockford, IL). Immun-Blot PVDF Membrane used for Western blotting and 10% Ready Tris-HCl Mini-gels were purchased from Bio-Rad Laboratories (Hercules, CA). Primers and probes for quantitative real-time PCR were custom-synthesized by Biosearch Technologies, Inc. (Novato, CA). TaqMan One Step PCR Master Mix Reagents Kits were purchased from Applied Biosystems (Foster City, CA). Kodak X-Omat blue film was from Eastman Kodak (Rochester, NY). Dulbecco’s modified Eagle’s medium (DMEM) was purchased from Mediatech Inc. (Herndon, VA). Penicillin/streptomycin and trypsin were from Invitrogen Corporation (Carlsbad, CA). All tissue culture plastic ware was purchased from Corning (Corning, NY). All chemicals were reagent-grade.
Cell cultures
Rat L6 and mouse C2C12 skeletal muscle cells, both originally developed by Yaffe [1968] and Yaffe and Saxel [1977], were purchased from the American Type Culture Collection (Manassas, VA). Both myogenic cell lines have been extensively used in muscle wasting research. L6 and C2C12 cells were grown and maintained in high-glucose DMEM supplemented with 10% FBS, 100 U/ml of penicillin, and 100 μg/ml of streptomycin in a 10% CO2 humidified atmosphere at 37°C. Since the extent of myoblast fusion into myotubes declines with increasing passage, cells were used at low passage (between passages 2 and 4) for all experiments to maintain the differentiation potential of the cultures. Cells grown to approximately 80% confluence in T75 culture flasks were trypsinized and seeded into 10 cm culture dishes or 6 or 12 -well culture plates for experiments. The cells were grown in the presence of 10% FBS until they reached about 90% confluence at which time the medium was replaced with DMEM containing 2% FBS for the induction of differentiation into myotubes. During this time, the myoblasts fused to form elongated, multi-nucleated myotubes. After 7 days, when greater than 90% of the cells had fused into myotubes, the cultures were treated with 10 μM cytosine arabinoside for 3 days to remove any dividing myoblasts. L6 and C2C12 myotube cultures were then treated with different concentrations of dexamatheasone, corticosterone, or RU38486 dissolved in 0.1% ethanol for 24 h (except when myotube diameters were measured and treatment was extended to 48 h) as described in Results. Control myotubes were treated with solvent (0.1% ethanol).
Measurement of myotube diameter
Myotube cultures were photographed under a phase contrast microscope at 100X magnification after 24 and 48 h treatment. The diameters were measured in a total of 60 myotubes from at least 10 random fields using ImageJ software (NIH, Frederick, MD). The measurements were conducted in a “blinded” fashion on coded pictures with the investigator being unaware of the group (control, dexamethasone- or corticosterone-treated myotubes) from which the cultures originated. Results were expressed as per cent of the diameter in the control group.
Measurement of protein degradation
Rates of protein degradation were determined by measuring the release of TCA-soluble radioactivity from proteins prelabeled with [3H]-tyrosine as described previously (9,10). After completing differentiation, myotubes were labeled with 1.0 μCi/ml of L-[3,5-3H]-tyrosine for 48 h in DMEM containing 2% FBS. Cells were then treated for 24 h with glucocorticoids and RU38486 in DMEM containing 2 mM unlabeled tyrosine. After treatment, the culture medium was transferred into a microcentrifuge tube containing 100 μl of bovine serum albumin (10 mg/ml) and TCA was added to a final concentration of 10% (w/v). Samples were incubated at 4°C for 1 h followed by centrifugation for 5 min. The supernatant was used for determination of TCA-soluble radioactivity. The protein precipitates were dissolved with a tissue solubilizer (Solvable™). Cell monolayers were washed with ice-cold PBS and solubilized with 0.5 M NaOH containing 0.1% Triton X-100. Radioactivity in the cell monolayer and TCA soluble and insoluble fractions were measured using a Packard TRI-CARB 1600 TR liquid scintillation analyzer. Protein degradation was expressed as the percentage protein degraded over the 24 h period and was calculated as 100 times the TCA-soluble radioactivity in the medium divided by the TCA-soluble plus the TCA-insoluble radioactivity in the medium plus the cell layer (i.e., myotube) radioactivity [Hong and Forsberg, 1995; Wang et al., 1998].
Measurement of protein synthesis
In order to determine whether the effect of glucocorticoids on protein degradation resulted from a generalized effect on protein turnover or was specific for protein degradation, the effect of glucocorticoids on protein synthesis was examined. Rates of protein synthesis were determined by measuring the incorporation of TCA insoluble radioactivity into cellular proteins prelabeled with [3H]-tyrosine, as described previously [Wang et al., 1998]. Myotubes were treated for 48 h with glucocorticoids. During the last hour of incubation, myotubes were incubated with 2.0 μCi/ml of L-[3,5-3H]-tyrosine in DMEM (2% FBS) containing 200 mM unlabeled tyrosine. After incubation, the medium was removed and the cell layers were harvested in 10% TCA. The TCA-precipitated proteins were washed several times and then solubilized with 0.1 N NaOH containing 1% Triton X-100. Radioactivity and protein levels were measured in aliquots from the solubilized proteins, as described above and below, respectively, and protein synthesis was expressed as cpm/mg protein.
Preparation of Total Cell Lysates and Nuclear Extracts
Total cell lysates were prepared by harvesting the myotubes directly in RIPA buffer (50 mM Tris-HCl, 150 mM NaCl, 0.5% sodium deoxycholate, 0.1% SDS, and 1% Nonidet P-40) containing Protease Inhibitor Cocktail Tablets (Roche Applied Science, Indianapolis, IN). After scraping the lysates into epindorf tubes, the samples were briefly sonicated using a Sonic Dismembrator (Fisher Scientific, Model 100) followed by centrifugation at 14,000 x g for 10 minutes at 4°C. Nuclear extracts were prepared from differentiated myotubes using the NE-PER® Nuclear and Cytoplasmic Extraction Reagents according to the manufacturer’s instructions. Concentrations of soluble proteins in the supernatants of the nuclear extracts and total cell lysates were determined by using the BCA Protein Assay Reagent Kit with bovine serum albumin as standard. Cell lysates were stored at −80°C until analyzed.
Western blotting
Western blotting was performed to determine protein levels of GR in myotubes. Aliquots containing 50 μg of cellular protein, prepared as described above, were boiled in Laemmli sample buffer containing 2-mercaptoethanol. Proteins were separated by sodium dodecyl sulphate (SDS)-polyacrylamide gel electorphoresis using 10% Tris-HCl gels and then transferred electrophoretically at 2000 mA for 1 h in transfer buffer (25 mM Tris, 192 mM glycine, pH 8.3, containing 20% methanol) onto Immun-Blot PVDF membranes at 4°C. The membranes were blocked with TTBS buffer (50 mM Tris-HCl, 130 mM NaCl and 0.05% Tween-20, pH 7.4) containing 5% nonfat dry milk for 1 h at room temperature. The membranes were then incubated overnight with a 1:1000 dilution of GR antibody (rabbit polyclonal antibody raised against amino acids 121-420 of human GR; Santa Cruz Biotechnology, Santa Cruz, CA). This antibody detects both the GRα and β isoforms of rat and mouse GR (Santa Cruz Biotechnology, technical notes). Following incubation with the primary antibody, the membranes were washed with TTBS 3 times and incubated for 1h at room temperature with a goat anti-rabbit horseradish peroxidase-conjugated secondary antibody (Santa Cruz Biotechnology) at a 1:10,000 dilution. Following an additional three washes with TTBS, immunoreactive protein bands were visualized by using the Western Lightning ™ Kit for enhanced chemiluminescence followed by exposure to Kodak X-Omat blue film. The identity of the bands on the Western blots was confirmed by including precision molecular weight standards during electrophoresis. Blots were stripped using Restore™ Western Blot Stripping Buffer according to the manufacturers instructions and reprobed with α-tubulin antibody to confirm equal loading.
