

Abstract
A number of dramatic breakthroughs in the neurobiology of addiction have occurred in the past 40 years. Two domains will be highlighted: the neurocircuitry of addiction and the molecular biology of addiction targets. The neurobiological substrates for the reinforcing effects of drugs of abuse have been largely identified both at the initial site of action and in the circuitry involved. In human imaging studies, decreases in dopaminergic function have been identified as a key element of addiction, lending support for research on the role of dopamine in addiction. Three novel areas currently are emerging: the role of deficits in frontal cortex functioning, changes in the brain neurocircuitry that convey long-term vulnerability to relapse, and the role of nondopaminergic systems in the neuroadaptations associated with the development of drug dependence. Parallel to these functional changes have been major advances in our understanding of the molecular biology of addiction; the greatest contribution has been in the understanding of the molecular mechanisms of opioid action. This paper reviews the major developments in our understanding of the molecular biology of the endogenous opioid system and the use of genomics to advance our knowledge of the function and regulation of opioid receptors and endorphins.
The Past and the Present
A number of dramatic breakthroughs in the neurobiology of addiction have occurred in the past 40 years with the support of the National Institute on Drug Abuse (NIDA). Two domains will be highlighted here: the neurocircuitry of addiction and the molecular biology of addiction targets. Advances in several areas have dominated the field in the neurocircuitry of addiction. Behaviorists in the United States laid the groundwork for the development of the animal models of addiction. The neurobiological substrates for the reinforcing effects of drugs of abuse have been largely identified both at the initial site of action (receptor, reuptake site, etc.) as well as the circuitry involved (mesocorticolimbic dopamine system and endogenous opioid systems). In human imaging studies, decreases in dopaminergic function have been identified as a key common element of addiction, lending support to what became a strong program on the role of dopamine in addiction (Volkow & Fowler, 2000).
However, as we move into current research, three novel areas are emerging. The role of deficits in frontal cortex functioning has become an area of intense investigation. The changes in the brain neurocircuitry that convey long-term vulnerability to relapse are being studied. The role of nondopaminergic systems in the neuroadaptations associated with the development of drug dependence has also moved to the forefront.
Parallel to these functional changes in the neurobiology of addiction have been major advances in our understanding of the molecular biology of addiction. Perhaps one of the greatest contributions has been in the understanding of the molecular mechanisms of opioid action. Dr. Simon’s contribution to these reflections will be to review some of the major developments (“breakthroughs”) in our understanding of the molecular biology and biochemistry of opiate action and the endogenous opioid system. Clearly, the list of breakthroughs discussed is not exhaustive, and the topics chosen are those deemed most important by Dr. Simon and within his areas of interest and expertise.
Conceptual Framework for Addiction
Drug addiction, or substance dependence, is a chronically relapsing disorder characterized by: (a) compulsion to seek and take the drug, (b) loss of control in limiting intake, and (c) emergence of a negative emotional state (e.g., dysphoria, anxiety, irritability) when access to the drug is prevented. Clinically, the occasional but limited use of an abusable drug is distinct from escalated drug use and the emergence of chronic drug dependence. An important goal of current neurobiological research is to understand the neuropharmacological and neuroadaptive mechanisms within specific neurocircuits that mediate the transition from occasional, controlled drug use and the loss of behavioral control over drug-seeking, and drug-taking that defines chronic addiction. Drug addiction has been conceptualized as a disorder that moves from impulsivity to compulsivity in a collapsed cycle of addiction comprised of three stages: preoccupation/anticipation, binge intoxication, and withdrawal/negative affect (Figure 1; Koob, 2004; Koob, Sanna, & Bloom, 1998).
Figure 1.
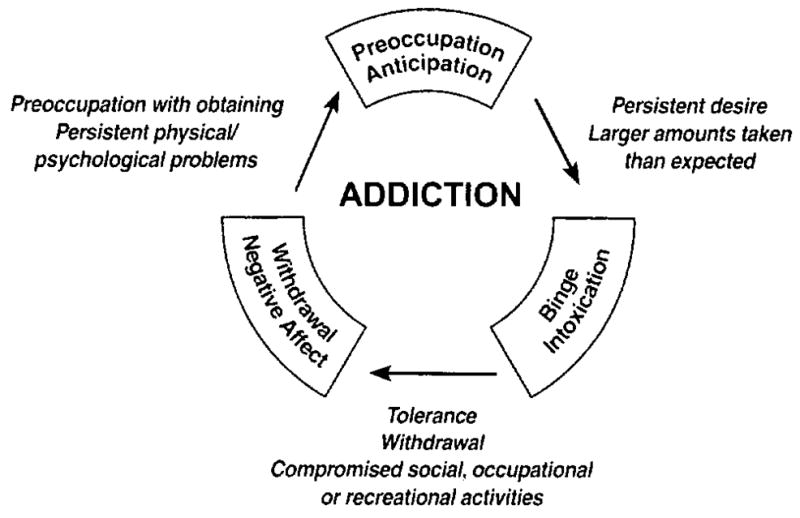
Diagram describing the addiction cycle—preoccupation/anticipation, binge/intoxication, and withdrawal/negative affect—from a psychiatric perspective with the different criteria for substance dependence incorporated from the DSM-IV.
