

Abstract
Solvation of molecules in water is at the heart of a myriad of molecular phenomena and of crucial importance to understanding such diverse issues as chemical reactivity or biomolecular function. Complementing well-established approaches, it has been shown that laser spectroscopy in the THz frequency domain offers new insights into hydration from small solutes to proteins. Upon introducing spatially-resolved analyses of the absorption cross section by simulations, the sensitivity of THz spectroscopy is traced back to characteristic distance-dependent modulations of absorption intensities for bulk water. The prominent peak at ≈200 cm-1 is dominated by first-shell dynamics, whereas a concerted motion involving the second solvation shell contributes most significantly to the absorption at about 80 cm-1 ≈2.4 THz. The latter can be understood in terms of an umbrella-like motion of two hydrogen-bonded tetrahedra along the connecting hydrogen bond axis. Thus, a modification of the hydrogen bond network, e.g., due to the presence of a solute, is expected to affect vibrational motion and THz absorption intensity at least on a length scale that corresponds to two layers of solvating water molecules. This result provides a molecular mechanism explaining the experimentally determined sensitivity of absorption changes in the THz domain in terms of distinct, solute-induced dynamical properties in solvation shells of (bio)molecules—even in the absence of well-defined resonances.
Keywords: ab initio molecular dynamics, hydrogen-bonding, solvation, infrared spectroscopy, vibrational modes
Water, often referred to as the “matrix of life” (1, 2), is a liquid that features many eccentricities (1, 3). As such water is the native solvent for biological processes. In recent years, however, the role of water has been revealed to be far more active than just being an inert solvent (2). Water molecules at protein/water interfaces, dubbed “hydration water” or “interfacial water,” have been shown not just to thermodynamically stabilize the native structure of biomacromolecules, but also to enhance their dynamical flexibility needed for efficient enzymatic catalysis and especially for the folding process that leads to the active structure of such molecules (4–7). Solvation was shown to be a key feature for protein function (8), with the coupling between protein and water dynamics (9) as an important ingredient to understand protein folding and binding. Hydration water has been in the focus of numerous theoretical and experimental studies (2), ranging from Raman and IR spectroscopy (10), femtosecond nonlinear spectroscopy (11), inelastic neutron scattering (12), femtosecond fluorescence spectroscopy (13), and molecular dynamics (MD) simulations (14, 15), to name but a few.
The intermolecular vibrational modes in the frequency window from 1 THz up to about 10 THz (16, 17), corresponding to ≈30–300 cm-1, have been proven recently to be a unique tool to study the hydration dynamics around solutes (18–21). Using THz laser spectroscopy, water dynamics has been shown to be influenced within an “extended dynamical hydration shell” (20) of about 6–7 Å for small solutes like carbohydrates (18) by changes in the absolute absorption intensity as probed at 2.4 THz (i.e., ≈80 cm-1). For proteins (20), this shell extends even up to ≈20 Å and is affected by e.g., folding state (21). The long-ranged sensitivity of THz absorption to dynamical properties of water at interfaces results from the presence of numerous processes in water occurring on picosecond time scales, e.g., hydrogen bond (HB) rearrangements and rotational relaxation. However, a full interpretation of the insights revealed by THz spectroscopy remains a daunting task.
Due to the importance of understanding the dynamics of solvation water it is thus mandatory to understand liquid water itself in the THz domain. The experimental far IR spectrum at THz frequencies (22, 23) features two pronounced maxima at approximately 200 and 650 cm-1. Roughly speaking the 200 cm-1 peak is related to hindered longitudinal translations of water molecules in the HB network (i.e., HB stretching vibrations) with a distinct collective character. The intermolecular vibrational modes belonging to frequencies above 300 cm-1 stem from librational (i.e., hindered rotational) motion (24, 25). In Raman spectra, additional resonances around 60 cm-1 are attributed to HB bending (24, 25) and contribute only weakly to the IR absorption. Based on this gross picture, the frequency range probed by the aforementioned THz experiments is therefore expected to be dominated in an intricate way by HB network motion.
In this study we have used MD simulations to address the nature of the underlying intermolecular motions. Unfortunately, the low-frequency absorption regime governed by intermolecular vibrations continues to be a challenge to force field based MD simulations (26–28). Although the IR spectrum of ambient water is qualitatively well described by force fields above ≈300 cm-1, the significant resonance at 200 cm-1 is either absent or emerges as a weak feature in such simulations (26–28). Polarizable force fields improve selected aspects of vibrational spectra, but also face difficulties to reproduce the prominent 200 cm-1 peak (27, 28). In contrast, pioneering ab initio MD (AIMD) simulations of the IR spectrum of heavy water (29) were able to reproduce the 200 cm-1 feature and assigned this to antisymmetric HB stretching modes. This discrepancy in the IR absorption cross section obtained from (polarizable) force fields versus AIMD studies has been explained (30, 31) in terms of intermolecular charge fluctuations in locally tetrahedral HB environments. These studies thus underline the collective character of low-frequency modes in conjunction with electronic polarization effects beyond single molecules (30, 31).