Measurement of mRNA levels
Atrogin-1, MuRF1, and GR mRNA levels were determined by real-time PCR, as described in detail recently [Wei et al., 2005]. Primers for atrogin-1, MuRF1, and GR were designed for rat (L6 myotubes) and mouse (C2C12 myotubes) using Primer Express 1.5 (Applied Biosystems, Foster City, CA). Cellular RNA was extracted from cultured myotubes grown in 6-well plates by the acid guanidinium thiocyanate-phenol-chloroform method [Chomczynski and Sacchi, 1987] using TRI REAGENT® (Molecular Research Center, Inc., Cincinnati, OH). Multiplex real-time PCR was performed for quantitation of mRNA expression with simultaneous amplification of 18S RNA as endogenous controls to normalize the mRNA concentrations. TaqMan analysis using the One Step PCR Master Mix Reagents Kit and subsequent calculations were performed with an ABI PRISM® 7700 Sequence Detection System (Applied Biosystems). For each sample, 100 ng of total RNA was subjected to real-time PCR (in duplicate) according to the protocol provided by the manufacturer. The sequences of the forward and reverse primer oligonucleotides, and the double-labeled TaqMan oligonucleotide probes for the mRNA’s were as follows, respectively: rat MuRF1; 5′-CGA CTC CTG CCG AGT GAC C-3′, 5′-GCG TCA AAC TTG TGG CTC AG-3′, and 5′-AGG AAA ACA GCC ACC AGG TGA AGG AGG-3′; rat Atrogin-1; 5′-CTT TCA ACA GAC TGG ACT TCT CGA-3′, 5′-CAG CTC CAA CAG CCT TAC TAC GT-3′, and 5′-TCG CAT CCT GGA TTC CAG AAG ATT CAA C-3′; mouse MuRF1; 5′-TGT CTG GAG GTC GTT TCC G-3′, 5′-TGC CGG TCC ATG ATC ACT T-3′, and 5′-TGC CCC TCG TGC CGC CAT-3′; rat GR; 5′-GAA TGA CTT GGG CTA CCC ACA-3′, 5′-GAA GCC GAA AGT CTG TTT CCC-3′, and 5′-CAG GGC CAA CTT GGC CTT TCC TCT-3′; mouse GR; 5′-ACA GCA ACG GGA CCA CCT C-3′, 5′-AAT GGC ATC CCG AAG CTT C-3′, and 5′-CAA ACT CTG CCT GGT GTG CTC CGA-3′. To measure mouse atrogin-1 mRNA levels, rat atrogin-1 primers were used, as these primers were validated to cross-react with mouse (C2C12) RNA (V. Petkova, unpublished observation). Three or four individual wells were used for each experimental group. mRNA levels in control myotubes were arbitrarily set to 1.0.
Statistical analysis
Results are presented as means ± SEM. Statistical analysis was performed by ANOVA followed by Tukey’s or Student-Newman-Keul’s test, as appropriate. p< 0.05 was considered statistically significant. Most experiments were repeated at least three times to provide evidence of reproducibility.
RESULTS AND DISCUSSION
In several previous reports, treatment of cultured L6 or C2C12 myotubes with dexamethasone was used as an in vitro model of muscle atrophy. The concentration of dexamethasone varied substantially in those studies with effects of dexamethasone induced by concentrations ranging from 10-50 nM [Thompson et al., 1999; Du et al., 2000; Marinovic et al., 2006] to 100 μM [Stitt et al., 2004; Latres et al., 2005]. No comparison of the effects of different concentrations of dexamethasone on protein degradation in the two cell lines has been reported previously. Here, we compared protein breakdown rates in L6 and C2C12 myotubes treated with different concentrations of dexamethasone (Fig 1A and B). Basal protein breakdown rates were similar (approximately 20-22%/24 h) in the two cell lines. Also, the sensitivity and responsiveness to dexamethasone were similar in L6 and C2C12 myotubes with a significant increase in protein degradation induced by 10 nM dexamethasone and a maximal effect (approximately 15-20%) noticed for 100 nM dexamethasone. The results are similar to previous reports in which treatment with dexamethasone stimulated protein degradation in both L6 [Wang et al., 1998; Evenson et al., 2005; Li et al., 2005] and C2C12 myotubes [Thompson et al., 1999; Sacheck et al., 2004; Stitt et al., 2004; Latres et al., 2005].
Fig 1. Glucocorticoids increase protein degradation in cultured L6 and C2C12 myotubes in a dose-dependent manner.

Proteins in differentiated myotubes were prelabeled for 48 h with [3H]-tyrosine and the myotubes were then treated for 24 h with different concentrations of dexamethasone (A and B) or corticosterone (C and D). Control myotubes were treated with solvent (0.1% ethanol). Protein degradation was determined by measuring release of TCA-soluble radioactivity into the culture medium as described in Materials and Methods. Results are means ± SEM with n=6 per group. *p<0.05 vs control (0 dexamethasone or corticosterone).
The present observations are important because they suggest that protein degradation is regulated by dexamethasone to a similar extent in the two cell lines and that dexamethasone can induce proteolysis at concentrations corresponding to expected physiological corticosterone levels. Serum concentrations of corticosterone in rats and mice vary from 0.06 to 0.40 μM under normal conditions and from 0.9 to 3.0 μM during stress [Raina and Jeejeebhoy, 1998; Voisin et al., 1998; Faggioni et al., 2000; Fischer et al., 2001]. Since dexamethasone is an artificial glucocorticoid with a potency approximately 25 times greater than that of the naturally occurring corticosterone [Orth et al., 1992], concentrations up to about 120 nM are in the physiological range. Hence, dexamethasone concentrations used in many previous studies corresponded to supraphysiological concentrations of corticosterone, at least when comparing with normal serum concentrations of corticosterone.
It should be noted, however, that comparing in vitro concentrations of glucocorticoids with physiological circulating concentrations needs to be done with caution. First, although normal ranges of corticosterone serum concentrations in rats have been established, it is not known what the corresponding muscle tissue concentrations are. Second, the in vitro experiments in the present study were conducted in the presence of serum (2% FBS), which contains albumin that is known to bind steroids [Milgrom, 1990], so even the free hormone concentration directly acting upon the cultured myotubes is not precisely known.
