

Abstract
Background
Sequencing of the approximately 1.7 billion bases of the zebrafish genome is currently underway. To date, few high resolution genetic maps exist for the zebrafish genome, based mainly on single nucleotide polymorphisms (SNPs) and short microsatellite repeats. The desire to construct a higher resolution genetic map led to the construction of a database of tandemly repeating elements within the zebrafish Zv8 assembly.
Description
Exact tandem repeats with a repeat length of at least three bases and a copy number of at least 10 were reported. Repeats with a total length of 250 or fewer bases and their flanking regions were masked for known vertebrate repeats. Optimal primer pairs were computationally designed in the regions flanking the detected repeats. This database of exact tandem repeats can then be used as a resource by molecular biologists with interests in experimentally testing VNTRs within a zebrafish population.
Conclusions
A total of 116,915 repeats with a base length of at least three nucleotides were detected. The longest of these was a 54-base repeat with fourteen tandem copies. A significant number of repeats with a base length of 18, 24, 27 and 30 were detected, many with potentially novel proline-rich coding regions.
Detection of exact tandem repeats in the zebrafish genome leads to a wealth of information regarding potential polymorphic sites for VNTRs. The association of many of these repeats with potentially novel yet similar coding regions yields an exciting potential for disease associated genes. A web interface for querying repeats is available at http://bioinformatics.louisville.edu/zebrafish/. This portal allows for users to search for a repeats of a selected base size from any valid specified region within the 25 linkage groups.
Background
Zebrafish genome
Zebrafish are studied as model vertebrate organism with an early embryonic development similar to human development and genetics [1]. The zebrafish genome is approximately 1.7 billion bases long, spread out over 25 linkage groups [2], which is roughly half the size of the human genome. The rate of single nucleotide polymorphisms in the zebrafish genome is approximately one in 200 bases, compared to one per 1,000 bases in humans [3]. While this seems like a rare event, given that the zebrafish genome is 1.7 billion bases in length and the human genome is 3.2 billion bases long, that equates to eight million differences across the zebrafish genome and three million differences across the human genome. Several maps have been made for the zebrafish genome [4-7], including a low density map of 2,035 SNPs and 178 insertion/deletion (indel) events [3], a high density map of candidate SNPS [8], dinucleotide microsatellites [9], and mutant loci [10].
Exact tandem repeats
Genomes can be thought of as an alphabet with four terminal symbols, or nucleotide bases, represented by A, C, G, T. The size of a genome can vary widely, ranging from a few thousand bases for viral genomes to tens of billions of bases for complex eukaryotic plants [11]. Across the genome of a particular species, a number of differences in the DNA sequences help to differentiate among individuals in the population.
Large regions of a genome may have undergone duplication events resulting in the repetition of the pattern in tandem, one right after another. For instance, if the pattern CAG is duplicated 10 times, it results in the sequence:
CAGCAGCAGCAGCAGCAGCAGCAGCAGCAG
This can be rewritten as (CAG)10, where CAG is the base pattern, and 10 is the number of times the pattern is repeated, also known as the copy number. In this example, CAG is a trinucleotide repeat, since the base sequence is three nucleotides long.
Tandemly repeated elements are prime locations for large scale polymorphisms within a population. As a result, microsatellite markers have been used to create high-density maps for a number of organisms, including the human [12], mouse [13] and rat [14].
In the human genome, at least 35 different human diseases have been associated with differences in repeat copy numbers [15]. Triplet repeats in particular are well studied, causing over 20 human neurological disorders and diseases associated with differences in the copy number of the repeat [15]. Expansion in the copy number of trinucleotide repeats expansion has been shown to play a major role in human disease, due to their disruption within the coding (exonic) and noncoding (intronic) regions of genes. Examples of diseases associated with the expansion of the trinucleotide CAG include Huntington's Disease, Smith's Disease, Kennedy's Disease, and Machado-Joseph Disease [16-18]. The repeat CAG within a coding region results in polyglutamine expansion, causing the protein to bind with GAPDH, thus affecting its ability to function in producing energy [19]. Other diseases, such as myotonic dystrophy, Fragile X syndrome, and Friedreich's ataxia [16,17] have been shown to be involved with trinucleotide expansions as well.
For some of these triplet repeat associated diseases, a small variation in the copy number can differentiate a healthy individual from an affected one. For instance, with Kennedy's Disease, a normal individual will have 21 tandem copies of a CAG repeat within an androgen receptor gene, while an individual with Kennedy's Disease will have 40-52 copies of the CAG repeat. Other triplet repeat associated diseases can result in a vast difference in the copy number. One instance of this is Fragile X, where a normal individual has 6-50 copies of a CCG repeat within the FMR1 gene and an affected individual may have 1,000 or more copies of CCG. In addition, dynamic mutations in the copy number of the tetramers CCTG [20] and TCAT [21] along with the pentamer ATTCT [22] and dodecamer CCCCGCCCCGCG [23] have been characterized within the human genome.
While the occurrence of short tandem repeats (STRs) of less than five base pairs within genomes have been well studied and used to create genetic maps [24-27] and looking for polymorphic markers within a population [28-30], less is known about the occurrence of polymorphic tandem repeats of longer lengths. Forensic tests use known polymorphic STRs to differentiate between different individuals [31]. In many cases, the pool of known polymorphic STRs is only a small subset existing within a species, leaving many STRs to be virtually unknown. Therefore, an efficient method for detecting regions susceptible to polymorphisms within a population is desired. Since STRs are prime candidates, a method for determining occurrences of short tandem repeats given a reference sequence is presented. Once the tandem repeats are discovered, the flanking regions can be mined for uniform PCR primers which can be used to experimentally test for variability in the copy number. Those regions which are then validated as having polymorphisms within the population can be used as regions for genetic markers.
The use of simple sequence repeats (SSRs) as markers for the construction of a genetic map within zebrafish has been previously proposed [32] by looking at a small subset of dinucleotide repeats. In the study by Goff et al., primer pairs were constructed for 25 different sequences, and 16 of the 17 pairs that produced products showed polymorphisms between two distantly related zebrafish lines, or a rate of 94%. Another study was able to map 2,000 microsatellite markers to the zebrafish genome by looking at simple sequence length polymorphisms (SSLPs) within CA dinucleotide repeats [9]. Now that a draft genome is available for the zebrafish, it is possible to look for tandem repeats in the genome beyond the traditional dinucleotide repeats.
Tandem repeat/microsatellite programs
A number of programs have previously been written to detect repeats within genomes, including both interspersed repeats [33] and microsatellite repeats [34-36]. Databases of short period microsatellite repeats have been created for prokaryotic [37] and eukaryotic genomes [38-40]. However, none of these present the user with a method to biologically test whether or not these repeats have variable copy numbers in a population. The most popular biological approach to microsatellite DNA typing that has been used is PCR amplification. Therefore, a method to both detect regions of exact tandem repeats and PCR primers within the flanking sequence is proposed.
Methods to determine the location of markers such as the dinucleotide CA/TG repeats have been previously described by constructing PCR primers from comparative genomes which are then tested against the genome of interest. In addition, it is desired to detect whether or not polymorphic copy numbers exist for a particular locus, which can be accomplished by running the results of PCR amplification for a number of different individuals through an agarose gel.
Project overview
This study detected exact tandem repeats of various length and copy number within the zebrafish genome. Once these regions were located, 500 bases flanking the beginning and ending of the repeat region were reported. These regions were additionally masked for known repeats using RepeatMasker. Primers were then identified in the flanking regions. These primers can then be used within PCR reactions to detect whether or not polymorphisms exist within a zebrafish population. Those regions where polymorphisms in the copy number exist can then be used to produce a higher resolution genetic map.