In the following, we introduce a consistent framework for a systematic decomposition of the IR absorption spectrum and intermolecular motion down to the THz regime in terms of correlations in time and space. In particular, analysis techniques are introduced which are able to spatially resolve the contributions of intermolecular motion and cross-correlation at given frequencies. These space-resolved methodologies in conjunction with an extensive set of AIMD trajectories of (light) liquid water allow us to assign the low-frequency modes at 2.4 THz to specific collective molecular motions that extend up to several Angstroms away from reference molecules.
Methodology: Spatial Decompositions of IR Spectra
The isotropic IR absorption coefficient per unit length is obtained as usual in linear response from
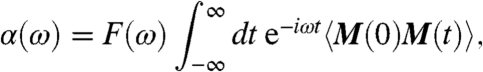 |
[1] |
which has been shown to work well for anharmonic HB systems (32). The prefactor reads F(ω) = (1/4πϵ0)(2πβω2/3Vcn(ω)) and M(t) is the total dipole moment of the sample of volume V with refractive index n(ω) at temperature T = 1/kBβ. The total dipole moment can be decomposed into a sum of molecular dipole moments,  , obtained from the centers of maximally localized Wannier functions (see Sec. 7.2 in ref. 33). This approach allows for the decomposition of M(0)M(t),
, obtained from the centers of maximally localized Wannier functions (see Sec. 7.2 in ref. 33). This approach allows for the decomposition of M(0)M(t),
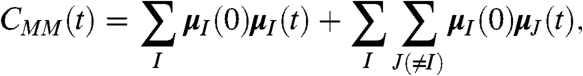 |
[2] |
in terms of self- and distinct-terms, i.e., auto- and cross-correlations of the molecular dipoles, respectively, and its generalization used to separate solute/solvent contributions (34, 35).
At variance with this established approach, we define here a smooth local dipole density
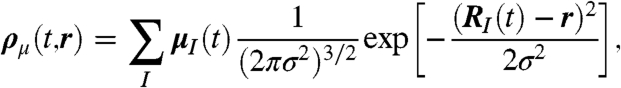 |
[3] |
where RI(t) is the Ith center-of-mass and σ is a suitable regularization parameter. The total dipole moment, M(t) = ∫Vdrρμ(t,r), can thus be decomposed smoothly in space, allowing one to introduce a spatially resolved total dipole autocorrelation function
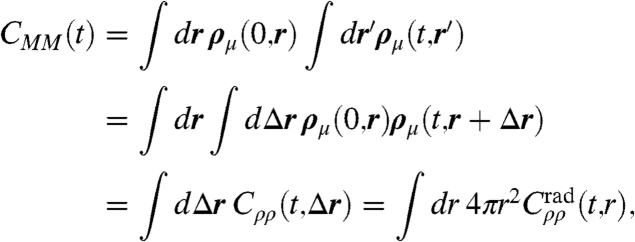 |
[4] |
which is done numerically upon introducing a spatial grid; here r′ = r + Δr is used, and in the last step isotropy has been imposed. Based on this transformation, a spatially-resolved IR absorption coefficient α(ω,r) can be introduced
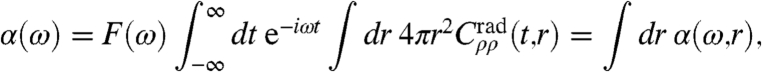 |
[5] |
which allows one to disentangle the spectra systematically at each frequency ω in terms of radially dependent contributions, r∈[0,rmax]; this continuous decomposition is similar to the discrete scheme introduced earlier (31) for analyzing the intermolecular contribution to dipole correlations only. In particular, absorption due to the self-term contributes exclusively for r → 0, whereas delocalized, collective modes will manifest themselves for r≫0 in terms of positive or negative α(ω,r) values at a fixed frequency ω, corresponding to correlated and anticorrelated motion, respectively. Furthermore, one obtains full spatial resolution α3D(ω,r,θ,ϕ) when employing Cρρ(t,Δr) in Eq. 5 in conjunction with a molecule-centered, moving coordinate system.