The influence of different concentrations of corticosterone, including concentrations in the range of normal rat and mouse serum levels, on protein degradation in cultured L6 and C2C12 myotubes is not known. Here, we found that treatment of the two cell lines with corticosterone at concentrations well within the range of estimated physiological concentrations resulted in increased protein degradation (Fig 1C and D). A significant increase in protein degradation was noticed at a corticosterone concentration of 0.1 μM in both cell lines consistent with a similar sensitivity to corticosterone in L6 and C2C12 myotubes. The maximal effect was seen at a corticosterone concentration of 1 μM and was somewhat greater in the L6 myotubes (approximately 20% increase in protein degradation) than in the C2C12 myotubes (approximately 10% increase in protein degradation). Taken together, the results shown in Fig 1 suggest that glucocorticoid-induced protein degradation in cultured L6 and C2C12 myotubes is not specific for the artificial glucocorticoid dexamethasone but can be induced by the naturally occurring corticosterone as well, even at concentrations corresponding to normal serum levels of the hormone.
We next examined whether the increase in protein degradation caused by dexamethasone and corticosterone was associated with reduced size of the myotubes. Treatment of the cultured muscle cells with glucocorticoids did not significantly affect myotube diameter after 24 h but decreased the diameter of L6 and C2C12 myotubes by approximately 25% and 30%, respectively, after treatment for 48 h (Fig 2A and B). These results suggest that the increase in protein degradation caused by dexamethasone and corticosterone results in myotube atrophy.
Fig 2. Treatment of cultured L6 and C2C12 myotubes with glucocorticoids results in myotube atrophy.
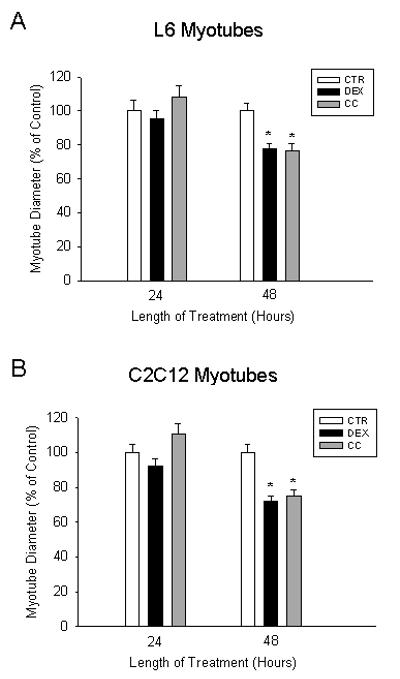
Differentiated myotubes were treated with vehicle (0.1% ethanol), 1 μM dexamethasone (DEX), or 1 μM corticosterone (CC) for 24 and 48 h. Myotube diameters from randomly selected fields were quantified using an image analysis program as described in Methods. Diameters were expressed as percent of control. Results are means ± SEM with n=60 per group. *p<0.05 vs control (CTR).
In order to determine whether reduced protein synthesis may also have contributed to the reduction in myotube size, the response to glucocorticoids of protein synthesis was examined. In L6 cells, dexamethasone induced a slight but statistically significant decrease in protein synthesis, while corticosterone did not alter protein synthesis rates (Fig 3A), n C2C12 cells, glucocorticoids had no effect on protein synthesis (Fig 3B). Although reduced muscle protein synthesis after treatment with glucocorticoids has been reported in several studies [Schakman et al., 2008], variable and apparently conflicting results have also been reported. For example, studies in L6 myotubes and myoblasts demonstrated that dexamethasone either increased or decreased protein synthesis depending upon the concentration of hormone used [Orzechowski et al., 2003], or the length of time of treatment [Roeder et al., 1986]. Other studies showed that short-term treatment (hrs) with dexamethasone reduced protein synthesis in C2C12 myotubes [Desler et al., 1996] and L6 myoblasts [Shah et al., 2000]. Thus, the effect of dexamethasone on protein synthesis appears to depend upon the concentration of glucocorticoid used, the length of time of treatment, and the differentiation state of the cells (myoblasts vs myotubes). Regardless, the results in the present study suggest that the increase in protein degradation induced by glucocorticoids is the primary cause of myotube atrophy, at least under the present experimental conditions.
Fig 3. Treatment of cultured L6 and C2C12 myotubes with glucocorticoids does not alter protein synthesis.

Myotube proteins were prelabeled for 1 h with [3H]-tyrosine following a 48 h treatment with 1 μM dexamethasone (DEX) or 1 μM corticosterone (CC). Control myotubes (CTR) were treated with solvent (0.1% ethanol). Protein synthesis was determined by measuring the incorporation of TCA-insoluble radioactivity into cellular proteins as described in Materials and Methods. Results are means ± SEM with n=4 to 8 per group. *p<0.05 vs control.
Previous studies suggest that muscle proteolysis in various catabolic conditions, including sepsis [Tiao et al., 1994, 1996, 1997; Wray et al., 2003; Klaude et al., 2007], burn injury [Fang et al., 1995; Pereira et al., 2005], and cancer [Williams et al., 1999; Acharyya and Guttridge, 2007], at least in part reflects increased ubiquitin-proteasome-dependent protein breakdown. Recent observations suggest that the ubiquitin ligases atrogin-1 and MuRF1 are particularly important for the regulation of the ubiquitin-proteasome system in atrophying muscle [Bodine et al al., 2001; Gomes et al., 2001; Wray et al., 2003] and in dexamethasone-treated myotubes [Sacheck et al., 2004; Stitt et al., 2004; Evenson et al., 2005; Latres et al., 2005].
It is not known from previous studies whether atrogin-1 and MuRF1 are regulated similarly by dexamethasone and corticosterone in L6 and C2C12 myotubes. In the present study, treatment with dexamethasone (50 nM or 1 μM) or corticosterone (0.1 or 1 μM) resulted in an approximately 2- to 2.5-fold increase in atrogin-1 mRNA levels in L6 and C2C12 myotubes (Fig 4A and B). Interestingly, the sensitivity to corticosterone with regards to atrogin-1 expression was lower in C2C12 than in L6 myotubes. Thus, atrogin-1 mRNA levels were significantly increased by 0.1 μM corticosterone in L6 myotubes whereas in the C2C12 myotubes, a corticosterone concentration of 10 μM was required to induce an increase in atrogin-1 mRNA levels. Increased expression of atrogin-1 in dexamethasone-treated L6 [Evenson et al., 2005; Yang et al., 2005a] and C2C12 [Sacheck et al., 2004; Stitt et al., 2004; Latres et al., 2005] myotubes was observed in previous reports as well, supporting the important role of this ubiquitin ligase in glucocorticoid-regulated muscle wasting. The present findings expand previous observations by showing that corticosterone, like dexamethasone, increases the expression of atrogin-1 in cultured myotubes.
Fig 4. Atrogin-1 and MuRF1 mRNA levels are differentially regulated by glucocorticoids in cultured L6 and C2C12 myotubes.