Multiple sources of reinforcement can be identified in these three stages (Koob & Le Moal, 2001). These include the positive reinforcement of the binge intoxication stage, the negative reinforcement of drug taking to avoid a negative emotional state (e.g., dysphoria, anxiety, and irritability) that emerges when access to the drug is prevented during the withdrawal/negative affect stage, and the conditioned reinforcement associated with the preoccupation/anticipation (craving) stage (Koob & Le Moal, 2001). We will explore the neurobiological mechanisms of addiction involved in these stages of the addiction cycle, and how research has progressed from a focus on the neurochemical changes associated with the acute reinforcing effects of drugs of abuse to understanding the neuroadaptative changes that occur during the transition from taking drugs to addiction. A parallel evolution has been the exploration of first the neurocircuitry changes, and then the molecular changes, responsible for the neuroadaptations that mediate the development of addiction.
Animal Models of Drug Abuse and Dependence
Much of the recent progress in understanding the mechanisms of addiction has derived from the study of animal models of addiction on specific drugs such as opiates, stimulants, and alcohol (Shippenberg & Koob, 2002). While no animal model of addiction fully imitates the human condition, animal models do permit investigation of specific elements of the process of drug addiction. Such elements can be defined by models of different systems, models of psychological constructs, such as positive and negative reinforcement, and models of different stages of the addiction cycle. Initially, animal models of addiction focused on the synaptic sites and transductive mechanisms (how these sites convey changes in excitability) in the nervous system on which drugs act initially to produce their positive reinforcing effects. Well-validated animal models exist for the positive reinforcing effects of drugs of abuse with high predictive validity. Drugs self-administered by animals correspond well with those with high abuse potential in humans, and intravenous drug self-administration is considered an animal model predictive of abuse potential. Other validated measures of the acute rewarding effects of drugs of abuse include brain stimulation reward and conditioned place preference.
Animal models of components of the negative reinforcing effects of dependence have subsequently been developed, and animal models of craving are being used to explore how the nervous system adapts to drug use. Measures of the negative reinforcement associated with withdrawal have evolved, where either extended access to the drug is provided or the animals are made dependent with chronic exposure. Dependent animals increase the amount of drug self-administration, increase the motivation to take drugs, and show an increase in drug seeking. These models provide validity model for compulsive drug use that characterizes the withdrawal/negative affect stage of the addiction cycle and the transition to addiction. Other measures of the withdrawal/negative affect stage include anxiety-like decreases in reward as measured by increases in threshold for brain stimulation reward and place aversions to a withdrawal stimulus. Increases in brain reward thresholds have been observed following withdrawal from psychomotor stimulants, opiates, ethanol, D9-tetrahydrocannabinol (THC).
Human studies have shown that the presentation of drugs themselves, or stimuli previously associated with drug delivery or drug withdrawal, or the state of stress, increase the likelihood of relapse as well as self-reports of craving and motivation to engage in drug-taking. Animal models have been developed that reflect all three stimuli important for relapse: ingestion of the drug itself, cues associated with the drug, and exposure to stressors. As such, animal models of craving can be divided into two domains: drug-seeking induced by stimuli paired with drug-taking, and drug-seeking induced by an acute stressor or a state of stress. Drug-seeking can be reinstated in animals that have acquired drug self-administration and then have been subjected to extinction of responding for the drug by the use of drug stimuli, cues paired with or predicting drug taking, and stress induction (McFarland & Kalivas, 2001; Shaham, Shalev, Lu, De Wit, & Stewart, 2003).
Neurocircuitry of drug reward and anti-reward relevant to addiction
A key element of drug addiction is how the brain reward system changes with the development of addiction, and one must understand the neurobiological bases for acute drug reward to understand how these systems change as addiction develops. A principle focus of research on the neurobiology of the positive reinforcing effects of drugs of abuse has been the origins and terminal areas of the mesocorticolimbic dopamine system, and evidence for the importance of this system in psychostimulant reward is compelling (Koob, 1992). The psychostimulant reward circuit has been broadened to include other drugs, as well as the many neural inputs and outputs that interact with the dopamine projection from the ventral tegmental area to the basal forebrain and includes dopamine-independent components of drug reward (Figure 2).
Figure 2.

Early neurocircuitry diagram of drug reward for opiates (Wise, 1980). “Suggested sites of potential interaction of opiates with brain reward circuitry. Opiate receptor fields are shaded in the region of the striatal dopamine terminal field, the tegmental dopamine cell region, and the region of the locus coeruleus, which is thought to inhibit reward circuitry, perhaps by an inhibitory synapse on the dopamine cells themselves. Opiates inhibit locus coeruleus firing; their actions in the tegmentum and striatum are not yet understood, and may be either pre- or post-synaptic in either region. Thus, opiates may act on, or either afferent or efferent to, the dopamine cells implicated in reward function.” [Taken with permission from Wise, 1980.]