Extending this approach, the vibrational density of states I(ω) can be analyzed in space by introducing a local density ρv(t,r) of the mass-weighted velocities  of atoms at positions ri(t) analogous to Eq. 3 as
of atoms at positions ri(t) analogous to Eq. 3 as 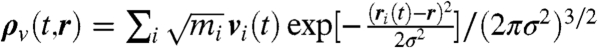 . Radially and spatially resolved functions I(ω,r) and I3D(ω,r,θ,ϕ), respectively, are defined in a similar way as introduced for the IR spectrum but without the prefactor F(ω). At variance to the IR spectrum approach, the commonly known vibrational density of states contributes only to I(ω,r ≈ 0) and I3D(ω,r ≈ 0,θ,ϕ) because it does not include cross-correlations of particle velocities.
. Radially and spatially resolved functions I(ω,r) and I3D(ω,r,θ,ϕ), respectively, are defined in a similar way as introduced for the IR spectrum but without the prefactor F(ω). At variance to the IR spectrum approach, the commonly known vibrational density of states contributes only to I(ω,r ≈ 0) and I3D(ω,r ≈ 0,θ,ϕ) because it does not include cross-correlations of particle velocities.
A complementary decomposition scheme is established by the idea of auto-correlating the dipole moment within a finite region,
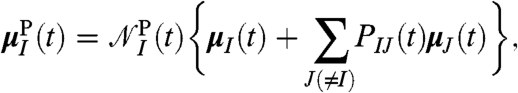 |
[6] |
around a (moving) molecule I serving as reference.
The considered region is thus a sphere, properly embedded in the bulk environment, that contains all J-molecules. Using Fermi’s cutoff function, PIJ(t) = 1/{1 + exp[(RIJ(t) - R0)/D]}, allows for a continuous selection of the set of “neighboring particles” {J}, both in time and space, with RIJ(t) = |RI(t) - RJ(t)| being the intermolecular center-of-mass distance at time t; R0 determines the cutoff radius, D measures the sharpness. The prefactor 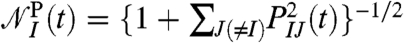 normalizes with respect to the (fractional) number of contributing J-molecules. Based on this projection, a distance-dependent absorption coefficient is defined
normalizes with respect to the (fractional) number of contributing J-molecules. Based on this projection, a distance-dependent absorption coefficient is defined
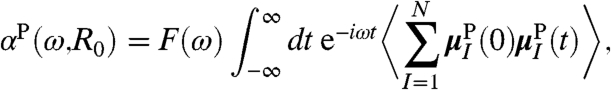 |
[7] |
after summing over all equivalent reference molecules {I}.
It is stressed that 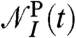 is constructed as to properly take into account the growing number of contributing molecules upon increasing R0. The meaning of the IR spectrum αP(ω,R0) is most easily revealed by considering two limiting cases. In the “bulk limit” all molecules are effectively taken into account, i.e., PIJ(t) = 1 in Eq. 6, which is obtained in the analysis by increasing R0 far beyond the average nearest-neighbor distance,
is constructed as to properly take into account the growing number of contributing molecules upon increasing R0. The meaning of the IR spectrum αP(ω,R0) is most easily revealed by considering two limiting cases. In the “bulk limit” all molecules are effectively taken into account, i.e., PIJ(t) = 1 in Eq. 6, which is obtained in the analysis by increasing R0 far beyond the average nearest-neighbor distance, 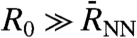 , such that
, such that 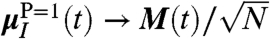 in Eq. 7 and one finds
in Eq. 7 and one finds 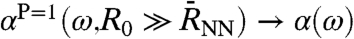 as defined in Eq. 1. In contrast, the “single molecule limit,” PIJ(t) = δIJ obtained via
as defined in Eq. 1. In contrast, the “single molecule limit,” PIJ(t) = δIJ obtained via 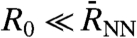 , yields
, yields 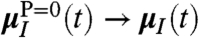 in Eq. 7, i.e.,
in Eq. 7, i.e., 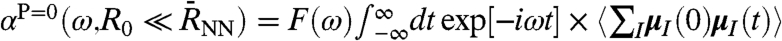 , and thus only the self-term of Eq. 2 contributes. Most interesting is obviously the intermediate regime, i.e., from
, and thus only the self-term of Eq. 2 contributes. Most interesting is obviously the intermediate regime, i.e., from 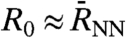 up to several intermolecular distances
up to several intermolecular distances 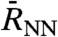 , where αP(ω,R0) quantifies the distinct responses of successive hydration shells. Thus, monitoring changes of αP(ω,R0) upon increasing R0 from small to intermediate distances might reveal how the solvation environment induces characteristic modulations of the IR absorbance around the reference molecule, starting from the individual polarized molecule via interfacial water towards bulk water at preselected frequencies ω.