Differentiated myotubes were treated for 24 h with different concentrations of dexamethasone (DEX) or corticosterone (CC) followed by determination of mRNA levels for atrogin-1 (A and B) or MuRF1 (C and D) by real-time PCR. mRNA levels in myotubes treated with solvent (0.1% ethanol; control) were arbitrarily set to 1.0. Results are means ± SEM with n=3 or 4 per group. Similar results were seen in 3 repeated experiments. *p<0.05 vs control (CTR).
MuRF1 mRNA levels were increased 2- to 3-fold by dexamethasone and corticosterone in L6 myotubes (Fig 4C). In sharp contrast, MuRF1 expression was not influenced by the hormones in C2C12 myotubes (Fig 4D). The unresponsiveness to both dexamethasone and corticosterone of MuRF1 expression in the C2C12 myotubes was observed in repeated experiments. This observation is supported by previous studies indicating that MuRF1 is not strongly regulated by glucocorticoids in C2C12 myotubes. For example, Stitt et al [2004] reported that whereas treatment of cultured C2C12 myotubes for 24 h with different concentrations (1, 10, or 100 μM) of dexamethasone resulted in a relatively robust upregulation of atrogin-1 mRNA levels, MuRF1 mRNA was not influenced by 1 or 10 μM dexamethasone and was only slightly increased after treatment of the myotubes with 100 μM dexamethasone. A similar relative unresponsiveness of MuRF1 to dexamethasone in C2C12 myotubes was reported by Sacheck et al [2004]. In their study, treatment of cultured C2C12 myotubes with 400 ng/ml (1 μM) of dexamethasone for 6 h resulted in a 5-fold increase in atrogin-1 mRNA levels but only a 2-fold increase in MuRF1 mRNA levels. Notably, in the study by Sacheck et al [2004], “dexamethasone” treatment consisted of a combined treatment with dexamethasone (400 ng/ml) and triiodothyronine (T3; 100 ng/ml) because other experiments in the same study indicated that the effects of dexamethasone were potentiated by T3. The combined treatment may be a reason why “dexamethasone” sitmulated the expression of atrogin to a greater extent (5-fold) than the approximately 2- to 2.5-fold increase that was observed in the present and other studies [Sultan et al., 2006] and why MuRF1 mRNA levels were increased at all.
Interestingly, there is evidence to suggest that the downregulation of atrogin-1 and MuRF1 by anabolic factors may also be differentially regulated in C2C12 myotubes. For example, in the report by Sacheck et al [2004], treatment of C2C12 myotubes with insulin or IGF-1 for 6 h resulted in an approximately 70% decrease in atrogin-1 expression but did not reduce MuRF1 mRNA levels. The mechanism behind the unresponsiveness of MuRF1 to glucocorticoids (as well as insulin and IGF-1) in C2C12 myotubes is not known at present. Although the two cell lines studied in the present report originated from different species (L6 myotubes being a rat cell line and C2C12 myotubes being a mouse cell line) it is not likely that the unresponsiveness of MuRF1 in C2C12 noticed here and in other reports [Sacheck et al., 2004; Stitt et al., 2004] reflected the species difference because increased MuRF1 expression in skeletal muscle has been reported in various catabolic conditions in both rats [Wray et al., 2003; Lang et al., 2006] and mice [Bodine et al., 2001; Cai et al., 2004].
Another potential mechanism whereby glucocorticoids induce different responses in L6 and C2C12 myotubes may be different abundance of the GR in the two cell lines. In order to examine that possibility, we determined GR concentrations by Western blotting. C2C12 myotubes did not express lower levels of the GR but actually expressed higher concentrations of the hormone receptor (Fig 5A). As expected, treatment with dexamethasone or corticosterone resulted in reduced levels of the GR in both cell lines, consistent with a negative feed-back effect [Vujcic et al., 2007]. Similar to previous reports in other cell types [Erdeljan et al., 2005], the glucocorticoid-induced downregulation of the GR protein levels was associated with reduced concentrations of GR mRNA (Fig 5B), suggesting that glucocorticoids downregulate the expression of the GR at the transcriptional level.
Fig 5. The glucocorticoid receptor (GR) is expressed in cultured L6 and C2C12 myotubes.

Differentiated myotubes were treated for 24 h with 1 μM dexamethasone (DEX) or 1 μM corticosterone (CC). Control myotubes (CTR) were treated with solvent (0.1% ethanol). (A) GR protein levels were determined by Western blotting in total cell lysates showing a glucocorticoid-induced decrease in GR levels. The GR bands represent the GRα isoform with a molecular weight of 95 kDa. α-Tubulin levels were determined as loading controls. (B) GR mRNA levels were determined by real-time PCR as described in Materials and Methods. GR mRNA levels in control myotubes were arbitrarily set to 1.0. Results are means ± SEM with n=3 per group. *p<0.05 vs control. (C) GR protein levels in myotube nuclear extracts showing a glucocorticoid-induced translocation of GR from the cytoplasm into the nucleus.
Glucocorticoids bind to the GR in the cytoplasm followed by dimerization and translocation of the receptor into the nucleus. In the nucleus, the GR binds to glucocorticoid response elements of glucocorticoid-responsive genes, resulting in transcriptional regulation [Heitzer et al., 2007]. In the present experiments, treatment of both L6 and C2C12 myotubes with dexamethasone or corticosterone resulted in increased nuclear translocation of the GR (Fig 5C). Taken together, the results in Fig 5 suggest that a less pronounced response to glucocorticoids in C2C12 compared to L6 myotubes (with regards to MuRF1 expression and corticosterone-induced protein degradation) does not reflect a lower concentration of the GR or different regulation of the GR by glucocorticoids in C2C12 myotubes.
Importantly, in a recent study we found that treatment of cultured C2C12 myotubes with TNFα or IL-1β activated the MuRF1 promoter [Cai et al., 2004] suggesting that MuRF1 may be sensitive to catabolic factors other than glucocorticoids in C2C12 myotubes. Indeed, additional experiments in the same study supported a model in which proinflammatory cytokines upregulate MuRF1 expression and cause muscle atrophy through an NF-kB-dependent mechanism. Thus, the present results and previous reports suggest that although both L6 and C2C12 myotubes can be used to examine glucocorticoid-induced protein degradation, there may be significant differences between the two cell lines with regards to the involvement of MuRF1 in the response to glucocorticoids.
Glucocorticoids typically exert their metabolic effects secondary to binding to the GR although other, GR-independent, mechanisms have been described as well [Sawart and Cabillic, 1985; Falkenstein et al., 2000; Borski et al., 2002]. The role of GR-binding in glucocorticoid-induced metabolic changes can be tested by using the GR antagonist RU38486 [Konagaya et al., 1986]. This drug also binds to and blocks the progesterone receptor [Philibert, 1984] but because skeletal muscle does not express the progesterone receptor [Perrot-Applanat et al., 1985], effects of RU38486 in skeletal muscle are usually considered to reflect GR inhibition [Konagaya et al., 1986]. RU38486 was used in several previous reports both from our [Hall-Angeras et al., 1991; Fang et al., 1995; Tiao et al., 1996] and other laboratories [Lang et al., 2006] to examine the role of glucocorticoids in muscle wasting.