In addition to the mesocorticolimbic dopamine system and its projections, specific components of the basal forebrain have more recently been identified with the hedonic neuroadaptations to acute drug reward and have focused on elements of the extended amygdala (Koob, 2003). As the neural circuits for the reinforcing effects of drugs of abuse have evolved, the role of neurotransmitters/neuromodulators also have evolved, and four of those systems are discussed below: mesolimbic dopamine, opioid peptide, gamma-aminobutyric acid (GABA), and serotonin (Table 1). Significant evidence exists for both dopamine-dependent and -independent neurocircuitry in drug reward with a focus on the nucleus accumbens and amygdala (Koob, 1992).
Table 1.
Neurotransmitters Implicated in the Motivational Effects of Withdrawal from Drugs of Abuse
| ↓ Dopamine … “dysphoria” | ↑ Dynorphin … “dysphoria” |
| ↓ Serotonin … “dysphoria” | ↑ CRF … stress |
| ↓ GABA … anxiety, panic attacks | ↑ Norepinephrine … stress |
| ↓ Opioid peptide … “dysphoria” | ↑ Glutamate … hyperexcitability |
All major drugs of abuse produce dysphoria during withdrawal, and animal models show an elevation of reward thresholds as measured by brain stimulation reward. Such effects have been observed with amphetamines, cocaine, opiates, alcohol, nicotine, and THC. A logical explanation for the neuroadaptative mechanisms responsible for the negative motivational effects of drug withdrawal (the “dark side”) may involve disruption of the same neural systems implicated in the positive reinforcing effects of drugs of abuse (Table 1) (Koob & Le Moal, 2005).
As such, these effects may reflect changes in the activity of reward neurotransmitter systems in the midbrain and forebrain implicated in the positive reinforcing effects of drugs. Examples of such changes at the neurochemical level include decreases in dopaminergic and serotonergic transmission in the nucleus accumbens during drug withdrawal as measured by in vivo microdialysis, increased sensitivity of opioid receptor transduction mechanisms in the nucleus accumbens during opiate withdrawal, decreased GABAergic and increased N-methyl-D-aspartate (NMDA) glutamatergic transmission during alcohol withdrawal, and differential regional changes in nicotinic receptor function. The decreases in reward neurotransmitters have been hypothesized to contribute significantly to the negative motivational state associated with acute drug abstinence and may trigger long-term biochemical changes that contribute to the clinical syndrome of protracted abstinence and vulnerability to relapse (Koob & Le Moal, 2001).
However, different neurochemical systems involved in stress modulation also may be engaged within the neurocircuitry of the brain reward and stress systems in an attempt to overcome the chronic presence of the drug and to restore normal function despite its presence. Chronic administration of drugs with dependence potential also dysregulate both the hypothalamic-pituitary-adrenal (HPA) axis and the brain stress system mediated by corticotropin-releasing factor (CRF) (Heinrichs & Koob, 2004). CRF is a 41 amino acid polypeptide with a wide distribution throughout the brain but with high concentrations of cell bodies in the paraventricular nucleus of the hypothalamus, the basal forebrain, and notably the extended amygdala and brainstem. Central administration of CRF mimics the behavioral response to activation and stress in rodents. Administration of competitive CRF receptor antagonists generally has the opposite effects. Common responses include an activated HPA stress response, elevated adrenocorticotropic hormone and corticosteroids, and an activated brain stress response with activated amygdala CRF during acute withdrawal from all major drugs of abuse. Also, CRF receptor antagonists selectively block the increase in drug-seeking observed in extended access and dependence (Koob, 2008).
Acute withdrawal from drugs of abuse also may increase the release of norepinephrine in the bed nucleus of the stria terminalis (BNST) and decrease levels of neuropeptide Y (NPY) in the central and medial nuclei of the amygdala (Heilig, Koob, Ekman, & Britton, 1994; Koob & Le Moal, 2005).
These results suggest not only a change in function of neurotransmitters associated with the acute reinforcing effects of drugs (dopamine, opioid peptides, serotonin, and GABA) during the development of dependence, but also recruitment of the brain arousal and stress systems (glutamate, CRF, and norepinephrine) and dysregulation of the NPY brain anti-stress system. These changes would represent a “between-system” neuroadaptation. Reward mechanisms in dependence are compromised by disruption of neurochemical systems involved in processing natural rewards and by recruitment of the anti-reward systems that represent neuroadaptation to the chronic exposure of the brain reward neurocircuitry to drugs of abuse (Koob & Le Moal, 2008).
The neuroanatomical entity termed the extended amygdala thus may represent a common anatomical substrate for some aspects of acute drug reward and a common neuroanatomical substrate for the negative effects on reward function produced by stress that help drive compulsive drug administration. The extended amygdala receives numerous afferents from limbic structures, such as the basolateral amygdala and hippocampus, and sends efferents to the medial part of the ventral pallidum and a large projection to the lateral hypothalamus, further defining the specific brain areas that interface classical limbic (emotional) structures with the extrapyramidal motor system (Koob, 2003).