, where αP(ω,R0) quantifies the distinct responses of successive hydration shells. Thus, monitoring changes of αP(ω,R0) upon increasing R0 from small to intermediate distances might reveal how the solvation environment induces characteristic modulations of the IR absorbance around the reference molecule, starting from the individual polarized molecule via interfacial water towards bulk water at preselected frequencies ω.
A further approach draws on the phenomenon that on average, any water molecule finds itself close to four neighbors in a roughly tetrahedral arrangement subject to fluctuations and defects (3, 29–31, 36, 37). This suggests to define four symmetry-adapted internal symmetric and antisymmetric stretching (ss, as) coordinates
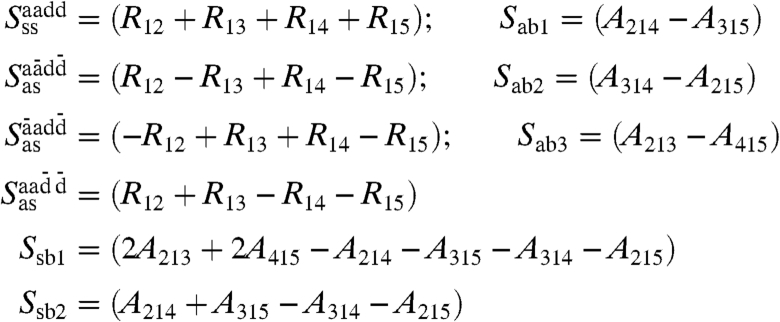 |
[8] |
as well as five such bending coordinates (sb, ab). Here RIJ is the center-of-mass distance between I and nearest-neighbor J whereas AJIK denotes the angle of J and K relative to the center-of-mass of the tetrahedron around I. Furthermore, in order to account for possible influences due to accepted and donated HBs the convention is introduced that waters 2 and 3 accept (a) a HB from 1 whereas 4, 5 donate (d) a HB to 1 according to the patterns encoded in the superscripts; bars denote a counterphase stretching of the corresponding HB.
In the same spirit, the second shell can be included after extending this tetrahedron by attaching a second tetrahedron via a HB to one of the corners of the former, i.e., with the additional water molecules 6, 7, and 8 being hydrogen bonded to 2. Using the introduced labeling the following intermolecular tetrahedron modes
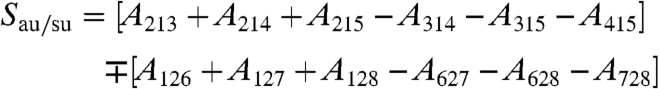 |
[9] |
are constructed which antisymmetrically/symmetrically correlate the umbrella motion (au/su) of the two tetrahedra with respect to the interconnecting HB axis. The corresponding spectral densities of these modes, 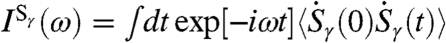 , are obtained as usual from the mode velocities,
, are obtained as usual from the mode velocities, 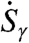 . Note that these procedures are related in spirit to, but clearly different from, earlier analysis techniques (29–31).
. Note that these procedures are related in spirit to, but clearly different from, earlier analysis techniques (29–31).
Results and Discussion
Spatial Unfolding of Computed IR Spectra.
In Fig. 1 we compare the IR spectrum computed for (light) liquid water to experimental data, 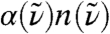 , as a function of frequency in wave numbers
, as a function of frequency in wave numbers 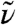 . To emphasize the statistical quality of the presented results we show the raw spectrum obtained from AIMD without applying any additional smoothing or signal extraction algorithm. As expected, the overall agreement is promising on the frequency and intensity scales set by the full spectrum (upper inset), apart from the well known red-shift (30) of the intramolecular OH-stretching band at high frequencies. More importantly, the difference between measured and computed H2O peak maximum of the 200 cm-1 resonance and the subsequent minimum both amount to ≈40 cm-1 (main) so that close accord is observed also at low frequencies where intermolecular motion determines the spectrum. In order to carefully assess the impact of quantum effects on liquid water in the THz frequency regime the experimental H2O spectrum is compared to that of pure heavy water (main). The differences are rather small and mainly sharpen up the peak around 200 cm-1 for H2O. In addition, we explore possible influences of anomalous quantum behavior of isotopic mixtures of liquid water (38) by precise measurements of the change in THz absorption
. To emphasize the statistical quality of the presented results we show the raw spectrum obtained from AIMD without applying any additional smoothing or signal extraction algorithm. As expected, the overall agreement is promising on the frequency and intensity scales set by the full spectrum (upper inset), apart from the well known red-shift (30) of the intramolecular OH-stretching band at high frequencies. More importantly, the difference between measured and computed H2O peak maximum of the 200 cm-1 resonance and the subsequent minimum both amount to ≈40 cm-1 (main) so that close accord is observed also at low frequencies where intermolecular motion determines the spectrum. In order to carefully assess the impact of quantum effects on liquid water in the THz frequency regime the experimental H2O spectrum is compared to that of pure heavy water (main). The differences are rather small and mainly sharpen up the peak around 200 cm-1 for H2O. In addition, we explore possible influences of anomalous quantum behavior of isotopic mixtures of liquid water (38) by precise measurements of the change in THz absorption 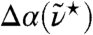 of H/D isotopic mixtures as a function of D2O concentration at a fixed frequency interval centered at
of H/D isotopic mixtures as a function of D2O concentration at a fixed frequency interval centered at 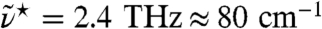 (lower inset). Within our experimental accuracy the recorded absorption changes turn out to be linear with increasing D2O mole fraction. Altogether, these experiments validate the basic approximation of using classical mechanics in conjunction with Eq. 1 in order to simulate the dynamics of liquid water for the purpose of the analyses to follow.