In order to test the role of the GR in dexamethasone- and corticosterone-induced changes in protein degradation and ubiquitin ligase expression in the present study, we treated myotubes with RU38486. Treatment of L6 myotubes with 10 μM RU38486 alone did not influence protein degradation but blunted the dexamethasone- and corticosterone-induced increase in protein degradation (Fig 6A). In contrast to the finding in L6 myotubes, treatment of C2C12 myotubes with RU38486 alone stimulated protein degradation (Fig 6B). Although RU38486 usually does not exert an agonist effect after binding to the GR, agonist effects have been reported previously [Schulz et al., 2002; Yang et al., 2005b]. When dexamethasone-treated C2C12 myotubes were exposed to RU38486, the increased protein degradation was not reduced (third and fourth bar in Fig 6B). The increase in protein degradation caused by RU38486 alone makes interpretation of this observation difficult and it is not known if the lack of inhibition by RU38486 reflected RU38486-induced increase in protein degradation or GR-independent effects of dexamethasone in C2C12 myotubes. The fact that RU38486 reduced protein degradation in corticosterone-treated C2C12 myotubes (last two bars in Fig 6B), however, suggests that glucocorticoids induce protein degradation in C2C12 myotubes at least in part secondary to binding to the GR receptor, similar to the mechanism in glucocorticoid-treated L6 myotubes.
Fig 6. Protein degradation is differentially regulated by the glucocorticoid receptor antagonist RU38486 in cultured L6 and C2C12 myotubes.

Differentiated L6 (A) and C2C12 (B) myotubes were treated for 24 h with 1 μM dexamethasone (DEX) or 1 μM corticosterone (CC) in the absence or presence of 10 μM RU38486 (RU). Other myotubes were treated with 10 μM RU38486 alone. Control myotubes were treated with solvent (0.1% ethanol). Results are means ± SEM with n=6 per group. *p<0.05 vs control; #p<0.05 vs dexamethasone; +p<0.05 vs corticosterone.
Similar to the effects on protein degradation, RU38486 alone did not influence the expression of atrogin-1 in L6 myotubes but upregulated atrogin-1 mRNA levels approximately 2.5-fold in C2C12 myotubes, almost identical to the effect of dexamethasone (Fig 7A and B). Atrogin-1 mRNA levels in dexamethasone- and corticosterone-treated L6 myotubes were reduced by RU38486 but were not significantly influenced by the glucocorticoid receptor antagonist in glucocorticoid-treated C2C12 myotubes.
Fig 7. Atrogin-1 and MuRF1 mRNA levels are differentially regulated by the glucocorticoid receptor antagonist RU38486 in cultured L6 and C2C12 myotubes.

Differentiated myotubes were treated for 24 h with 1 μM dexamethasone (DEX) or 1 μM corticosterone (CC) in the absence or presence of 10 μM RU38486 (RU). Other myotubes were treated with 10 μM RU38486 alone. Control myotubes (CTR) were treated with solvent (0.1% ethanol). After treatments, mRNA levels for atrogin-1 (A and B) and MuRF1 (C and D) were determined by real-time PCR. Results are means ± SEM with n=3 or 4 per group. *p<0.05 vs control; #p<0.05 vs dexamethasone; +p<0.05 vs corticosterone.
The effects of RU38486 on MuRF1 mRNA levels in control and glucocorticoid-treated myotubes are shown in Fig 7C and D. The regulation by RU38486 of MuRF1 expression in L6 myotubes (Fig 7C) was similar to the regulation of atrogin-1 expression (see Fig 7A). In C2C12 myotubes, RU38486 did not influence MuRF1 mRNA levels in control or glucocorticoid-treated C2C12 myotubes (Fig 7D).
The reason why the effects of RU38486 were different in L6 and C2C12 myotubes is not known at present. An agonist effect of RU38486 combined with a relative inability to block glucocorticoid-induced changes (as noticed in the C2C12 myotubes in the present study) has been reported in other cell types as well [Schulz et al., 2002; Zhang et al., 2003] and may reflect the involvement of GR-independent signaling pathways induced by glucocortiocids [Sawart and Cabillic, 1985; Falkenstein et al., 2000; Borski et al., 2002]. Results from experiments in other cell types suggest that GR-independent responses to glucocorticoid treatment may at least in part be mediated by membrane-associated receptors and activation of various intracellular signaling pathways [Lackner et al., 1998; Qiu et al., 2001].
It should be noted that although results in the present study suggest that an agonist effect of RU38486 may be specific for the C2C12 myotubes, in recent studies we found that RU38486 had an agonist effect on the expression of the nuclear cofactor p300 and the transcription factor C/EBPβ in L6 myotubes and did not prevent the dexamethasone-induced increase in p300 and C/EBPβ levels [Yang et al., 2005a,b]. Thus, it is possible that different effects of glucocorticoids are mediated by different mechanisms in the same cell type as exemplified by the inability of RU38486 to prevent dexamethasone-induced p300 and C/EBPβ expression in L6 myotubes [Yang et al., 2005a,b] and inhibition of glucocorticoid-induced protein degradation and expression of atrogin-1 and MuRF1 by RU38486 in L6 myotubes in the present study.
Although the present results support the use of dexamethasone-treated L6 and C2C12 myotubes as in vitro models of muscle wasting, the study has several potential limitations that need to be considered. First, the effects of dexamethasone or corticosterone alone were determined. Even if glucocorticoids may be important regulators of muscle breakdown [Hasselgren, 1999; Menconi et al., 2007], there is evidence that muscle proteolysis in various catabolic conditions is regulated by multiple factors in addition to glucocorticoids, in particular the proinflammatory cytokines TNFα and IL-1β [Argiles et al., 2005]. The observation that MuRF1 was upregulated in C2C12 myotubes by TNFα and IL-1β in recent experiments [Cai et al., 2004] but not by glucocorticoids in the present study illustrates the importance of catabolic factors other than glucocorticoids in the development of muscle wasting. Interestingly, previous studies suggest that glucocorticoids and TNF interact at the cellular level to induce muscle wasting [Hall-Angeras et al., 1990]. Other studies suggest that most of the catabolic effects of TNF are secondary to TNF-induced release of glucocorticoids from the adrenal glands [Warren et al., 1988; Zamir et al., 1992], further supporting the use of myotubes treated with glucocorticoids as a model of muscle wasting.
An additional limitation of the present study is the fact that only L6 and C2C12 myotubes were studied. Although several other cell lines have been used to examine the regulation of muscle protein degradation, for example L8 myotubes [Hong and Forsberg, 1995], most previous muscle wasting-related studies, both from our and other laboratories, used cultured L6 or C2C12 myotubes. In addition, by using L6 (a rat cell line) and C2C12 (a mouse cell line) myotubes, potential species-specific differences in glucocorticoid-induced muscle wasting could be assessed.
Additionally, we determined total protein degradation in the present study, despite the fact that previous reports suggest that myofibrillar proteins are preferentially degraded in catabolic muscle [Tiao et al., 1994, 1996]. In previous reports, however, evidence was found that measurement of total protein degradation (determined as release of TCA-soluble radioactivity from myotube proteins pre-labeled with radioactive tyrosine or phenylalanine) in dexamethasone-treated myotubes reflected myofibrillar protein degradation [Wang et al., 1998; Thompson et al., 1999; Sacheck et al., 2004].