Construction and content
Sequence data
For the zebrafish genome sequencing project performed at the Sanger Centre, DNA was taken from ~1,000 zebrafish embryos [2]. The eighth assembly of the zebrafish genome (Zv8) dated 12-Dec-2008 was downloaded from Ensembl's FTP site [41] in the form of supercontigs. For the Zv8 assembly, there are 1,481,241,295 bases of finished sequence data, and 123,873,047 bases of unfinished sequences. The Zv8 assembly was derived from 9,816 clones assembled into 11,623 scaffolds as well as 6,882,050 reads from whole genome shotgun sequencing of a single Tuebingen, double haploid zebrafish.
Repeat detection
Exact tandem repeats were detected using a method implemented as a perl script. Input into the script requires the sequence file in fasta format, the minimum and maximum repeat length to find, and a minimum repeat periodicity. Using these four inputs, tandem repeats can be detected for any sequence.
The algorithm proceeds across the length of the sequence, S, using a sliding window approach. The window size, w, is chosen according to the periodicity currently being considered. A substring S'i,w is taken from S, where i is the beginning location of the substring of length w. If S'i,w is equal to the previous substring found, then the repeat copy number is increased and the window proceeds to the next substring.
If S'i,w is not equal to the previous repeat, then it is tested to confirm that it is valid. In order to be a valid repeat, it is tested to ensure it does not contain a repeat of a smaller period. For instance, if w = 12, then S'i,w is tested to make sure that a repeat of length 1, 2, 3, 4, or 6 is not contained within it. As an example, if S'i,w = ACTAACTAACTA, then this would not be a valid repeat, since it contains the subrepeat (ACTA)3. All repeats found with a copy number greater than the user specified value are stored. Once all repeats have been detected, the repeats found are analyzed for overlaps due to frame shifting of the window. In the cases where overlaps are present, the repeat with the largest copy number is chosen as the repeat for this region.
After the repeats have been identified, the 500 bases (if available) upstream and downstream the repeat are reported. These regions will be used in the primer design step. At the conclusion of the program, information is printed concerning the base repeat, the copy number, beginning and ending location of the repeat, the whole repeat sequence, and the upstream and downstream regions.
This algorithm runs in C*n time, where C is the number of repeat lengths to search, and n is the length of the sequence. Since C is a constant generally computed beforehand, this factors to O(n) time complexity.
Sequences used
In order to narrow down the search space, repeats with a length of at least three bases and a copy number of at least ten exact tandem repeats were detected. For instance, the repeat sequence AAGCT has a length of five; if AAGCT is found repeated at least 10 consecutive times (for instance, AAGCT AAGCT AAGCT AAGCT AAGCT AAGCT AAGCT AAGCT AAGCT AAGCT), then it is reported.
PCR primers
After exact tandem repeat regions were detected, the 5' and 3' flanking regions were extracted and masked for low complexity and known repeat families using RepeatMasker [2] and MaskerAid [33]. PCR primers were detected within the flanking regions using MPrime 1.3 [42]. The resulting primer pairs can be used in PCR experiments to determine if polymorphisms in the copy number for this particular repeat occur in a given population. MPrime reports information concerning primer location, end annealing and self annealing scores, GC concentration, melting temperature, product size, and sequence similarity scores each of which is stored in a MySQL database for further use.
Database structure
A relational database to store the zebrafish repeats was constructed using MySQL version 5.0.19. The database Zv8 is available with "select" privileges at the server http://kbrin.a-bldg.louisville.edu/ under the user "zebrafish" and password "daniorerio". Ten tables were created, primarily for genomic contig information, transcript location, and repeat identification. An entity-relationship (E-R) diagram of the database is provided in Fig. 1. A brief description of the content of each table follows.
Figure 1.
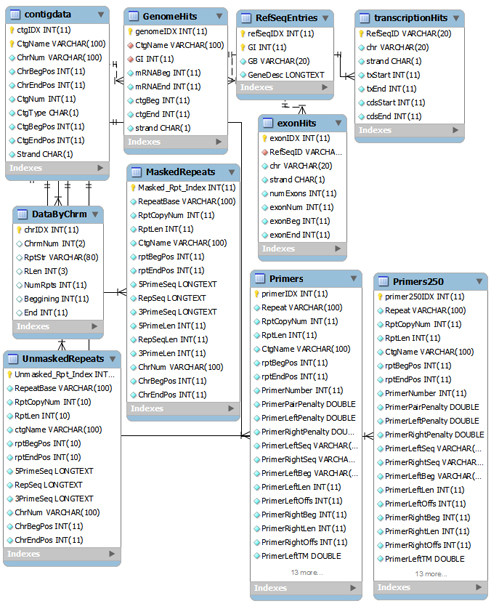
Entity-relationship model for zebrafish repeats database.
contigdata contains information on assembled clones for the Zv8 assembly of the zebrafish genome, including their length and mapping location in relationship to a zebrafish linkage group. This information is taken directly from the Zv8_scaffold.agp file available from the Ensembl ftp site.
RefSeqEntries is a table of basic details of zebrafish RefSeq entries including RefSeqID, GenBank accession (GB), GenBank identifier (GI), and a brief description (GeneDesc). No sequence information for the RefSeq entries is stored locally within the Zv8 database since it can be readily accessed externally.
transcriptionHits contains coarse-level information about the transcription as well as translation start and stop of a RefSeq entry within a zebrafish linkage group. This information can then be used to quickly determine if any feature (such as a repeat) is genic or intergenic in nature.
GenomeHits contains the same information as transcriptionHits provided at a contig level as opposed to a linkage group level.
exonHits is a table providing exon-level information. This table contains information as far as the exon beginning and ending location and the exon number is concerned. exonHits can thus be used to determine if a feature is intronic or exonic in nature.
UnmaskedRepeats houses information for each detected repeat, including the contig in which it is found (ctgName), its location (rptBegPos, rptEndPos), its length and copy number (RptLen, RptCopyNum) and flanking sequence information (5PrimeSeq, 3PrimeSeq) which can be used to detect amplification primers.
MaskedRepeats contains the same information as UnmaskedRepeats with the exception that the flanking sequences have been masked for known repetitive elements and simple repeats using RepeatMasker as described in the previous section.
DataByChrm is a summary table taken from UnmaskedRepeats that can quickly be indexed when searching for repeats by linkage group.
Primers is a table containing information on amplification primers that have been detected using MPrime 1.3 in the flanking regions of the repeat masked entries of MaskedRepeats. Information contained in this table includes thermodynamic properties and parameters used in designing the primers.
Primers250 is a subset of Primers where the repeat region itself is 250 bases or fewer in length. The primers and corresponding repeats reported in this table are those most likely to be useful for studies of copy number polymorphisms, since they can be sequenced in a single run using traditional Sanger sequencing techniques.
Utility
A list of the number of repeats found with base length three or more with a copy number of at least ten are given in Table 1. To no surprise, the number of tandem repeats is dominated by bases with a length of 3-, 4-, and 5-mers. The majority of these repeats are AT-rich, with the base repeat AAT and its reverse complement ATT accounting for 87% of all triplet tandem repeat loci (Additional file 1, Table S1). The repeats AGAT and its reverse complement ATCT compose 41% of all quadruplet tandem repeats (Additional file 1, Table S2) while 64% of all pentamer repeats are represented by the either the base sequence AATAT or its complement TTATA (Additional file 1, Table S3).
Table 1.