(lower inset). Within our experimental accuracy the recorded absorption changes turn out to be linear with increasing D2O mole fraction. Altogether, these experiments validate the basic approximation of using classical mechanics in conjunction with Eq. 1 in order to simulate the dynamics of liquid water for the purpose of the analyses to follow.
Fig. 1.

Absolute experimental THz absorption spectra, 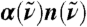 , for H2O (blue) and D2O (green) at 20 °C from Fourier transform spectroscopy compared to the AIMD H2O spectrum (red) obtained from Eq. 1; thick red line shows smoothened AIMD data to guide the eye. In the upper inset the full AIMD IR spectrum,
, for H2O (blue) and D2O (green) at 20 °C from Fourier transform spectroscopy compared to the AIMD H2O spectrum (red) obtained from Eq. 1; thick red line shows smoothened AIMD data to guide the eye. In the upper inset the full AIMD IR spectrum, 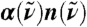 , is compared to the standard experimental H2O data (23). For mixtures of light and heavy water with increasing mole fraction of heavy water, x(D2O), the change in absorbance with respect to pure H2O,
, is compared to the standard experimental H2O data (23). For mixtures of light and heavy water with increasing mole fraction of heavy water, x(D2O), the change in absorbance with respect to pure H2O, 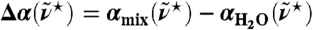 , is shown in the lower inset at 20 °C; The difference of the integrated THz absorption coefficient between 2.1 and 2.8 THz (centered at 2.4 THz) was measured as a function of D2O fraction (see Methods).
, is shown in the lower inset at 20 °C; The difference of the integrated THz absorption coefficient between 2.1 and 2.8 THz (centered at 2.4 THz) was measured as a function of D2O fraction (see Methods). 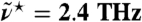 ≈80 cm-1.
≈80 cm-1.
Secondly, the radially resolved spatial decomposition of 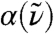 , i.e.,
, i.e., 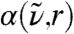 as introduced in Eq. 5, reveals pronounced distance-dependent correlations and anticorrelations as a function of frequency
as introduced in Eq. 5, reveals pronounced distance-dependent correlations and anticorrelations as a function of frequency 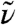 according to Fig. 2A. Notably, and in accord with Eq. 5, the total IR absorption spectrum as shown in Fig. 1 is restored from Fig. 2A upon integration along the correlation distance r. In the decomposed spectrum the positive auto-correlations seen for about r < 1 Å are found to dominate the IR spectrum at all frequencies, whereas distinct frequency-dependent cross-correlations are observed beyond. Motions of the nearest-neighbors up to r ≈ 3.5 Å around a given water molecule at r = 0 Å induce dipole cross-correlations within the first solvation shell in qualitative accord with earlier observations (31). Note that the computed center-of-mass radial distribution function gcom(r) has its first intermolecular maximum and minimum at r ≈ 2.7 Å and ≈3.3 Å, respectively, and a second maximum at ≈4.4 Å. For the OH-stretching band around 3,200 cm-1 a strong correlation peak is observed in the IR spectrum with a maximum close to ≈3 Å, followed by a faint anticorrelation around 4.5 Å. Joint analysis of the corresponding vibrational correlations in (B) can reveal its nature: the radially-resolved IR spectrum in (A) includes the electronic contributions to cross-correlations via the (fully polarizable) water dipoles obtained from Wannier analysis of the electronic degrees of freedom, whereas the radially-resolved vibrational density of states in Fig. 2B is exclusively sensitive to the underlying motion of the atom positions. This one-to-one comparison discloses that at 3,200 cm-1 there is almost no correlated particle motion for separations exceeding about r ≈ 1 Å, whereas pronounced dipole correlations do exist in the first shell followed by weak anticorrelations in the second shell. Notably, the positive (red) and negative (blue) dipole correlations observed in Fig. 2A in the first shell, r ≈ 3 Å, at the intramolecular vibrations, i.e., at ≈3,200 and 1,650 cm-1 respectively, vanish upon replacing the full electronic structure as embodied in the Wannier representation by classical point-charge electrostatics. This finding clearly supports that the observed dipole correlations in the first solvation shell are largely imprinted by correlated electronic polarization effects and not due to correlated particle motion at intramolecular frequencies. This result not only supports earlier findings (39, 40) concerning the OH-stretching IR band (where this mode, however, is termed delocalized or collective) but also provides the space-resolved decay of both the underlying dipole and particle correlations.