Animal models of “craving” involve the use of drug-primed reinstatement, cue-induced reinstatement, or stress-induced reinstatement in animals that acquire drug self-administration and then are subjected to extinction of responding for the drug. Most evidence from animal studies suggests that drug-induced reinstatement is localized to a medial prefrontal cortex/nucleus accumbens/ventral pallidum circuit mediated by the neurotransmitter glutamate. In contrast, neuropharmacological and neurobiological studies using animal models for cue-induced reinstatement involve the basolateral amygdala as a critical substrate with a possible feed-forward mechanism through the prefrontal cortex system involved in drug-induced reinstatement. Stress-induced reinstatement of drug-related responding in animal models appears to depend on activation of both CRF and norepinephrine in elements of the extended amygdala (central nucleus of the amygdala and BNST) (McFarland & Kalivas, 2001; Shaham et al., 2003). In human imaging studies, another common element of drug dependence is decreased function in the orbitofrontal/medial prefrontal cortex as measured both by neuropsychological tests and imaging (Volkow & Li, 2004).
In summary, three neurobiological circuits have been identified as important to the study of the neurobiological changes associated with the development and persistence of drug dependence (Koob & Le Moal, 2006). The acute reinforcing effects of drugs of abuse that comprise the binge/intoxication stage of the addiction cycle most likely involve actions localized to a nucleus accumbens-amygdala reward system, dopamine inputs from the ventral tegmental area, local opioid peptide circuits, and opioid peptide inputs in the arcuate nucleus of the hypothalamus. In contrast, the symptoms of acute motivational withdrawal important for addiction, such as dysphoria and increased anxiety associated with the withdrawal/negative affect stage, most likely involve decreases in function of the reward system and recruitment of brain stress neurocircuitry. The preoccupation/anticipation (or “craving”) stage involves key afferent projections to the extended amygdala and nucleus accumbens, specifically the prefrontal cortex (for drug-induced reinstatement) and the basolateral amygdala (for cue-induced reinstatement). Compulsive drug-seeking behavior is hypothesized to be driven by ventral striatal-ventral pallidal-thalamic-cortical loops.
Molecular Approaches to Addiction
Many scientists feel that the modern age of drug abuse research began in the early 1970s with the discovery of the opioid receptors. This discovery was made simultaneously by three laboratories, including that of Eric J. Simon, (Pert & Snyder 1973; Simon, Hiller, & Edelman, 1973; Terenius, 1973). The discovery that opiate analgesics such as morphine or etorphine, as well as antagonists such as naloxone and diprenorphine, bind stereospecifically to animal and human brain homogenates, provided the initial evidence for the existence of these receptors. This was quickly followed by evidence indicating that these stereospecific binding sites are functional receptors. This discovery led to a rapid increase in molecular research on opiates and to several additional important discoveries in the opiate field. One was the finding that there are three types of opioid receptors, mu (MOP), delta (DOP), and kappa (KOP). All three have distinct distributions in the brain, properties, and actions. The Simon laboratory pioneered the purification of these receptors, making pure proteins available to investigators for structural and functional studies (Gioannini, Howard, Hiller, & Simon, 1985). Such pure receptors were reconstituted with pure G-proteins and phospholipids to form an active complex, able to bind both opioid agonists and antagonists with high affinity in vitro (Gioannini et al., 1993).
Scientists posed the obvious question: Why are there receptors in the brain that bind morphine and synthetic drugs? Why have these receptors survived eons of evolution? What is their real physiological function? This led to the next important breakthrough, the discovery of substances in the brain and other tissues with morphine-like activity, often called endorphins (a term coined by Simon). These substances were determined to be peptides (short chains of amino acids, ranging in size from 5 amino acids for enkephalins to 32 amino acids for beta-endorphin). Table 3 lists the known opioid peptides and their amino acid sequences, expressed in the single-letter nomenclature of amino acids, such as Y for Tyrosine, G for glycine, etc. (the complete list of amino acid single-letter sequences is readily available in any biochemistry textbook for those who are interested).
Table 3.
Endogenous Opioid Peptides
| • Met-enkephalin | YGGFM |
| • Leu-enkephalin | YGGFL |
| • Heptapeptide | YGGFMRF |
| • Octapeptide | YGGFMRGL |
| • Beta-endorphin |
YGGFMTSEKSQTPLVTL FKNAIIKKNAYKKGE |
| • Dynorphin A | YGGFLRRIRPKLKWDNQ |
| • Dynorphin B | YGGFLRROFKVVT |
| • Neoendorphin | YGGFLRKYPK |
What is the value of knowing the DNA structures (nucleotide sequences) of genes and the amino acid sequences of proteins? DNA cloning has led to significant advances in techniques that have had a huge impact on biomedical research. Some of these techniques are explained here, with a focus on their applications to the study of opioids (Table 4).
Table 4.
List of Techniques Derived from DNA Cloning
|
A series of exciting and outstanding studies led to an understanding of how these opioid peptides are biosynthesized (see Simon & Hiller, 1994). All of them are made via three large precursor proteins, called opioimelanocortin, proenkephalin, and prodynorphin. Opiomelanocortin is of particular interest because it is the precursor for several important and seemingly unrelated biologically active molecules, namely, b-endorphin, adrenocorticotropic hormone, and several melanocyte-stimulating hormones.