Frequency of tandem repeats within the zebrafish genome Zv8 assembly by base length
| Repeat Len | # | Repeat Len | # | Repeat Len | # |
| 1-mers | N/A | 19-mers | 5 | 37-mers | 3 |
| 2-mers | N/A | 20-mers | 6 | 38-mers | 1 |
| 3-mers | 37,383 | 21-mers | 7 | 39-mers | 2 |
| 4-mers | 67,313 | 22-mers | 9 | 40-mers | 0 |
| 5-mers | 11,767 | 23-mers | 7 | 41-mers | 2 |
| 6-mers | 93 | 24-mers | 18 | 42-mers | 0 |
| 7-mers | 1 | 25-mers | 4 | 43-mers | 1 |
| 8-mers | 10 | 26-mers | 7 | 44-mers | 2 |
| 9-mers | 1 | 27-mers | 40 | 45-mers | 2 |
| 10-mers | 5 | 28-mers | 5 | 46-mers | 5 |
| 11-mers | 5 | 29-mers | 4 | 47-mers | 0 |
| 12-mers | 6 | 30-mers | 117 | 48-mers | 1 |
| 13-mers | 3 | 31-mers | 10 | 49-mers | 1 |
| 14-mers | 16 | 32-mers | 5 | 50-mers | 1 |
| 15-mers | 6 | 33-mers | 1 | 51-mers | 1 |
| 16-mers | 16 | 34-mers | 2 | 52-mers | 0 |
| 17-mers | 5 | 35-mers | 2 | 53-mers | 1 |
| 18-mers | 21 | 36-mers | 9 | 54-mers | 1 |
The results in Table 1 indicate a drastic drop off when the base repeat reaches a length of six. In addition, there appear to be spikes in the data when the base repeat length is a multiple of three, particularly with repeats with a base length of 18, 24, 27, and 30. Further analysis indicates that of the 191 detected exact tandem repeats of length 18, 24, 27, or 30, only 18 of these are within 10 kb of another such repeat. Further stringency finds that fourteen are within 500 bp, with six being direct overlaps due to a staggered repeat. Fig. 2 visually illustrates the linkage group localization of these repeats while Fig. 3 shows the distribution among linkage groups. A complete list of all 191 detected repeats is given in Additional file 2, Table S4 (length 18), Additional file 2, Table S5 (length 24), Additional file 2, Table S6 (length 27), and Additional file 2, Table S7 (length 30).
Figure 2.
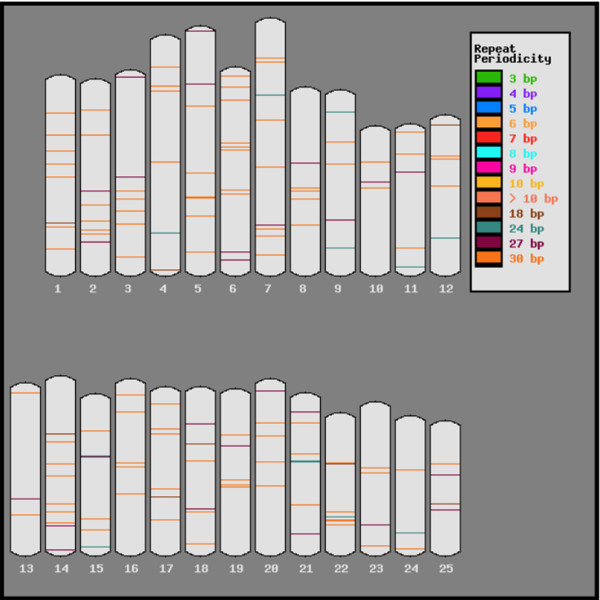
Linkage group localization for repeats with a base length of 18, 24, 27 and 30.
Figure 3.
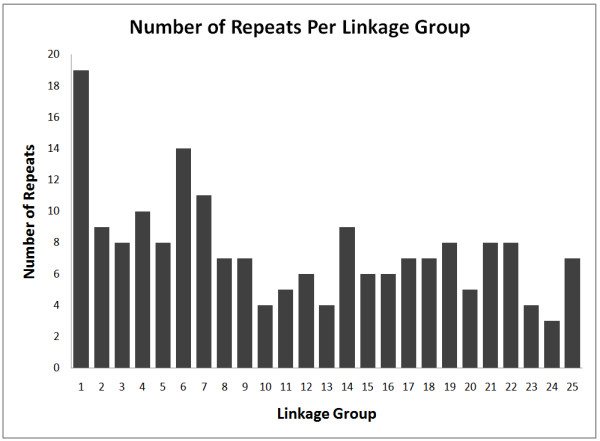
Distribution of repeats of base length 18, 24, 27 and 30 among linkage groups.
There is a single repeat with a base of 54 found within the Zv8 assembly. This is repeated fourteen times. This repeat is:
(CGAGTTCTTATCAGCTGTGTTGTCGCG
CGCGTACTGAATAGCGGTGTTGTCGCA)14
This repeat occurs in an intronic region of the gene with RefSeq ID NM_199842 (GenBank:41054140).
Repeats with multiple of 3 base length
Repeats with a multiple of three base lengths are candidates for genic regions due to their ability to maintain the current reading frame while potentially providing for a repetitive amino acid sequence. Tandem repeats within a transcribed region are known to be affiliated with a plethora of genetic diseases. Such repeats can be classified into Type I (exonic) or Type II (intronic) expansions. The type of expansion characterizes not only the location of the repeats, but the underlying pathology as well [16].
Repeats occurring within transcribed regions were studied using the RefSeq gene sequence track from the December 2008 Goldenpath assembly of the zebrafish genome (corresponding to the Zv8 assembly) from the Table Browser of the UCSC genome browser [43]. A total of 131,383 exon locations were noted from 15,417 mapped loci. Each repeat location was compared against the RefSeq loci to determine if the repeat is close to a known gene (within 1,000 bp of the 5' or 3' end), overlaps a known gene region, or is contained within a known transcriptional unit. A total of 2,397 tandem repeats were found to be close to a known gene with nearly all of these (2,393) having a base repeat length of either three, four or five. These repeats are A+T rich. Forty-three tandem repeats were found to overlap intron/exon boundaries. Twenty-eight of these have a base length of three while eleven have a base length of four, three have a base length of five and one has a base length of 30. Forty-nine exonic tandem repeats were found, 30 with a base length of three, 15 with a base length of four, three with a base length of five, and one with a base length of 30 (Additional file 3, Table S8). Further analysis shows the majority of these repeats (35/49) are found in the 3' UTR. Two are found in the 5' UTR. The remaining twelve repeats are found within coding exons. There were 26,909 intronic tandem repeats detected. 7,624 of these intronic tandem repeats are of base length three, 15,410 are length four, while 2,752 are of base length five. For the 191 repeats with a base size of 18, 24, 27 or 30, forty-eight were found to occur within intronic regions; one within an exonic region; and one in an intron/exon boundary. The remaining 141 repeats appear to occur in intergenic regions.
Repeats with a base length of 18, 24, 27 or 30 were further characterized by looking at both the nucleic acid composition as well as the six possible protein translations. Out of the 21 repeats with a base of 18, eighteen can be translated into the consensus protein sequence PP[E|V]LPD (Table 2). This is particularly surprising since the majority are located in intergenic regions and are coded by six unique nucleotide sequences. For the repeats with a base length of 24, thirteen out of 18 can be translated into the consensus protein sequence PGPPPQLHA from one of five nucleotide sequences (Table 3). All but six of the repeats of base length 27 fall into one of three categories, translating into the consensus patterns V[L|M][IVNSK]S[C|G]VVAR, [L|P]A[L|Q]PAP[P|H]R[R|L|P], or L[P|A]E[W|R]PPPPE (Table 4). For the repeats of base length 30, one hundred and three can be translated into the perfectly conserved consensus pattern APAPERPPVS using one of seven different nucleotide sequences. One of these instances, in fact, is within a hypothetical protein sequence. An additional five instances translate into the peptide AP[A|V]QLPPVPP while eight translate to QLTRWPTPVL using one of two nucleotide patterns (Table 5).
Table 2.