according to Fig. 2A. Notably, and in accord with Eq. 5, the total IR absorption spectrum as shown in Fig. 1 is restored from Fig. 2A upon integration along the correlation distance r. In the decomposed spectrum the positive auto-correlations seen for about r < 1 Å are found to dominate the IR spectrum at all frequencies, whereas distinct frequency-dependent cross-correlations are observed beyond. Motions of the nearest-neighbors up to r ≈ 3.5 Å around a given water molecule at r = 0 Å induce dipole cross-correlations within the first solvation shell in qualitative accord with earlier observations (31). Note that the computed center-of-mass radial distribution function gcom(r) has its first intermolecular maximum and minimum at r ≈ 2.7 Å and ≈3.3 Å, respectively, and a second maximum at ≈4.4 Å. For the OH-stretching band around 3,200 cm-1 a strong correlation peak is observed in the IR spectrum with a maximum close to ≈3 Å, followed by a faint anticorrelation around 4.5 Å. Joint analysis of the corresponding vibrational correlations in (B) can reveal its nature: the radially-resolved IR spectrum in (A) includes the electronic contributions to cross-correlations via the (fully polarizable) water dipoles obtained from Wannier analysis of the electronic degrees of freedom, whereas the radially-resolved vibrational density of states in Fig. 2B is exclusively sensitive to the underlying motion of the atom positions. This one-to-one comparison discloses that at 3,200 cm-1 there is almost no correlated particle motion for separations exceeding about r ≈ 1 Å, whereas pronounced dipole correlations do exist in the first shell followed by weak anticorrelations in the second shell. Notably, the positive (red) and negative (blue) dipole correlations observed in Fig. 2A in the first shell, r ≈ 3 Å, at the intramolecular vibrations, i.e., at ≈3,200 and 1,650 cm-1 respectively, vanish upon replacing the full electronic structure as embodied in the Wannier representation by classical point-charge electrostatics. This finding clearly supports that the observed dipole correlations in the first solvation shell are largely imprinted by correlated electronic polarization effects and not due to correlated particle motion at intramolecular frequencies. This result not only supports earlier findings (39, 40) concerning the OH-stretching IR band (where this mode, however, is termed delocalized or collective) but also provides the space-resolved decay of both the underlying dipole and particle correlations.
Fig. 2.

(A) Top: Radially-resolved IR absorption spectrum, 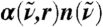 , of liquid H2O from AIMD obtained from Eq. 5. (B) Bottom: Corresponding radially-resolved vibrational correlations,
, of liquid H2O from AIMD obtained from Eq. 5. (B) Bottom: Corresponding radially-resolved vibrational correlations, 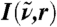 , see text.
, see text.
This picture changes drastically at lower frequencies, i.e., in the domain where intermolecular motion dictates the dynamics and thus the spectra. Below about 1,000 cm-1 the very broad IR bands in Fig. 2A feature significant negative cross-correlations in the first solvation shell and possibly also in the second shell below 100 cm-1. Even more pronounced is the spatial extent of concerted particle motions that contribute to the vibrational cross-correlations, i.e., for r > 1 Å, which can be both strongly positive and negative on the scale of several Angstroms into the bulk according to Fig. 2B. This result not only indicates a qualitative change of mechanism but clearly quantifies the collective nature and delocalized character of these low-frequency modes, which are shown here to involve systematic correlations of particle motion beyond the first solvation shell. This scenario is distinctly different from the purely intramolecular vibrational motion revealed in Fig. 2B for frequencies above 1,000 cm-1, where the longer-ranged correlations clearly detected in the radially-resolved IR spectrum in Fig. 2A are induced by coupled fluctuations of the electrons at neighboring water molecules.
Contributions of Solvation Shells to IR Absorption.