Once these precursors and their structures were known, there was a race to clone them. The term cloning is an unfortunate term. It refers here to the purification and nucleotide sequencing of the DNA of a gene. The structure of the protein can then be inferred by using the universal genetic code to translate nucleotide triplets (codons) into amino acids. The structure of the full gene with its long introns can also be readily attained. The DNA sequences were successfully determined for the genes encoding the three opioid peptide precursors, the first of which was the opiomelanocortin gene (Noda et al., 1982).
More recently, the gene and protein sequences for the three types of opioid receptors also have been identified. The first to be cloned was the delta receptor (Evans, Keith, Morrison, Magendzo, & Edwards, 1992; Kieffer, Befort, Ruff, & Hirth, 1992). All three types have since been cloned, and no genes for additional opioid receptor types have subsequently been discovered. The knowledge of the sequence of the “reading frame” of the genes and the resulting knowledge of the amino acid sequence of the receptor proteins confirmed previous pharmacological studies that had suggested opioid receptors are members of the large family of G protein-coupled receptors and have the typical structure of such receptors.
Site-directed mutations and deletions
The DNA of a gene can be manipulated by creating mutations, replacing or deleting nucleotides that change the code for an amino acid, and eliminating a portion of the gene. Thus, the codon for a single amino acid in a crucial area of the molecule can be altered or a portion of the DNA deleted, such as the DNA coding for the carboxyl tail of an opioid receptor. Such altered genes then can be introduced into cells in culture or into animals, and the effects of these changes on the phenotype can be studied.
Construction of chimera
One can now take part of the DNA encoding one molecule (e.g., the mu opioid receptor) and another portion of the DNA encoding another molecule (e.g., the delta opioid receptor) and create a chimera. One can then ask questions about which properties and functions come from one or the other receptor.
Knockout and transgenic animals
It is possible to generate knockout or transgenic animals. Mice are most commonly used for this purpose. Knockout mice are those that have only an inactive form of the gene under study and therefore lack an active protein (e.g., one or more of the opioid receptors or peptides). A transgenic mouse has a gene that has been transferred and is not normally expressed (e.g., a gene for the human opioid receptor or a molecule it does not normally make or a gene that has been manipulated by mutation or deletion). The phenotype of such mice can now be probed by behavioral, electrophysiological, pharmacological, and metabolic measurements to determine the impact of the presence or absence of the gene in question (see Kieffer & Gaveriaux-Ruff, 2002).
Microarrays to study gene expression
Microarrays, arrays of thousands of genes of interest on a tiny chip, have recently become available. Only those genes that anneal with messenger RNA (the gene product that is the intermediate for protein synthesis) from the tissue under study are expressed in that tissue and under the given physiological or pathological conditions. This allows changes in the level of gene expression in various physiological or pathological conditions to be determined. For example, one can ask what brain genes are turned up, turned down, or turned on or off, when a dependent rat is withdrawn from a drug or when an animal is given cues that lead to craving for a drug.
Study of single nucleotide polymorphisms and other genetic variants
Many mutations (alleles) exist in human and animal cells and often do not drastically change the phenotype but rather could produce subtle changes. The reason for the relatively mild effects of such mutants is either because the altered amino acid is in a portion of the gene not essential for the function of the protein or because the change in the nucleotide does not result in a change in the amino acid. Such natural mutations are called single nucleotide polymorphisms (SNPs). Many laboratories are now attempting to determine whether such SNPs have subtle effects on phenotype and thus may contribute, for example, to the striking differences observed between individuals in their sensitivity to the addicting effects of abused drugs.
Species differences and evolutionary changes in gene structure
The complete sequencing of the human genome and that of many other creatures, animals, microorganisms, and plants, has made possible the study of changes in gene structure as never before. These advances have already improved understanding of species’ differences and of the workings of evolution. Such genomic approaches will likely further our understanding, prevention, and treatment of human diseases, including drug addiction.
Studies of gene expression and promoter regulation by transcription factors
Finally, studies in which levels of gene expression and promoter regulation by proteins called transcription factors are measured can now be conducted. Every gene has a promoter region, where gene activity is turned on or off, and where, when on, the level of gene transcription is regulated. Understanding how gene expression is regulated is of considerable importance. These sophisticated experiments are best performed in cell culture, although studies in animals are becoming more and more possible. Numerous transcription factors have already been discovered, resulting in a better understanding of how gene transcription and replication are controlled. The cloning of genes has led to numerous experimental approaches that were heretofore impossible, and many of these are powerful techniques that are already providing information we never dreamed of being able to obtain.
Signal transduction
The final topic to be discussed is the exciting research area known as signal transduction. These investigations probe the chemical reactions that allow the signal generated by a ligand (e.g., opiate drug or opioid peptide) binding to a receptor to be transduced to the site at which the action of the ligand or drug takes place. Numerous laboratories have made much progress in our understanding of signal transduction by opioid receptors. One example of progress in this area is the study of opioid receptors (and other G protein-coupled receptors) where there is activation of protein-phosphorylating enzymes, called mitogen-activated protein (MAP) kinases. This ability had previously been thought to be restricted to hormone and growth factor receptors. These MAP kinases are known to be involved in changes in gene expression, via phosphorylation of transcription factors, clearly important for the type of adaptations seen in drug dependence. The Simon laboratory and that of Susan George were able to show independently that MAP kinase activation involves phosphorylation of tyrosine residues on the delta and mu opioid receptors, respectively (Kramer, Andria, Esposito, & Simon, 2000; Pak, O’Dowd, Wang, & George, 1999). An understanding of signal transduction pathways could lead to very specific interference with a particular step, such as a step involved in the development of tolerance, physical dependence, or craving for drugs.