Repeats of length 18 with each of the six possible protein translations
| GP | Repeat Base | Possible Protein Translations | # | |||||
|---|---|---|---|---|---|---|---|---|
| 1 | ACCCTCCAGAGCTGCCAG | PPELPD | VWQLWR | PSRAAR | LQSCQT | GLAALE | GSSGGS | 10 |
| 1 | AGCTCTGGAGGGTCTGGC | PPELPD | VWQLWR | PSRAAR | LQSCQT | GLAALE | GSSGGS | 3 |
| 1 | ACTGGAGGGTCTGGCAGC | PPVLPD | VWQHWR | PSSAAR | LQCCQT | ALEGLA | GSTGGS | 1 |
| 1 | AGCTCTGGCGGGTCTGGC | PPELPD | VWQLWR | PARAAR | RQSCQT | GLAALA | SGSSGG | 1 |
| 1 | ACCCTCCAGTGCTGCCAG | PPVLPD | VHQHWR | PSSAAR | LQCCQT | GLAALE | SGSTGG | 2 |
| 1 | ACCCGCCAGAGCTGCCAG | PPELPD | VWQLWR | PARAAR | RQSCQT | GLAALA | GSSGGS | 1 |
| 2 | ACGCCGCAGCCAGAGTCG | CGVDSG | AASTLA | RRRLWL | ARVDAA | QPESTP | SQSRRR | 1 |
| 3 | ATCGTGGCCCCCTCGTCC | RPSWPP | VHRGPL | SIVAPS | RGPRWT | RGGHDG | EGATMD | 1 |
| 4 | CCCTGTGGTGCTGTGTGT | CGAVCP | VVLCVP | WCCVSL | GTHSTT | QGHTAP | RDTQHH | 1 |
GP: grouping; #: number of instances found in the Zv8 assembly. Note the repeat base has been reordered by lexicographical order.
Table 3.
Repeats of length 24 with each of the six possible protein translations
| GP | Repeat Base | Possible Protein Translations | # | |||||
|---|---|---|---|---|---|---|---|---|
| 1 | ACGCTCCAGGCCCTCCGCAGCTCC | PGPPQLHA | AWSCGGPG | SRPSAAPR | QALRSSTL | ERGAAEGL | SVELRRAW | 6 |
| 1 | AGCGTGGAGCTGCGGAGGGCCTGG | PGPPQLHA | AWSCGGPG | SRPSAAPR | QALRSSTL | ERGAAEGL | SVELRRAW | 4 |
| 1 | ACGCCCCAGGCCCTCCGCAGCTCC | PGPPQLHA | AWSCGGPG | PRPSAAPR | QALRSSTP | GRGAAEGL | GVELRRAW | 1 |
| 1 | AGCTGCGGAGGGCCTGGGGCGTGG | PGPPQLHA | AWSCGGPG | PRPSAAPR | QALRSSTP | LRGAAELG | GVELRRAW | 1 |
| 1 | ACGCTCCAGGCCCTCGGCAGCTGC | PGPRQLHA | TLQALGSC | SRPSAAAR | GACSCRGP | ERAAAELG | SVQLPRAW | 1 |
| 2 | AAGCCCGAGGCGACGCCATTGGAG | GLLQWRRL | EATPLEKP | RRRHWRSP | GDAIGEAR | SGFSNGVA | RASPMASP | 1 |
| 2 | AAGGCCGAGGCGACGCCATTGGAG | GLLQWRRL | EATPLEKA | RRRHWRRP | GDAIGEGR | SAFSNGVA | MASPRPSP | 1 |
| 3 | AAGCGGATTTTTGACGCGCGAGTG | *SGFLTRE | EADF*RAS | KRIFDARV | LARQKSAS | HSRVKNPL | TRASKIRF | 1 |
| 4 | AAGCGCCGGTGAGCCCTCGCCCTC | ALEGEGSP | RLRARAHR | A*GRGLTG | R*ALALKR | AGEPSPSS | PVSPRPQA | 1 |
| 5 | AAGCTCAGGCGGCGGCCATTCAGG | GGGHSGSS | AAAIQEAQ | RRPFRKLR | *AS*MAAA | PELPEWPP | LSFLNGRR | 1 |
GP: grouping; #: number of instances found in the Zv8 assembly. Note the repeat base has been reordered by lexicographical order.
Table 4.
Repeats of length 27 with each of the six possible protein translations
| GP | Repeat Base | All Possible Protein Translations | # | |||||
|---|---|---|---|---|---|---|---|---|
| 1 | AACACAGCTGATAAGAACTCGCGCGAC | VLISCVVAR | RATTQLIRT | SRDNTADKN | FLSAVLSRE | SSYQLCCRA | LARQHS**E | 2 |
| 1 | AACACAGCTGATCAGTACGCGCGCGAC | VLISCVVAR | RATTQLIST | ARDNTADQY | Y*SAVLSRA | RTDQLCCRA | RARQHS*SV | 1 |
| 1 | AGCTGTGTTGTCGCGCGCGTTATGATC | VMISCVVAR | RATTQLIIT | ARDNTADHN | L*SAVLSRA | RYDQLCCRA | RARQHS*S* | 2 |
| 1 | AGCTGTGTTGTCGCGCGCGTTCTGATC | VLISCVVAR | RATTQLIRT | ARDNTADQN | F*SAVLSRA | RSDQLCCRA | RARQHS*SE | 1 |
| 1 | AATAGCGGTGTTGTCGCGCGCGTTCTG | VLNSGVVAR | RATTPLFRT | ARDNTAIQN | F*IAVLSRA | RSE*RCCRA | RARQHRYSE | 1 |
| 1 | AACACCGCTCTTCAGTACGCGCGCGAC | VLKSGVVAR | RATTPLFST | ARDNTALQY | Y*RAVLSRA | RTEERCCRA | RARQHRSSV | 1 |
| 1 | AGCTGTGTTGTCGCGCGAGTTCTTATC | VLISCVVAR | RATTQLIRT | SRDNTADKN | FLSAVLSRE | SSYQLCCRA | LARQHS**E | 3 |
| 1 | AACACAGCTGATAAGAACGCGCGCGAC | VLISCVVAR | RATTQLIRT | ARDNTADKN | FLSAVLSRA | RSYQLCCRA | RARQHS**E | 1 |
| 1 | AACACCGCTACTCAGTACGCGCGCGAC | VLSSGVVAR | RATTPLLST | ARDNTATQY | Y*VAVLSRA | RTE*RCCRA | RARQHRYSV | 1 |
| 1 | AACACAGCTGATCAGAACGCGCGCGAC | VLISCVVAR | RATTQLIRT | ARDNTADQN | VLSRAF*SA | RSDQLCCRA | RARQHS*SE | 1 |
| 1 | AACACAGCTGACAAGAACTCGCGCGAC | VLVSCVVAR | RATTQLTRT | SRDNTADKN | FLSAVLSRE | SSCQLCCRA | LARQHS*QE | 1 |
| 2 | ACCCAGGCTCCTCGCCCTGCCGGCGCC | LALPAPPRL | SPCRRHPGS | RPAGATQAP | RSLGGAGRA | GAWVAPAGR | EPGWRRQGE | 1 |
| 2 | ACCCAGACGTCTCGCCCTGCCGGCGCC | LALPAPPRR | SPCRRHPDV | RPAGATQTS | RRLGGAGRA | DVWVAPAGR | TSGWRRQGE | 1 |
| 2 | ACGTCTGGGTGGCGCCGGCAGGGCGAG | LALPAPPRR | SPCRRHPDV | RPAGATQTS | RRLGGAGRA | DVWVAPAGR | TSGWRRQGE | 1 |
| 2 | ACGTCTGGGTGGCGCCGGCTGGGCGAG | LAQPAPPRR | SPSRRHPDV | RPAGATQTS | RRLGGAGWA | DVWVAPAGR | TSGWRRLGE | 1 |
| 2 | ACAGGCCTCCAGCCCAGCCGGCTCCCC | PAQPAPHRP | GSRLGWRPV | SPAGSPQAS | GGLWGAGWA | LGWRPVGSR | PAGLEACGE | 1 |
| 3 | AATGGCCGCCGCCTCCTGAGCTTCCTG | LPEWPPPPE | SSGGGGHSG | LRRRRPFRK | S*MAAAS*A | AQEAAAIQE | FLNGRRLLS | 1 |
| 3 | AAGCTCAGGAGGCGGCGGCCATTCAGG | LPEWPPPPE | SSGGGGHSG | LRRRRPFRK | S*MAAAS*A | AQEAAAIQE | FLNGRRLLS | 2 |
| 3 | AGCTCAGGCGGCGGCGGCCATTCAGGG | LPEWPPPPE | SSGGGGHSG | LRRRRPFRE | P*MAAAA*A | AQAAAAIQG | SLNGRRRLS | 1 |
| 3 | AGCGAGCTCGGGAGGCGGCGGCCATTC | LAEWPPPPE | SSGGGGHSA | LGRRRPFSE | R*MAAASRA | AREAAAIQR | SLNGRRLPS | 1 |
| 3 | AATGGCCGCCGCCGCCTGAGCTTCCTG | LPEWPPPPE | SSGGGGHSG | LRRRRPFRK | S*MAAAA*A | AQAAAAIQE | FLNGRRRLS | 3 |