Finally, it is not known from the present study which cellular proteolytic pathway was stimulated by the glucocorticoids. Previous reports suggest, however, that treatment of cultured myotubes with dexamethasone stimulates both calcium-calpain- and ubiquitin-proteasome-dependent proteolysis [Hong and Forsberg, 1995; Wang et al., 1998], similar to the upregulated expression and activity of the calpain and ubiquitin-proteasome systems in vivo in various catabolic conditions [Tiao et al., 1994; Wray et al., 2003; Wei et al., 2005].
Although several muscle wasting-related responses to dexamethasone and corticosterone were similar in L6 and C2C12 myotubes in the present study, the effects of the glucocorticoids were not identical. Most notably, the regulation by glucocorticoids of MuRF1 and the role of the GR were different in L6 and C2C12 myotubes. These differences should be taken into account when cultured myotubes are used in future studies to further explore mechanisms of muscle wasting.
Acknowledgments
Supported in part by NIH grants R01 DK37908 (POH), R01 NR08545 (POH) and R01 DK062307 (SL).
REFERENCES
- Acharyya S, Guttridge DC. Cancer cachexia signaling pathways continue to emerge yet much still points to the proteasome. Clin Cancer Res. 2007;13:1356–1361. doi: 10.1158/1078-0432.CCR-06-2307. [DOI] [PubMed] [Google Scholar]
- Argiles JM, Busquets S, Lopez-Soriano FJ. The pivotal role of cytokines in muscle wasting during cancer. Int J Biochem Cell Biol. 2005;37:2036–2046. doi: 10.1016/j.biocel.2005.03.014. [DOI] [PubMed] [Google Scholar]
- Bodine SC, Latres E, Baumheuter S, Lai VK, Nunez L, Clarke BA, Poueymiron WT, Panaro FJ, Na E, Dharmarajan K, Pan ZQ, Valenzuela DM, Dechiara TM, Stitt TN, Yancopoulos GD, Glass DJ. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science. 2001;294:1704–1708. doi: 10.1126/science.1065874. [DOI] [PubMed] [Google Scholar]
- Borski RJ, Hyde GN, Fruchtman S. Signal transduction mechanisms mediating rapid, nongenomic effects of cortisol on prolactin release. Steroids. 2002;67:539–548. doi: 10.1016/s0039-128x(01)00197-0. [DOI] [PubMed] [Google Scholar]
- Cai D, Frantz JD, Tawa NE, Melendez PA, Oh BC, Lidov HG, Hasselgren PO, Frontera WR, Lee J, Glass DJ, Shoelson SE. IKKβ/NF-kB activation causes severe muscle wasting in mice. Cell. 2004;119:285–298. doi: 10.1016/j.cell.2004.09.027. [DOI] [PubMed] [Google Scholar]
- Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-choloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- Dellavalle A, Sampaolesi M, Tonlorenzi R, Tagliafico E, Sacchetti B, Perani L, Innocenzi A, Galvez BG, Messina G, Morosetti R, Li S, Belicchi M, Peretti G, Chamberlain JS, Wright WE, Torrente Y, Ferrari S, Bianco P, Cossu G. Pericytes of human skeletal muscle are myogenic precursors distinct from satellite cells. Nat Cell Biol. 2007;9:255–267. doi: 10.1038/ncb1542. [DOI] [PubMed] [Google Scholar]
- Desler MM, Jones SJ, Smith CW, Woods TL. Effects of dexamethasone and anabolic agents on proliferation and protein synthesis and degradation in C2C12 moyogenic cells. J Anim Sci. 1996;74:1265–1273. doi: 10.2527/1996.7461265x. [DOI] [PubMed] [Google Scholar]
- Du J, Mitch WE, Wang X, Price SR. Glucocorticoids induce proteasome C3 subunit expression in L6 muscle cells by opposing the suppression of its transcription by NF-kB. J Biol Chem. 2000;275:19661–19666. doi: 10.1074/jbc.M907258199. [DOI] [PubMed] [Google Scholar]
- Erdeljan P, Andrews MH, MacDonald JF, Matthews SG. Glucocorticoids and serotonin alter glucocorticoid receptor mRNA levels in fetal guinea-pig hippocampal neurons, in vitro. Reprod Fertil Dev. 2005;17:743–749. doi: 10.1071/rd05043. [DOI] [PubMed] [Google Scholar]
- Evenson AR, Fareed MU, Menconi MJ, Mitchell JC, Hasselgren PO. GSK-3β inhibitors reduce protein degradation in muscles from septic rats and in dexamethasone-treated myotubes. Int J Biochem Cell Biol. 2005;37:2226–2238. doi: 10.1016/j.biocel.2005.06.002. [DOI] [PubMed] [Google Scholar]
- Faggioni R, Moser A, Feingold KR, Grunfeld C. Reduced leptin levels in starvation increase susceptibility to endotoxic shock. Am J Pathol. 2000;156:1781–1787. doi: 10.1016/S0002-9440(10)65049-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falkenstein E, Tillman HC, Christ M, Feuring M, Wehling M. Multiple actions of steroid hormones – a focus on rapid, nongenomic effects. Pharmacol Rev. 2000;52:513–556. [PubMed] [Google Scholar]
- Fang CH, James JH, Fischer JE, Hasselgren PO. Influence of burn injury on protein metabolism in different types of skeletal muscle and the role of glucocorticoids. J Am Coll Surg. 1995;180:33–42. [PubMed] [Google Scholar]
- Fischer DR, Sun X, Williams AB, Gang G, Pritts TA, James JH, Molloy M, Fischer JE, Paul RJ, Hasselgren PO. Dantrolene reduces serum TNFα and corticosterone levels and muscle calcium, calpain gene expression, and protein breakdown in septic rats. Shock. 2001;15:200–207. doi: 10.1097/00024382-200115030-00007. [DOI] [PubMed] [Google Scholar]
- Gomes MD, Lecker SH, Jagoe RT, Navon A, Goldberg AL. Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proc Natl Acad Sci USA. 2001;98:14440–14445. doi: 10.1073/pnas.251541198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall-Angeras M, Angeras U, Zamir O, Hasselgren PO, Fischer JE. Interaction between corticosterone and tumor necrosis factor stimulated protein breakdown in rat skeletal muscle, similar to sepsis. Surgery. 1990;108:460–466. [PubMed] [Google Scholar]
- Hall-Angeras M, Angeras U, Zamir O, Hasselgren PO, Fischer JE. Effect of the glucocorticoid receptor antagonist RU38486 on muscle protein breakdown in sepsis. Surgery. 1991;109:468–473. [PubMed] [Google Scholar]
- Hasselgren PO. Glucocorticoids and muscle catabolism. Curr Opin Nutr Metab Care. 1999;2:201–205. doi: 10.1097/00075197-199905000-00002. [DOI] [PubMed] [Google Scholar]