One dramatic example comes from the cancer field (Schiffer, 2007). Cancer cells in chronic myelogenous leukemia (CML) contain a protein-phosphorylating enzyme (kinase) that is not present in exactly the same form in normal host cells. A drug has been developed that specifically inhibits this cancer cell enzyme with little or no effects on host cell kinases. This drug is called Gleevec and is remarkably effective in CML, with little or no side effects. The effects on CML and some other cancers have been so dramatic that some physicians have called this treatment a cure.
Clearly this is the future, to try to develop highly specific treatments for all human diseases that produce minimal secondary effects because of the specificity of action. Signal transduction pathways are just one of the places to look for possible targets.
Where we are going
With the advances discussed above, the field is moving into new areas that promise significant progress in identifying, preventing, and treating addiction. Many key questions remain. What neurobiological processes (system, cellular, and molecular) convey vulnerability to the transition from drug use to abuse and addiction, with a particular emphasis on adolescent exposure? What neurobiological processes convey vulnerability for relapse? How do environmental and genetic factors facilitate the changes in neurobiological processes that convey vulnerability to addiction—and protection from addiction? How can the brain best “recover” to eliminate abuse and addiction and to eliminate relapse (behavioral therapies, pharmacotherapies)?
Finally, while no exact marker necessarily predicts addiction, two salient changes in imaging in established and unrecovered substance-dependent individuals that cut across different drugs are decreases in orbitofrontal/prefrontal cortex function and decreases in brain dopamine D2 receptors. No biochemical markers are sufficiently specific to predict a given stage of the addiction cycle, but changes in certain intermediate early genes with chronic drug exposure in animal models show promise of long-term changes in specific brain regions that may be common to all drugs of abuse. Although no biological markers of substance abuse disorders are on the immediate horizon, many promising and continually evolving biological and neurobiological aspects of substance use disorders will eventually aid in the specific diagnoses of substance use, misuse, and dependence.
Much needs to be done in the area of molecular and biochemical approaches to drug addiction, although much has been achieved, as seen from this brief review. The big disappointment to scientists in this field is that 34 years into the modern era of drug abuse research, we still do not have a clear understanding of the cellular and molecular bases of the development and maintenance of the phenomena of addiction, such as tolerance, physical dependence, and psychic craving. The progress in biochemical approaches to our understanding of the enzymes involved in signal transduction pathways also has been quite impressive. Additional technologies, such as imaging and electrophysiology, which are not discussed here, have progressed rapidly as well. We feel cautiously optimistic that the molecular basis of some of the events that lead to drug addiction and abuse will be discovered within the next 5 to 10 years. What is the molecular basis of tolerance? Of dependence? What alterations in the brain cause craving for abused drugs to increase greatly in some persons who use drugs casually and not in others? What is the genetic basis for these individual differences? What is the biochemical basis of reward and of the motivational symptoms of withdrawal? Which parts of the pathways that are set into motion by the consumption of abused drugs can we intervene to block the development of addiction? These are some of the questions that should be answered, using the highly sophisticated technologies currently available.
Summary and Conclusions
The advances in the neurobiology of addiction are examined here from two domains. First, a heuristic framework is provided of the neurocircuitry of addiction. Second, advances in the molecular biology of the opioid system demonstrate the tremendous advances we have made in our understanding of the molecular basis of the action of the drugs of abuse. A conceptual structure for drug addiction that focuses on changes in reward system function that lead to excessive drug intake provides a framework with which to identify the neurobiological and neuroadaptive mechanisms involved in the neurobiology of drug addiction. The neurochemical systems implicated in the acute reinforcing effects of drugs of abuse include key elements of the basal forebrain linked by the mesocorticolimbic dopamine system, which include dopamine-dependent and -independent actions via dopamine, opioid peptides, GABA, and serotonin in the acute reinforcing effects of drugs of abuse. Neuroadaptive changes in the brain reward system during the development of dependence include decreases in these same drug reward neurochemical systems and recruitment of brain stress systems (CRF and norepinephrine; “dark side”) and dysregulation of brain anti-stress systems (NPY) in the extended amygdala that provide the negative motivational state associated with drug abstinence {Koob & Le Moal, 2008). Neurochemical systems have been implicated in animal models of relapse, including dopamine, opioid peptide, and glutamate, and in drug- and cue-induced relapse in prefrontal cortical and basolateral amygdala projections to the basal forebrain. The brain stress systems in the extended amygdala are directly implicated in stress-induced relapse.
Gaps in our knowledge center on identifying the molecular changes that contribute to the neuroadaptations within specific motivationally relevant neurocircuits associated with the transition to dependence, motivational withdrawal, protracted abstinence, and vulnerability to relapse. Nevertheless, the identification of specific neurochemical systems within specific components of the brain reward and stress systems that have an important role in mediating various sources of motivation for drug-seeking behavior provides a solid foundation for bridging the molecular/cellular gap.