| 3 | AAGCTCAGGAGGCGGCGGCCGTTCAGG | LPERPPPPE | SSGGGGRSG | LRRRRPFRK | S*TAAAS*A | AQEAAAVQE | FLNGRRLLS | 3 |
| 3 | AATGGCCGCCGCCGCCTGAGCTCCCTG | LPEWPPPPE | SSGGGGHSG | LRRRRPFRE | P*MAAAA*A | AQAAAAIQG | LNGRRRLSS | 1 |
| 3 | AACGGCCGCCGCCTCCTGAACTCCCTG | LPERPPPPE | SSGGGGRSG | FRRRRPFRE | P*TAAAS*T | VQEAAAVQG | LNGRRLLNS | 2 |
| 4 | AAGACCAGAGGGGAGCCGGCGGGGCTG | G*RPEGSRR | AGGAEDQRG | LKTRGEPAG | PRRLPSGLQ | PAGSPLVFS | PPAPLWSSA | 1 |
| 4 | CCCCTCTGGTCTCCTGCCCTGCCGGCT | GRRPEGSRQ | AGRAGDQRG | QETRGEPAG | PCRLPSGLL | PAGSPLVSC | LPAPLWSPA | 1 |
| 5 | AACTCTATTGAGTGTCAGGTCATCTCC | SSPTLLSVR | HLQLY*VSG | ISNSIECQV | DLTNRVGD | T*HSIELEM | PDTQ*SWR* | 1 |
| 6 | AAAGCAACAACTCCACCAACATCAGCT | QLHQHQLKQ | NSTNIS*SN | TPPTSAKAT | CCFS*CWWS | VALADVGGV | LL*LMLVEL | 1 |
| 7 | AACAGCAGAGCCATTCACTGACCCCAG | H*PQNSRAI | TDPRTAEPF | LTPEQQSHS | *MALLFWGQ | EWLCCSGVS | NGSAVLGSV | 1 |
| 8 | AAGGAGCCGGGCTGGGGCCGGCAGGGC | AGRARSRAG | PAGQGAGLG | RQGKEPGWG | APARLLALP | PQPGSLPCR | PSPAPCPAG | 1 |
GP: grouping; #: number of instances found in the Zv8 assembly. Note the repeat base has been reordered by lexicographical order.
Table 5.
Repeats of length 30 with each of the six possible protein translations
| GP | Repeat Base | All possible protein translations | # | |||||
|---|---|---|---|---|---|---|---|---|
| 1 | AGCCCCTGAGCGCCCTCCAGTGTCGGCTCC | APAPERPPVS | DTGGRSGAGA | RHWRALRGWS | RLQPLSALQC | GSSP*APSSV | TLEGAQGLEP | 32 |
| 1 | AGAGCGCCCGCCAGTGTCGGCTCCAGCCCC | APAPERPPVS | DTGGRSGAGA | RHWRALWGWS | RLQPQSARQC | GSSPRAPASV | TLAGALGLEP | 12 |
| 1 | ACACTGGAGGGCGCTCAGGGGCTGGAGCCG | APAPERPPVS | DTGGRSGAGA | RHWRALRGWS | RLQPLSALQC | GSSP*APSSV | TLEGAQGLEP | 16 |
| 1 | ACACTGGCGGGCGCTCTGGGGCTGGAGCCG | APAPERPPVS | DTGGRSGAGA | RHWRALWGWS | RLQPQSARQC | GSSPRAPASV | TLAGALGLEP | 12 |
| 1 | ACACTGGAGGGCGCTCTGGGGCTGGAGCCG | APAPERPPVS | DTGGRSGAGA | RHWRALWGWS | RLQPQSALQC | GSSPRAPSSV | TLEGALGLEP | 15 |
| 1 | AGAGCGCCCTCCAGTGTCGGCTCCAGCCCC | APAPERPPVS | DTGGRSGAGA | RHWRALWGWS | RLQPQSALQC | GSSPRAPSSV | TLEGALGLEP | 14 |
| 1 | ACACTGGCGGGCGCTCGGGGGCTGGAGCCG | APAPERPPVS | DTGGRSGAGA | RHWRALGGWS | RLQPPSARQC | GSSPRAPASV | TLAGARGLEP | 2 |
| 2 | ACTGGAGGCAGCTGGACTGGAGCCGGCGGG | APVQLPPVPP | AGGTGGSWTG | PAGLEAAGLE | RRDWRQLDWS | LQSSCLQSRR | SSPAASSPAG | 2 |
| 2 | ACAGGAGGCAGCTGGGCTGGAGCCGGCGGG | APAQLPPVPP | AGGTGGSWAG | PAGQEAAGLE | RRDRRQLGWS | LQPSCLLSRR | SSPAASCPAG | 1 |
| 2 | AGCTGCCTCCAGTCCCGCCGGCTCCTGCCC | APAQLPPVPP | AGGTGGSWAG | PAGLEAAGQE | RRDWRQLGRS | LLPSCLQSRR | SSPAGSCPAA | 1 |
| 2 | ACTGGAGGCAGCTGGGCAGGAGCCGGCGGG | APAQLPPVPP | AGGTGGSWAG | PAGLEAAGQE | RRDWRQLGRS | LLPSCLQSRR | SSPAGSCPAA | 1 |
| 3 | AAGATGGCCGACTCCAGTCCTCCAGCTCAC | QLTRWPTPVL | SSQDGRLQSS | AHKMADSSP | WRTGVGHLVS | GGLESAIL*A | EDWSRPSCEL | 7 |
| 3 | ACTGGAGTCGGCCATCTTGTGAGCTGGAGG | QLTRWPTPVL | SSQDGRLQSS | AHKMADSSP | WRTGVGHLVS | GGLESAIL*A | EDWSRPSCEL | 1 |
| 4 | AAACTGCCGCAAGGCTCCAAATACTTCTCC | KLPQGSKYFS | NCRKAPNTSP | TAARLQILLQ | LEKYLEPCGS | WRSIWSLAAV | GEVFGALRQF | 1 |
GP: grouping; #: number of instances found in the Zv8 assembly. Note the repeat base has been reordered by lexicographical order.
Repeat distribution
The distribution of repeats along each of the linkage groups was examined. Each linkage group was divided into twenty segments along their length with each segment representing 5% of the total length of the linkage group. Repeats were placed in a bin based upon their begin location. Repeat base lengths of 3 bp, 4 bp, 5 bp, and >5 bp were examined. A chi-square test was performed for each base length and linkage group combination independently (results not shown). For 19 degrees of freedom, the critical χ2 value for rejecting the null hypothesis of a uniform distribution with p = 0.95 is 30.14 and for p = 0.99, the value is 36.19. For triplet repeats, the null hypothesis is rejected for all 25 linkage groups with the highest differences occurring in bins 1 and 20, representing the telomeric regions of the linkage group. The null hypothesis is rejected for all 25 linkage groups for 4 base repeats with bins 1, 3, 18 and 20 showing the largest differences. Pentamer repeats also exhibit non-uniform distribution as well since the null hypothesis is rejected for all linkage groups. Repeats with a base length greater than 5 do not seem to have as many statistically significant hot spots since the null hypothesis is only rejected for eleven linkage groups at a level of p = 0.95. However, the bins showing the largest difference are bins 18 and 20 in the telomeric region at one end of the linkage group. These results are summarized in Fig. 4 which shows the distribution of repeats along the chromosome. As Fig. 4 indicates, there seems to be a strong preference for repeats in the telomeric regions of the linkage group. In fact, as the trend line indicates, the further away from the center of the linkage group, the more likely it is that a repeat will occur.