In order to understand this qualitatively different behavior in the intramolecular vs. intermolecular frequency windows, the distance-dependent IR absorption coefficient, Eq. 7, is employed, where the dipole fluctuations of all molecules within a smooth sphere of a fixed but arbitrary radius R0 contribute to the IR signal. This analysis therefore not only includes the self-correlations of all water dipoles around a reference molecule up to R0 but all their cross-correlations as well. This complements the results shown in Fig. 2A, where dipole auto-correlations (r → 0) are separated from the cross-correlations (r≫0). The integral 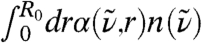 of the data shown in Fig. 2A may be compared to
of the data shown in Fig. 2A may be compared to 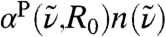 , however it must be noted that in the limiting case
, however it must be noted that in the limiting case 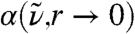 describes the local dipole density spectrum of an average point in space, while
describes the local dipole density spectrum of an average point in space, while 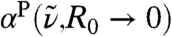 describes the proper single molecule limit. Since both quantities yield the same total spectrum
describes the proper single molecule limit. Since both quantities yield the same total spectrum 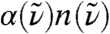 for
for 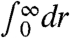 and as R0 → ∞, respectively, the approach to this limit must be different. Both spectral decompositions therefore provide rather independent information and can only be compared with care.
and as R0 → ∞, respectively, the approach to this limit must be different. Both spectral decompositions therefore provide rather independent information and can only be compared with care.
The IR spectra 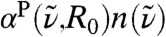 feature a strong and nonmonotonic intensity modulation as a function of R0, see inset of Fig. 3, which is found to exceed the correlations within the first solvation shell depending on frequency. In particular, the IR peak around
feature a strong and nonmonotonic intensity modulation as a function of R0, see inset of Fig. 3, which is found to exceed the correlations within the first solvation shell depending on frequency. In particular, the IR peak around 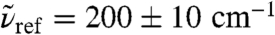 displays a steep maximum at R0 ≈ 2.7 Å corresponding to the first solvation shell according to the center-of-mass radial distribution function, gcom(r), followed by an only weakly modulated and mild increase beyond, approaching the bulk value for large R0 as shown in the main plot of Fig. 3.
displays a steep maximum at R0 ≈ 2.7 Å corresponding to the first solvation shell according to the center-of-mass radial distribution function, gcom(r), followed by an only weakly modulated and mild increase beyond, approaching the bulk value for large R0 as shown in the main plot of Fig. 3.
Fig. 3.

Relative intensities from distance-decomposed IR absorption spectra, 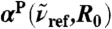 of liquid H2O from AIMD obtained from Eq. 7, as a function of R0 at selected reference frequencies
of liquid H2O from AIMD obtained from Eq. 7, as a function of R0 at selected reference frequencies 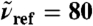 (blue, dots), 200 (green), and 1,630 cm-1 (red) integrated within a window of
(blue, dots), 200 (green), and 1,630 cm-1 (red) integrated within a window of 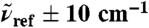 . The intensities are shown relative to the distance-decomposed absorption intensity at R0 = 7.5 Å, i.e.,
. The intensities are shown relative to the distance-decomposed absorption intensity at R0 = 7.5 Å, i.e., 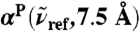 , where n(80 cm-1)αP(80 cm-1,7.5 Å) = 1,335 cm-1, n(200 cm-1)αP(200 cm-1,7.5 Å) = 2,654 cm-1, n(1,630 cm-1)αP(1,630 cm-1,7.5 Å) = 4,124 cm-1. Vertical arrows mark the successive maxima of the center-of-mass radial distribution function, gcom(r), stemming from contributions due to first, second, and third solvation shell. Horizontal dashed lines mark the usual integrated intensities obtained in the bulk, i.e.,
, where n(80 cm-1)αP(80 cm-1,7.5 Å) = 1,335 cm-1, n(200 cm-1)αP(200 cm-1,7.5 Å) = 2,654 cm-1, n(1,630 cm-1)αP(1,630 cm-1,7.5 Å) = 4,124 cm-1. Vertical arrows mark the successive maxima of the center-of-mass radial distribution function, gcom(r), stemming from contributions due to first, second, and third solvation shell. Horizontal dashed lines mark the usual integrated intensities obtained in the bulk, i.e., 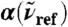 obtained from Eq. 1 again relative to
obtained from Eq. 1 again relative to 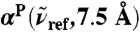 , corresponding to
, corresponding to 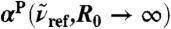 for infinitely large systems. Inset: Overview
for infinitely large systems. Inset: Overview 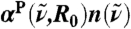 spectra as a function of
spectra as a function of 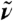 at selected distances R0: R0 → 0 (single molecule limit, orange), R0 = 2.7 Å (cyan), 4.1 Å (magenta), and R0 → ∞ (bulk limit, black). The vertical bars indicate the frequency windows used in the main figure with the same color code.
at selected distances R0: R0 → 0 (single molecule limit, orange), R0 = 2.7 Å (cyan), 4.1 Å (magenta), and R0 → ∞ (bulk limit, black). The vertical bars indicate the frequency windows used in the main figure with the same color code.