Table 2.
Neurotransmitters Implicated in the Motivational Effects of Withdrawal from Drugs of Abuse
| Neurotransmitter | Functional effect |
|---|---|
| ↓ Dopamine | “dysphoria” |
| ↓ Serotonin | “dysphoria” |
| ↓ γ-aminobutyric acid | anxiety, panic attacks |
| ↓ Neuropeptide Y | anti-stress |
| ↑ Dynorphin | “dysphoria” |
| ↑ Corticotropin-releasing factor | stress |
| ↑ Norepinephrine | stress |
Acknowledgments
Research was supported by National Institutes of Health grants AA06420 and AA08459 from the National Institute on Alcohol Abuse and Alcoholism, DA04043 and DA04398 from the National Institute on Drug Abuse, and DK26741 from the National Institute on Diabetes and Digestive and Kidney Diseases. Research also was supported by the Pearson Center for Alcoholism and Addiction Research at The Scripps Research Institute. Dr. Simon gratefully acknowledges the support of NIDA for over 40 years (DA00017), the former KO5 (DA 00364), which paid a portion of his salary for 5 years, as well as a Training Grant (T32-DA07254). He also wishes to pay tribute to the many students, postdoctoral fellows, and visiting professors he has been privileged to train and collaborate with, and whose input and hard work have been extremely important to the success of his laboratory. The authors gratefully acknowledge the editorial and research assistance of Mr. Michael A. Arends.
Biographies
George F. Koob, Ph.D., is Professor and Chairman of the Committee on the Neurobiology of Addictive Disorders at The Scripps Research Institute and Adjunct Professor in the Departments of Psychology and Psychiatry and the Skaggs School of Pharmacy and Pharmaceutical Sciences at the University of California, San Diego. He specializes on the neurobiology and theoretical constructs of drug addiction, reward, and stress.
Dr. Eric J. Simon is Professor of Psychiatry and Pharmacology at New York University Medical Center. Dr. Simon's laboratory has done pioneering research on the mode of action of narcotic analgesics. In 1973 his laboratory, and others, discovered the opiate receptors in the brain. He and his collaborators have continued to work on opiate receptors and endogenous opioid peptides and have published 250 papers in excellent journals. He coined the widely used term “endorphins.”
References
- Evans C, Keith D, Morrison H, Magendzo K, Edwards R. Cloning of a delta-opioid receptor by functional expression. Science. 1992;258:1952–1955. doi: 10.1126/science.1335167. [DOI] [PubMed] [Google Scholar]
- Gioannini TL, Fan LQ, Hyde L, Ofri D, Yao Y, Hiller JM, Simon EJ. Reconstitution of a purified mu-opioid binding protein in liposomes: Selective, high affinity, GTP-gamma S-sensitive mu-opioid agonist binding is restored. Biochemical and Biophysical Research Communications. 1993;194:901–908. doi: 10.1006/bbrc.1993.1906. [DOI] [PubMed] [Google Scholar]
- Gioannini TL, Howard AD, Hiller JM, Simon EJ. Purification of an active opioid binding protein from bovine striatum. Journal of Biological Chemistry. 1985;260:15117–15121. [PubMed] [Google Scholar]
- Heilig M, Koob GF, Ekman R, Britton KT. Corticotropin-releasing factor and neuropeptide Y: Role in emotional integration. Trends in Neurosciences. 1994;17:80–85. doi: 10.1016/0166-2236(94)90079-5. [DOI] [PubMed] [Google Scholar]
- Heinrichs SC, Koob GF. Corticotropin-releasing factor in brain: A role in activation, arousal, and affect regulation. Journal of Pharmacology and Experimental Therapeutics. 2004;311:427–440. doi: 10.1124/jpet.103.052092. [DOI] [PubMed] [Google Scholar]
- Kieffer B, Befort K, Ruff C, Hirth C. The delta-opioid receptor: Isolation of a cDNA by expression cloning and pharmacological characterization. Proceedings of the National Academy of Sciences USA. 1992;89:12048–12052. doi: 10.1073/pnas.89.24.12048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieffer B, Gaveriaux-Ruff C. Exploring the opioid system by gene knockout. Progress in Neurobiology. 2002;66:285–306. doi: 10.1016/s0301-0082(02)00008-4. [DOI] [PubMed] [Google Scholar]
- Koob GF. Drugs of abuse: Anatomy, pharmacology, and function of reward pathways. Trends in Pharmacological Sciences. 1992;13:177–184. doi: 10.1016/0165-6147(92)90060-j. [DOI] [PubMed] [Google Scholar]
- Koob GF. Neuroadaptive mechanisms of addiction: Studies on the extended amygdala. European Neuropsychopharmacology. 2003;13:442–452. doi: 10.1016/j.euroneuro.2003.08.005. [DOI] [PubMed] [Google Scholar]