Figure 4.
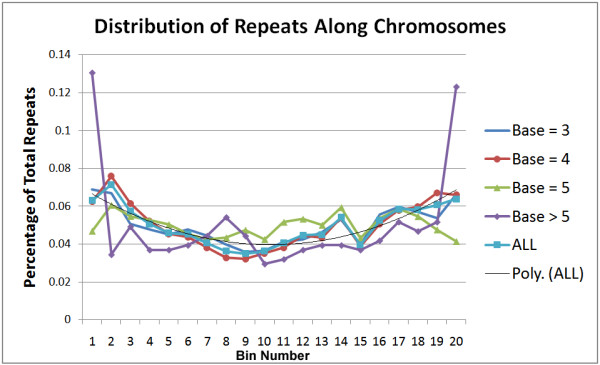
Distribution of repeats across linkage groups. The bin number in the x-axis represents increments of 5% across each linkage group, while the y-axis represents the percentage of all repeats falling within the respective bin.
Discussion
Comparison to other eukaryotic genomes
The most comprehensive resource for microsattelite repeats is the Tandem Repeats Database [44]. Our analysis of the exact tandem repeats in the zebrafish genome were compared to the human genome (hg19 build), mouse (mm8), rat (rn4), fugu (fr1), and tetraodon (tetNig1) genomes available in the tandem repeats database (Table 6). Based on these queries, it is shown that the detected exact tandem repeats within the zebrafish genome are much more prevalent, occurring at a rate of once out of every 12,659 based compared to the closest genome, mouse, which shows a frequency of 1/45,498 bases. For each of the genomes (with the exception of tetraodon), the most prevalent repeat base length is 4, with roughly twice as many tandem repeats as triplet base lengths. One interesting observation is that the number of hexamer repeats is highly variable and is found at a low frequency in the zebrafish. A comparison of the repeats of length 18, 24, 27, and 30 suggest the zebrafish is somewhat unique. It must be noted that this analysis should be taken with care, since the other genomes are older assemblies and the repeat detection mechanisms between Tandem Repeats Finder and our approach while similar, are not identical.
Table 6.
Comparison of exact tandem repeats within various eukaryotic genomes
| Base Size | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Genome | Build | Genome Size (MB) | All | 3 | 4 | 5 | 6 | 18, 24, 27, 30 | Long | Frequency |
| zebrafish | Zv8 | 1480 | 116,915 | 37,383 | 67,313 | 11,767 | 93 | 191 | 54 | 1/12,659 |
| mouse | Mm8 | 2600 | 57,145 | 16,022 | 33,430 | 5,186 | 2,066 | 17 | 68 | 1/45,498 |
| rat | Rn4 | 2800 | 41,422 | 16,746 | 22,077 | 1,213 | 930 | 35 | 70 | 1/67,597 |
| fugu | Fr1 | 395 | 3,785 | 997 | 2,375 | 366 | 33 | 0 | 48 | 1/104,359 |
| tetraodon | TetNig1 | 350 | 1,808 | 1,287 | 419 | 82 | 14 | 0 | 12 | 1/193,584 |
Long represents the longest base size repeat detected. Frequency calculated as the genome size divided by the number of total repeats.
Conserved repeat regions
While there is not any direct evidence that more than one of the exact repeats of length 18, 24, 27 or 30 are part of a coding region, the striking conservation at the protein level cannot be ignored, particularly since the nucleotide sequence in not necessarily conserved. Looking at the consensus peptide translations, in can be seen that many of these are proline-rich. Most of these contain the pattern PxxP, which has been shown to bind to SH3 domain containing proteins [45]. This indicates the potential of novel signaling and protein-interaction genes [46-48].
NCBI blastp analysis of the nr database searching for the repeat APAPERPPVS yields six hypothetical zebrafish proteins from the RefSeq XP division (GenBank:18954121, GenBank:189515106, GenBank:189514738, GenBank:12581872, GenBank:189532449, GenBank:125844672, GenBank:189518247) detected using GNOMON [49]. Most of the blastp hits yield hypothetical proteins. Annotated hits include a serum response binding protein with the repeat RPAVERPAVE (GI 89271263); inclusion membrane protein with the repeat APAPEAPAPE (GenBank:169667289); cell wall type 2 protein with the repeat KPPVEKPPVY (GenBank:2347094); and an outer membrane adhesion-like protein with the repeat APAPEPSPAP (GenBank:170774881). These annotations further underline the potential for these repeats to belong to novel proline-rich genes. Further research into these repeats is needed to prove conclusive biological evidence.
Since the zebrafish genome is currently not at a finished state, it must be noted that some of the tandem repeats detected may result from misassemblies. In addition, the repeat detection program as currently constructed only detects exact tandem repeats. If a repeat region is long enough, a stutter in the repeat base will result in two adjacent repeat regions being reported. It is highly likely that some repetitive regions will go undetected or will be presented as truncated using this method. The repeats detected and presented using this approach will not be a complete set of repeat regions but should yield a high percentage of all tandemly repeating elements with a repeat copy number of ten or more.
Since the tiling path for the Zv8 reference genome is a consensus from over 1,000 different animals, the consensus sequence may under- or over-represent the repetitive tandemly repeating regions within an individual. Further insight might be gathered by looking at individual trace files which could indicate repeats with variable copy numbers. However, gaps in the Zv8 tiling path are filled using a single animal that has been sequenced using whole genome sequencing methods. In addition, Zv8 provides 6.5-7× coverage, indicating that only commonly found variations would be detected within the trace files.
Conclusions
In this research, we detected exact tandem repeats within the zebrafish genome with a base length of at least three and a copy number of at least 10 with the end goal of detecting regions likely to be vulnerable to VNTRs. A total of 116,915 such regions were detected, along with primer pairs in the flanking regions that can be used for biological assays. The high incidence of repeats with a base length of 18, 21, 24, 27, and 30 suggests that many of these are involved in genic regions. While none of these are within known protein coding sequences, the high similarity of potential amino acid translations suggests novel coding sequences containing highly repetitive regions. Our results should serve as a resource for zebrafish molecular biologists interested in studying potential diseases associated with these repeats, as well as serve as a source for construction of a higher resolution genetic map than is currently available.
Availability and requirements
A web interface for querying repeats is available at http://bioinformatics.louisville.edu. This portal allows for users to search for a repeats of a selected base size from any valid specified region within the 25 linkage groups. The information returned is a graphical representation of the repeats as well as a link to a tab-delimited text file containing information concerning the linkage group, chromosomal repeat begin and end positions, repeat base size, repeat base sequence, copy number, and the highest scoring forward and reverse primer from the flanking sequence. In addition, the repeats are available for download as custom tracks for the UCSC [43] and FishMap [50] genome browsers. The MySQL Zv8 database can be accessed directly with select privledges from the MySQL server http://kbrin.a-bldg.louisville.edu with the username 'zebrafish' and the password 'daniorerio'.
Authors' contributions
ER was responsible for all aspects of this work, including project design, database design, analysis, and manuscript preparation. All authors read and approved of the final manuscript.
Supplementary Material
Tables containing information on the number of trinucleotide, quadruplet and pentamer tandem repeats detected in the zebrafish genome Zv8 assembly.
Tables containing information on the repeat instances with a base length size of 18, 24, 27 and 30 detected in the zebrafish genome Zv8 assembly.
Table containing information on the exact tandem repeat instances found within coding regions of RefSeq entries mapped in the zebrafish genome Zv8 assembly.
Acknowledgements
ER would like to thank members of the University of Louisville Bioinformatics Research Group (BRG) for providing important feedback. This work was supported by the National Institutes of Health [P20RR16481, P30ES014443]. Its contents are solely the responsibility of the author and do not necessarily represent the official views of NCRR, NIEHS, or NIH.