- Heitzer MD, Wolf IM, Sanchez ER, Witchel SF, DeFranco DB. Glucocorticoid receptor physiology. Rev Endocr Metab Disord. 2007;8:321–330. doi: 10.1007/s11154-007-9059-8. [DOI] [PubMed] [Google Scholar]
- Hong DH, Forsberg NE. Effects of dexamethasone on protein degradation and protease gene expression in rat L8 myotube cultures. Mol Cell Endocrinol. 1995;108:199–209. doi: 10.1016/0303-7207(95)03476-n. [DOI] [PubMed] [Google Scholar]
- Klaude M, Fredriksson K, Tjader I, Hammarqvist F, Ahlman B, Rooyackers O, Wernerman J. Proteasome proteolytic activity in skeletal muscle is increased in patients with sepsis. Clin Sci. 2007;112:499–506. doi: 10.1042/CS20060265. [DOI] [PubMed] [Google Scholar]
- Konagaya M, Bernard PA, Max SR. Blockade of glucocorticoid receptor binding and inhibition of dexamethasone-induced muscle atrophy in the rat by RU38486, a potent glucocorticoid antagonist. Endocrinology. 1986;119:375–380. doi: 10.1210/endo-119-1-375. [DOI] [PubMed] [Google Scholar]
- Lackner C, Daufeldt S, Wildt L, Allera A. Glucocorticoid-recognizing and – effector sites in rat liver plasma membrane. Kinetics of corticosterone uptake by isolated membrane vesicles. III. Specificity and stereospecificity. J Steroid Biochem Mol Biol. 1998;64:69–82. doi: 10.1016/s0960-0760(97)00141-6. [DOI] [PubMed] [Google Scholar]
- Lang CH, Huber D, Frost RA. Burn-induced increase in atrogin-1 and MuRF1 in skeletal muscle is glucocorticoid independent but downregulated by IGF-I. Am J Phsyiol. 2006;292:R328–R336. doi: 10.1152/ajpregu.00561.2006. [DOI] [PubMed] [Google Scholar]
- Latres E, Amini AR, Amini AA, Griffiths J, Martin FJ, Wei Y, Lin HC, Yancopoulos GD, Glass DJ. Insulin-like growth factor-1 (IGF-1) inversely regulates atrophy-induced genes via the phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin (PI3K/Akt/mTOR) pathway. J Biol Chem. 2005;280:2737–2744. doi: 10.1074/jbc.M407517200. [DOI] [PubMed] [Google Scholar]
- Li BG, Hasselgren PO, Fang CH. Insulin-like growth factor-I inhibits dexamethasone-induced proteolysis in cultured L6 myotubes through PI3K/Akt/GSK-3β and PI3K/Akt/mTOR-dependent mechanisms. Int J Biochem Cell Biol. 2005;37:2207–2216. doi: 10.1016/j.biocel.2005.04.008. [DOI] [PubMed] [Google Scholar]
- Marinovic AC, Zheng B, Mitch WE, Price SR. Tissue-specific regulation of ubiquitin (UbC) transcription by glucocorticoids: In Vivo and In Vitro analyses. Am J Physiol. 2006;292:F660–F666. doi: 10.1152/ajprenal.00178.2006. [DOI] [PubMed] [Google Scholar]
- Menconi M, Fareed M, O’Neal P, Poylin V, Wei W, Hasselgren PO. Role of glucocorticoids in the molecular regulation of muscle wasting. Crit Care Med. 2007;35:S602–S608. doi: 10.1097/01.CCM.0000279194.11328.77. [DOI] [PubMed] [Google Scholar]
- Milgrom E. Steroid Hormones. In: Baulieu EE, Kelly PK, editors. Hormones from Molecules to Disease. Chapman Hill; New York: 1990. p. 396. [Google Scholar]
- Orth DN, Kovacs WJ, DeBold CR. The adrenal cortex. In: Wilson JD, Foster DW, editors. Williams Textbook of Endocrinology. 8th edition WB Saunders Co; Philadelphia, PA: 1992. pp. 489–619. [Google Scholar]
- Orzechowski A, Jank M, Gajkowska B, Sadkowski T, Godlewski MM, Ostaszewski P. Dilineation of signalling pathway leading to antioxidant-dependent inhibition of dexamethasone-mediated muscle cell death. J Mus Res Cell Motility. 2003;24:33–53. doi: 10.1023/a:1024887431768. [DOI] [PubMed] [Google Scholar]
- Pereira C, Murphy K, Jeschke M, Herndon DN. Post burn muscle wasting and the effects of treatments. Int J Biochem Cell Biol. 2005;37:1948–1961. doi: 10.1016/j.biocel.2005.05.009. [DOI] [PubMed] [Google Scholar]
- Perrot-Applanat M, Logeat F, Groyer-Picard MT, Milgrom E. Immuno-cytochemical study of mammalian progesterone receptor using monoclonal antibodies. Endocrinology. 1985;116:1473–1484. doi: 10.1210/endo-116-4-1473. [DOI] [PubMed] [Google Scholar]
- Philibert D. RU38486: an original multi-faceted antihormone in vivo. In: Agarwal MK, editor. Adrenal steroid antagonism. de Gruyter; Hawthorne, NY: 1984. p. 77. [Google Scholar]
- Qiu J, Wang P, Jing Q, Zhang W, Li X, Zhong Y, Sun G, Pei G, Chen Y. Rapid activation of ERK ½ mitogen activated protein kinase by corticosterone in PC12 cells. Biochem Biophys Res Commun. 2001;287:1017–1024. doi: 10.1006/bbrc.2001.5691. [DOI] [PubMed] [Google Scholar]
- Raina N, Jeejeebhoy KN. Changes in body composition and dietary intake induced by tumor necrosis factor α and corticosterone – individually and in combination. Am J Clin Nutr. 1998;68:1284–1290. doi: 10.1093/ajcn/68.6.1284. [DOI] [PubMed] [Google Scholar]
- Roeder RA, Thorpe SD, Byers FM, Schelling GT, Gunn JM. Influence of anabolic agents on protein synthesis and degradation in muscle cells grown in culture. Growth. 1986;50:485–495. [PubMed] [Google Scholar]
- Sacheck JM, Ohtsuka A, McLary SC, Goldberg AL. IGF-I stimulates muscle growth by suppressing protein breakdown and expression of atrophy-related ubiquitin ligases, atrogin-1 and MuRF1. Am J Physiol. 2004;287:E591–E601. doi: 10.1152/ajpendo.00073.2004. [DOI] [PubMed] [Google Scholar]
- Sawart M, Cabillic Y. Specific binding of dexamethasone to plasma membranes from skeletal muscle. Biochim Biophys Acta. 1985;813:87–95. doi: 10.1016/0005-2736(85)90348-7. [DOI] [PubMed] [Google Scholar]
- Schakman O, Gilson H, Thissen JP. Mechanisms of glucocorticoid-induced myopathy. Review. J Endocrinol. 2008;197:1–10. doi: 10.1677/JOE-07-0606. [DOI] [PubMed] [Google Scholar]
- Schulz M, Eggert M, Baniahmad A, Dostert A, Heinzel T, Renkawitz R. RU486-induced glucocorticoid receptor agonism is controlled by the receptor N terminus and by corepressor binding. J Biol Chem. 2002;277:26238–26243. doi: 10.1074/jbc.M203268200. [DOI] [PubMed] [Google Scholar]