- Koob GF. Allostatic view of motivation: Implications for psychopathology. In: Bevins RA, Bardo MT, editors. Motivational factors in the etiology of drug abuse (series title: Nebraska symposium on motivation. Vol. 50. Lincoln NE: University of Nebraska Press; 2004. pp. 1–18. [PubMed] [Google Scholar]
- Koob GF. A role for brain stress systems in addiction. Neuron. 2008;59:11–34. doi: 10.1016/j.neuron.2008.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, Le Moal M. Drug addiction, dysregulation of reward, and allostasis. Neuropsychopharmacology. 2001;24:97–129. doi: 10.1016/S0893-133X(00)00195-0. [DOI] [PubMed] [Google Scholar]
- Koob GF, Le Moal M. Plasticity of reward neurocircuitry and the ‘dark side’ of drug addiction. Nature Neuroscience. 2005;8:1442–1444. doi: 10.1038/nn1105-1442. [DOI] [PubMed] [Google Scholar]
- Koob GF, Le Moal M. Neurobiology of addiction. London: Academic Press; 2006 . [Google Scholar]
- Koob GF, Le Moal M. Addiction and the brain antireward system. Annual Review of Psychology. 2008;59:29–53. doi: 10.1146/annurev.psych.59.103006.093548. [DOI] [PubMed] [Google Scholar]
- Koob GF, Sanna PP, Bloom FE. Neuroscience of addiction. Neuron. 1998;21:467–476. doi: 10.1016/s0896-6273(00)80557-7. [DOI] [PubMed] [Google Scholar]
- Kramer HK, Andria ML, Esposito DH, Simon EJ. Tyrosine phosphorylation of the delta-opioid receptor: Evidence for its role in mitogen-activated protein kinase activation and receptor internalization. Biochemical Pharmacology. 2000;60:781–792. doi: 10.1016/s0006-2952(00)00400-7. [DOI] [PubMed] [Google Scholar]
- McFarland K, Kalivas PW. The circuitry mediating cocaine-induced reinstatement of drug-seeking behavior. Journal of Neuroscience. 2001;21:8655–8663. doi: 10.1523/JNEUROSCI.21-21-08655.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noda M, Teranishi Y, Takahashi H, Toyosato M, Notake M, Nakanishi S, Numa S. Isolation and structural organization of the human preproenkephalin gene. Nature. 1982;297:431–434. doi: 10.1038/297431a0. [DOI] [PubMed] [Google Scholar]
- Pak Y, O’Dowd BF, Wang JB, George SR. Agonist-induced, G protein-dependent and -independent down-regulation of the mu opioid receptor. The receptor is a direct substrate for protein-tyrosine kinase. Journal of Biological Chemistry. 1999;274:27610–27616. doi: 10.1074/jbc.274.39.27610. [DOI] [PubMed] [Google Scholar]
- Pert CB, Snyder SH. Opiate receptor: Demonstration in nervous tissue. Science. 1973;179:1011–1014. doi: 10.1126/science.179.4077.1011. [DOI] [PubMed] [Google Scholar]
- Schiffer CA. BCR-ABL tyrosine kinase inhibitors for chronic myelogenous leukemia. New England Journal of Medicine. 2007;357:258–265. doi: 10.1056/NEJMct071828. [DOI] [PubMed] [Google Scholar]
- Shaham Y, Shalev U, Lu L, De Wit H, Stewart J. The reinstatement model of drug relapse: History, methodology and major findings. Psychopharmacology. 2003;168:3–20. doi: 10.1007/s00213-002-1224-x. [DOI] [PubMed] [Google Scholar]
- Shippenberg TS, Koob GF. Recent advances in animal models of drug addiction and alcoholism. In: Davis KL, Charney D, Coyle JT, Nemeroff C, editors. Neuropsychopharmacology: The fifth generation of progress. Philadelphia: Lippincott Williams and Wilkins; 2002. pp. 1381–1397. [Google Scholar]
- Simon EJ, Hiller JM. Opioid peptides and opioid receptors. In: Siegel GJEA, editor. Basic neurochemistry: Molecular, cellular, and medical aspects. New York: Raven Press; 1994. pp. 321–339. [Google Scholar]
- Simon EJ, Hiller JM, Edelman I. Stereospecific binding of the potent narcotic analgesic [3H]etorphine to rat brain homogenate. Proceedings of the National Academy of Sciences USA. 1973;70:1947–1949. doi: 10.1073/pnas.70.7.1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terenius L. Stereospecific interaction between narcotic analgesics and a synaptic plasma membrane fraction of rat cerebral cortex. Acta Pharmacoligica Toxicoligica. 1973;32:317–320. doi: 10.1111/j.1600-0773.1973.tb01477.x. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Fowler JS. Addiction, a disease of compulsion and drive: Involvement of the orbitofrontal cortex. Cerebral Cortex. 2000;10:318–325. doi: 10.1093/cercor/10.3.318. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Li TK. Drug addiction: The neurobiology of behavior gone awry. Nature Reviews Neuroscience. 2004;5:963–970. doi: 10.1038/nrn1539. [DOI] [PubMed] [Google Scholar]
- Wise RA. Action of drugs of abuse on brain reward systems. Pharmacology Biochemistry and Behavior. 1980;13(Suppl 1):213–223. doi: 10.1016/s0091-3057(80)80033-5. [DOI] [PubMed] [Google Scholar]