References
- Zebrafish FAQs. http://www.neuro.uoregon.edu/k12/FAQs.html
- Zebrafish (Danio rerio) Sequencing Project. http://www.sanger.ac.uk/Projects/D_rerio/
- Stickney HL, Schmutz J, Woods IG, Holtzer CC, Dickson MC, Kelly PD, Myers RM, Talbot WS. Rapid mapping of zebrafish mutations with SNPs and oligonucleotide microarrays. Genome Res. 2002;12:1929–1934. doi: 10.1101/gr.777302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler R, Rauch GJ, Baier H, van BF, Bross L, Dekens MP, Finger K, Fricke C, Gates MA, Geiger H, Geiger-Rudolph S, Gilmour D, Glaser S, Gnugge L, Habeck H, Hingst K, Holley S, Keenan J, Kirn A, Knaut H, Lashkari D, Maderspacher F, Martyn U, Neuhauss S, Neumann C, Nicolson T, Pelegri F, Ray R, Rick JM, Roehl H, Roeser T, Schauerte HE, Schier AF, Schonberger U, Schonthaler HB, Schulte-Merker S, Seydler C, Talbot WS, Weiler C, Nusslein-Volhard C, Haffter P. A radiation hybrid map of the zebrafish genome. Nat Genet. 1999;23:86–89. doi: 10.1038/12692. [DOI] [PubMed] [Google Scholar]
- Hukriede N, Fisher D, Epstein J, Joly L, Tellis P, Zhou Y, Barbazuk B, Cox K, Fenton-Noriega L, Hersey C, Miles J, Sheng X, Song A, Waterman R, Johnson SL, Dawid IB, Chevrette M, Zon LI, McPherson J, Ekker M. The LN54 radiation hybrid map of zebrafish expressed sequences. Genome Res. 2001;11:2127–2132. doi: 10.1101/gr.210601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postlethwait JH, Yan YL, Gates MA, Horne S, Amores A, Brownlie A, Donovan A, Egan ES, Force A, Gong Z, Goutel C, Fritz A, Kelsh R, Knapik E, Liao E, Paw B, Ransom D, Singer A, Thomson M, Abduljabbar TS, Yelick P, Beier D, Joly JS, Larhammar D, Rosa F, Westerfield M, Zon LI, Johnson SL, Talbot WS. Vertebrate genome evolution and the zebrafish gene map. Nat Genet. 1998;18:345–349. doi: 10.1038/ng0498-345. [DOI] [PubMed] [Google Scholar]
- Woods IG, Kelly PD, Chu F, Ngo-Hazelett P, Yan YL, Huang H, Postlethwait JH, Talbot WS. A comparative map of the zebrafish genome. Genome Res. 2000;10:1903–1914. doi: 10.1101/gr.10.12.1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guryev V, Koudijs MJ, Berezikov E, Johnson SL, Plasterk RH, van Eeden FJ, Cuppen E. Genetic variation in the zebrafish. Genome Res. 2006;16:491–497. doi: 10.1101/gr.4791006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimoda N, Knapik EW, Ziniti J, Sim C, Yamada E, Kaplan S, Jackson D, de SF, Jacob H, Fishman MC. Zebrafish genetic map with 2000 microsatellite markers. Genomics. 1999;58:219–232. doi: 10.1006/geno.1999.5824. [DOI] [PubMed] [Google Scholar]
- Geisler R, Rauch GJ, Geiger-Rudolph S, Albrecht A, van BF, Berger A, Busch-Nentwich E, Dahm R, Dekens MP, Dooley C, Elli AF, Gehring I, Geiger H, Geisler M, Glaser S, Holley S, Huber M, Kerr A, Kirn A, Knirsch M, Konantz M, Kuchler AM, Maderspacher F, Neuhauss SC, Nicolson T, Ober EA, Praeg E, Ray R, Rentzsch B, Rick JM, Rief E, Schauerte HE, Schepp CP, Schonberger U, Schonthaler HB, Seiler C, Sidi S, Sollner C, Wehner A, Weiler C, Nusslein-Volhard C. Large-scale mapping of mutations affecting zebrafish development. BMC Genomics. 2007;8:11. doi: 10.1186/1471-2164-8-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernal A, Ear U, Kyrpides N. Genomes OnLine Database (GOLD): a monitor of genome projects world-wide. Nucleic Acids Res. 2001;29:126–127. doi: 10.1093/nar/29.1.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dib C, Faure S, Fizames C, Samson D, Drouot N, Vignal A, Millasseau P, Marc S, Hazan J, Seboun E, Lathrop M, Gyapay G, Morissette J, Weissenbach J. A comprehensive genetic map of the human genome based on 5,264 microsatellites. Nature. 1996;380:152–154. doi: 10.1038/380152a0. [DOI] [PubMed] [Google Scholar]
- Dietrich WF, Miller J, Steen R, Merchant MA, mron-Boles D, Husain Z, Dredge R, Daly MJ, Ingalls KA, O'Connor TJ. A comprehensive genetic map of the mouse genome. Nature. 1996;380:149–152. doi: 10.1038/380149a0. [DOI] [PubMed] [Google Scholar]
- Jacob HJ, Brown DM, Bunker RK, Daly MJ, Dzau VJ, Goodman A, Koike G, Kren V, Kurtz T, Lernmark A. A genetic linkage map of the laboratory rat, Rattus norvegicus. Nat Genet. 1995;9:63–69. doi: 10.1038/ng0195-63. [DOI] [PubMed] [Google Scholar]
- Cleary JD, Pearson CE. The contribution of cis-elements to disease-associated repeat instability: clinical and experimental evidence. Cytogenet Genome Res. 2003;100:25–55. doi: 10.1159/000072837. [DOI] [PubMed] [Google Scholar]
- Reddy PS, Housman DE. The complex pathology of trinucleotide repeats. Curr Opin Cell Biol. 1997;9:364–372. doi: 10.1016/S0955-0674(97)80009-9. [DOI] [PubMed] [Google Scholar]
- Everett CM, Wood NW. Trinucleotide repeats and neurodegenerative disease. Brain. 2004;127:2385–2405. doi: 10.1093/brain/awh278. [DOI] [PubMed] [Google Scholar]
- Gunawardena S, Goldstein LS. Polyglutamine diseases and transport problems: deadly traffic jams on neuronal highways. Arch Neurol. 2005;62:46–51. doi: 10.1001/archneur.62.1.46. [DOI] [PubMed] [Google Scholar]
- Orru S, Ruoppolo M, Francese S, Vitagliano L, Marino G, Esposito C. Identification of tissue transglutaminase-reactive lysine residues in glyceraldehyde-3-phosphate dehydrogenase. Protein Sci. 2002;11:137–146. doi: 10.1110/ps.ps.17102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liquori CL, Ricker K, Moseley ML, Jacobsen JF, Kress W, Naylor SL, Day JW, Ranum LP. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science. 2001;293:864–867. doi: 10.1126/science.1062125. [DOI] [PubMed] [Google Scholar]
- Albanese V, Biguet NF, Kiefer H, Bayard E, Mallet J, Meloni R. Quantitative effects on gene silencing by allelic variation at a tetranucleotide microsatellite. Hum Mol Genet. 2001;10:1785–1792. doi: 10.1093/hmg/10.17.1785. [DOI] [PubMed] [Google Scholar]
- Matsuura T, Yamagata T, Burgess DL, Rasmussen A, Grewal RP, Watase K, Khajavi M, McCall AE, Davis CF, Zu L, Achari M, Pulst SM, Alonso E, Noebels JL, Nelson DL, Zoghbi HY, Ashizawa T. Large expansion of the ATTCT pentanucleotide repeat in spinocerebellar ataxia type 10. Nat Genet. 2000;26:191–194. doi: 10.1038/79911. [DOI] [PubMed] [Google Scholar]
- Lalioti MD, Scott HS, Buresi C, Rossier C, Bottani A, Morris MA, Malafosse A, Antonarakis SE. Dodecamer repeat expansion in cystatin B gene in progressive myoclonus epilepsy. Nature. 1997;386:847–851. doi: 10.1038/386847a0. [DOI] [PubMed] [Google Scholar]
- Collins JR, Stephens RM, Gold B, Long B, Dean M, Burt SK. An exhaustive DNA micro-satellite map of the human genome using high performance computing. Genomics. 2003;82:10–19. doi: 10.1016/S0888-7543(03)00076-4. [DOI] [PubMed] [Google Scholar]