- Shah OJ, Kimball SR, Jefferson LS. Among translational effectors, p70S6k is uniquely sensitive to inhibition by glucocorticoids. Biochem J. 2000;347:389–397. doi: 10.1042/0264-6021:3470389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stitt TN, Drujan D, Clarke BA, Panaro F, Timofeyva Y, Kline WO, Gonzalez M, Yancopoulos GD, Glass DJ. The IGF-I/PI3K/Akt pathway prevents expression of muscle atrophy-induced ubiquitin ligases by inhibiting FOXO transcription factors. Mol Cell. 2004;14:395–403. doi: 10.1016/s1097-2765(04)00211-4. [DOI] [PubMed] [Google Scholar]
- Sultan KR, Henkel B, Terlou M, Haagsman HP. Quantification of hormone-induced atrophy of large myotubes from C2C12 and L6 cells: atrophy-inducible and atrophy-resistant C2C12 myotubes. Am J Physiol. 2006;290:C650–C659. doi: 10.1152/ajpcell.00163.2005. [DOI] [PubMed] [Google Scholar]
- Thompson MG, Thom A, Partridge K, Garden K, Campbell GP, Calder G, Palmer RM. Stimulation of myofibrillar protein degradation and expression of mRNA encoding the ubiquitin-proteasome system in C2C12 myotubes by dexamethasone: Effect of the proteasome inhibitor MG-132. J Cell Physiol. 1999;181:455–461. doi: 10.1002/(SICI)1097-4652(199912)181:3<455::AID-JCP9>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Tiao G, Fagan J, Roegner V, Lieberman M, Wang JJ, Fischer JE, Hasselgren PO. Energy-ubiquitin-dependent muscle proteolysis during sepsis in rats is regulated by glucocorticoids. J Clin Invest. 1996;97:339–348. doi: 10.1172/JCI118421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiao G, Fagan JM, Samuels N, James JH, Hudson K, Lieberman M, Fischer JE, Hasselgren PO. Sepsis stimulates nonlysosomal, energy-dependent proteolysis and increases ubiquitin mRNA levels in rat skeletal muscle. J Clin Invest. 1994;94:2255–2264. doi: 10.1172/JCI117588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiao G, Hobler S, Wang JJ, Meyer TA, Luchette FA, Fischer JE, Hasselgren PO. Sepsis is associated with increased mRNAs of the ubiquitin-proteasome proteolytic pathway in human skeletal muscle. J Clin Invest. 1997;99:163–168. doi: 10.1172/JCI119143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tortorella LL, Pilch PF. C2C12 myocytes lack an insulin-responsive vesicular compartment despite dexamethasone-induced GLUT4 expression. Am J Physiol. 2002;283:E514–E524. doi: 10.1152/ajpendo.00092.2002. [DOI] [PubMed] [Google Scholar]
- Voisin L, Breuille D, Ruot B, Ralliere C, Rambourdin F, Dalle M, Obled C. Cytokine modulation by PX differently affects specific acute phase proteins during sepsis in rats. Am J Physiol. 1998;275:R1412–R1419. doi: 10.1152/ajpregu.1998.275.5.R1412. [DOI] [PubMed] [Google Scholar]
- Vujcic MT, Velickovic N, Ruzdijic S. Dexamethasone treatment affects nuclear glucocorticoid receptor and glucocorticoid response element binding activity in liver of rats (Rattus norvegicus) during aging. Comp Biochem Physiol B Biochem Mol Biol. 2007;148:463–469. doi: 10.1016/j.cbpb.2007.07.090. [DOI] [PubMed] [Google Scholar]
- Wang L, Luo GJ, Wang JJ, Hasselgren PO. Dexamethasone stimulates proteasome- and calcium-dependent proteolysis in cultured L6 myotubes. Shock. 1998;10:298–306. doi: 10.1097/00024382-199810000-00011. [DOI] [PubMed] [Google Scholar]
- Warren RS, Starnes HF, Alcock N, Calvano S, Brennan MD. Hormonal and metabolic response to recombinant human tumor necrosis factor in rat: in vitro and in vivo. Am J Physiol. 1988;255:E206–E212. doi: 10.1152/ajpendo.1988.255.2.E206. [DOI] [PubMed] [Google Scholar]
- Wei W, Fareed MU, Evenson A, Menconi MJ, Yang H, Petkova V, Hasselgren PO. Sepsis stimulates calpain activity in skeletal muscle by decreasing calpastatin activity but does not activate caspase-3. Am J Physiol. 2005;288:R580–R590. doi: 10.1152/ajpregu.00341.2004. [DOI] [PubMed] [Google Scholar]
- Williams AB, Sun X, Fischer JE, Hasselgren PO. The expression of genes in the ubiquitin-proteasome proteolytic pathway is increased in skeletal muscle from patients with cancer. Surgery. 1999;126:744–750. [PubMed] [Google Scholar]
- Wray CJ, Mammen JM, Hershko DD, Hasselgren PO. Sepsis upregulates the gene expression of multiple ubiquitin ligases in skeletal muscle. Int J Biochem Cell Biol. 2003;35:698–705. doi: 10.1016/s1357-2725(02)00341-2. [DOI] [PubMed] [Google Scholar]
- Yaffe D. Retention of differentiation potentialities during prolonged cultivation of myogenic cells. Proc Natl Acad Sci USA. 1968;61:477–483. doi: 10.1073/pnas.61.2.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaffe D, Saxel O. Serial passaging and differentiation of myogenic cells isolated from dystrophic mouse muscle. Nature. 1977;270:725–727. doi: 10.1038/270725a0. [DOI] [PubMed] [Google Scholar]
- Yang H, Mammen J, Wei W, Menconi M, Evenson A, Fareed M, Petkova V, Hasselgren PO. Expression and activity of C/EBPβ and δ are upregulated by dexamethasone in skeletal muscle. J Cell Physiol. 2005a;204:219–226. doi: 10.1002/jcp.20278. [DOI] [PubMed] [Google Scholar]
- Yang H, Menconi MJ, Wei W, Petkova V, Hasselgren PO. Dexamethasone upregulates the expression of the nuclear cofactor p300 and its interaction with C/EBPβ in cultured myotubes. J Cell Biochem. 2005b;94:1058–1067. doi: 10.1002/jcb.20371. [DOI] [PubMed] [Google Scholar]
- Zamir O, Hasselgren PO, Higashiguchi T, Frederick JA, Fischer JE. Tumor necrosis factor (TNF) and interleukin-1 (IL-1) induce muscle proteolysis through different mechanisms. Med Inflam. 1992;1:247–250. doi: 10.1155/S0962935192000371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Krishnamoorthy RR, Prasanna G, Narayan S, Clark A, Yorio T. Dexamethasone regulates endothelin-1 and endothelin receptors in human non-pigmented ciliary epithelial (HNPE) cells. Exp Eye Res. 2003;76:261–272. doi: 10.1016/s0014-4835(02)00323-8. [DOI] [PubMed] [Google Scholar]