- Menotti-Raymond M, David VA, Lyons LA, Schaffer AA, Tomlin JF, Hutton MK, O'Brien SJ. A genetic linkage map of microsatellites in the domestic cat (Felis catus) Genomics. 1999;57:9–23. doi: 10.1006/geno.1999.5743. [DOI] [PubMed] [Google Scholar]
- Miyao A, Zhong HS, Monna L, Yano M, Yamamoto K, Havukkala I, Minobe Y, Sasaki T. Characterization and genetic mapping of simple sequence repeats in the rice genome. DNA Res. 1996;3:233–238. doi: 10.1093/dnares/3.4.233. [DOI] [PubMed] [Google Scholar]
- Reed PW, Davies JL, Copeman JB, Bennett ST, Palmer SM, Pritchard LE, Gough SC, Kawaguchi Y, Cordell HJ, Balfour KM. Chromosome-specific microsatellite sets for fluorescence-based, semi-automated genome mapping. Nat Genet. 1994;7:390–395. doi: 10.1038/ng0794-390. [DOI] [PubMed] [Google Scholar]
- Andres AM, Soldevila M, Lao O, Volpini V, Saitou N, Jacobs HT, Hayasaka I, Calafell F, Bertranpetit J. Comparative genetics of functional trinucleotide tandem repeats in humans and apes. J Mol Evol. 2004;59:329–339. doi: 10.1007/s00239-004-2628-5. [DOI] [PubMed] [Google Scholar]
- Andres AM, Lao O, Soldevila M, Calafell F, Bertranpetit J. Dynamics of CAG repeat loci revealed by the analysis of their variability. Hum Mutat. 2003;21:61–70. doi: 10.1002/humu.10151. [DOI] [PubMed] [Google Scholar]
- Morell R, Liang Y, Asher JH Jr, Weber JL, Hinnant JT, Winata S, Arhya IN, Friedman TB. Analysis of short tandem repeat (STR) allele frequency distributions in a Balinese population. Hum Mol Genet. 1995;4:85–91. doi: 10.1093/hmg/4.1.85. [DOI] [PubMed] [Google Scholar]
- Decorte R, Cassiman JJ. Forensic medicine and the polymerase chain reaction technique. J Med Genet. 1993;30:625–633. doi: 10.1136/jmg.30.8.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goff DJ, Galvin K, Katz H, Westerfield M, Lander ES, Tabin CJ. Identification of polymorphic simple sequence repeats in the genome of the zebrafish. Genomics. 1992;14:200–202. doi: 10.1016/S0888-7543(05)80309-X. [DOI] [PubMed] [Google Scholar]
- Bedell JA, Korf I, Gish W. MaskerAid: a performance enhancement to RepeatMasker. Bioinformatics. 2000;16:1040–1041. doi: 10.1093/bioinformatics/16.11.1040. [DOI] [PubMed] [Google Scholar]
- Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27:573–580. doi: 10.1093/nar/27.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karaca M, Bilgen M, Onus AN, Ince AG, Elmasulu SY. Exact tandem repeats analyzer (E-TRA): a new program for DNA sequence mining. J Genet. 2005;84:49–54. doi: 10.1007/BF02715889. [DOI] [PubMed] [Google Scholar]
- Landau GM, Schmidt JP, Sokol D. An algorithm for approximate tandem repeats. J Comput Biol. 2001;8:1–18. doi: 10.1089/106652701300099038. [DOI] [PubMed] [Google Scholar]
- Sreenu VB, Alevoor V, Nagaraju J, Nagarajaram HA. MICdb: database of prokaryotic microsatellites. Nucleic Acids Res. 2003;31:106–108. doi: 10.1093/nar/gkg002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astolfi P, Bellizzi D, Sgaramella V. Frequency and coverage of trinucleotide repeats in eukaryotes. Gene. 2003;317:117–125. doi: 10.1016/S0378-1119(03)00659-0. [DOI] [PubMed] [Google Scholar]
- Li B, Xia Q, Lu C, Zhou Z, Xiang Z. Analysis on frequency and density of microsatellites in coding sequences of several eukaryotic genomes. Genomics Proteomics Bioinformatics. 2004;2:24–31. doi: 10.1016/S1672-0229(04)02004-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Missirlis PI, Mead CL, Butland SL, Ouellette BF, Devon RS, Leavitt BR, Holt RA. Satellog: A database for the identification and prioritization of satellite repeats in disease association studies. BMC Bioinformatics. 2005;6:145. doi: 10.1186/1471-2105-6-145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ensembl public FTP site. ftp://ftp.ensembl.org/pub/assembly/zebrafish/Zv8release/
- Rouchka EC, Khalyfa A, Cooper NGF. MPrime: Efficient large scale multiple primer and oligonucleotide design for customized gene microarrays. BMC Bioinformatics. 2005;6:175. doi: 10.1186/1471-2105-6-175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn RM, Karolchik D, Zweig AS, Wang T, Smith KE, Rosenbloom KR, Rhead B, Raney BJ, Pohl A, Pheasant M, Meyer L, Hsu F, Hinrichs AS, Harte RA, Giardine B, Fujita P, Diekhans M, Dreszer T, Clawson H, Barber GP, Haussler D, Kent WJ. The UCSC Genome Browser Database: update 2009. Nucleic Acids Res. 2009;37:D755–D761. doi: 10.1093/nar/gkn875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelfand Y, Rodriguez A, Benson G. TRDB--the Tandem Repeats Database. Nucleic Acids Res. 2007;35:D80–D87. doi: 10.1093/nar/gkl1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandropoulos K, Cheng G, Baltimore D. Proline-rich sequences that bind to Src homology 3 domains with individual specificities. Proc Natl Acad Sci USA. 1995;92:3110–3114. doi: 10.1073/pnas.92.8.3110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Beullens M, Zhang J, Zhou Y, Nicolaescu E, Lesage B, Hu Q, Wu J, Bollen M, Shi Y. Structure and function of the two tandem WW domains of the pre-mRNA splicing factor FBP21. J Biol Chem. 2009;284:25375–25387. doi: 10.1074/jbc.M109.024828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay BK, Williamson MP, Sudol M. The importance of being proline: the interaction of proline-rich motifs in signaling proteins with their cognate domains. FASEB J. 2000;14:231–241. [PubMed] [Google Scholar]
- Mok MK, Leung KW, Xie HQ, Guo AJ, Chen VP, Zhu JT, Choi RC, Tsim KW. A new variant of proline-rich membrane anchor (PRiMA) of acetylcholinesterase in chicken: expression in different muscle fiber types. Neurosci Lett. 2009;461:202–206. doi: 10.1016/j.neulet.2009.06.015. [DOI] [PubMed] [Google Scholar]
- Nagy A, Hegyi H, Farkas K, Tordai H, Kozma E, Banyai L, Patthy L. Identification and correction of abnormal, incomplete and mispredicted proteins in public databases. BMC Bioinformatics. 2008;9:353. doi: 10.1186/1471-2105-9-353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meli R, Prasad A, Patowary A, Lalwani MK, Maini J, Sharma M, Singh AR, Kumar G, Jadhav V, Scaria V, Sivasubbu S. FishMap: a community resource for zebrafish genomics. Zebrafish. 2008;5:125–130. doi: 10.1089/zeb.2008.0531. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables containing information on the number of trinucleotide, quadruplet and pentamer tandem repeats detected in the zebrafish genome Zv8 assembly.
Tables containing information on the repeat instances with a base length size of 18, 24, 27 and 30 detected in the zebrafish genome Zv8 assembly.
Table containing information on the exact tandem repeat instances found within coding regions of RefSeq entries mapped in the zebrafish genome Zv8 assembly.