The second frequency window, 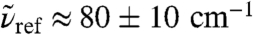 , is selected based on the results of previous studies in which the absorption at 2.4 THz ≈ 80 cm-1 revealed long-range effects in solvation dynamics of various molecules using THz laser spectroscopy (18, 20). Most interestingly, the distance-resolved IR spectra,
, is selected based on the results of previous studies in which the absorption at 2.4 THz ≈ 80 cm-1 revealed long-range effects in solvation dynamics of various molecules using THz laser spectroscopy (18, 20). Most interestingly, the distance-resolved IR spectra, 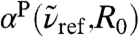 , uncover a strong and long-ranged spatial modulation at
, uncover a strong and long-ranged spatial modulation at 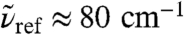 where no peak can be discerned in the bulk absorption spectrum,
where no peak can be discerned in the bulk absorption spectrum, 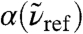 , see Fig. 1. In stark contrast to the 200 cm-1 resonance, the distance-dependent absorption at about 80 cm-1 is characterized by a shoulder at the first shell whereas the most pronounced absolute absorption is observed close to the second solvation shell as heralded by a significant and broad maximum at about R0 ≈ 4.1 Å. One can even detect the onset of another modulation of
, see Fig. 1. In stark contrast to the 200 cm-1 resonance, the distance-dependent absorption at about 80 cm-1 is characterized by a shoulder at the first shell whereas the most pronounced absolute absorption is observed close to the second solvation shell as heralded by a significant and broad maximum at about R0 ≈ 4.1 Å. One can even detect the onset of another modulation of 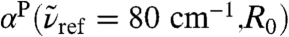 around R0 ≈ 6.7 Å which coincides with the third solvation shell according to the third maximum of the gcom(r). The intensity ratio between the maximum value found in the second shell and the bulk value, i.e.,
around R0 ≈ 6.7 Å which coincides with the third solvation shell according to the third maximum of the gcom(r). The intensity ratio between the maximum value found in the second shell and the bulk value, i.e., 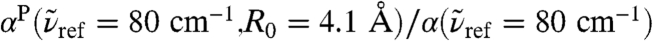 , features a clear increase in the interfacial region with respect to the bulk limit. Although this result cannot be compared directly for obvious reasons, it is noted that a similar absorbance increase has been measured by THz laser spectroscopy at 2.4 THz ≈ 80 cm-1 around solvated molecules (18, 20) and ascribed to an “extended dynamical solvation shell” (20). Overall, the long-ranged modulation of the distance-dependence in the THz frequency domain, stemming from intermolecular modes, is distinctly different from the essentially monotonous behavior as observed for intramolecular modes in the IR regime, as exemplified for the bending mode at ≈1,630 cm-1. This confirms our statement that the THz regime requires new concepts and cannot be described by those known from the infrared.
, features a clear increase in the interfacial region with respect to the bulk limit. Although this result cannot be compared directly for obvious reasons, it is noted that a similar absorbance increase has been measured by THz laser spectroscopy at 2.4 THz ≈ 80 cm-1 around solvated molecules (18, 20) and ascribed to an “extended dynamical solvation shell” (20). Overall, the long-ranged modulation of the distance-dependence in the THz frequency domain, stemming from intermolecular modes, is distinctly different from the essentially monotonous behavior as observed for intramolecular modes in the IR regime, as exemplified for the bending mode at ≈1,630 cm-1. This confirms our statement that the THz regime requires new concepts and cannot be described by those known from the infrared.
Characterizing Intermolecular Modes in the THz Domain.
Having explicitly demonstrated that the 200 cm-1 resonance is dominated by particle motion within the first solvation shell, the vibrational density of states corresponding to the tetrahedral Sγ coordinates as defined in Eq. 8 are used to analyze this band in Fig. 4. As expected, based on a multitude of previous analyses, this band is dominated by stretching motions of HBs. Here, we characterize these motions systematically in terms of symmetric stretching Sss at the lowest frequency ≈160 cm-1, antisymmetric stretching 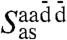 due to counterphase stretching motion of the two donor vs. acceptor HBs at the highest frequency ≈290 cm-1, and two degenerate antisymmetric stretches
due to counterphase stretching motion of the two donor vs. acceptor HBs at the highest frequency ≈290 cm-1, and two degenerate antisymmetric stretches  and
and 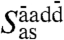 at intermediate frequency around 220 cm-1 which involve counterphase donor and acceptor motion; the two modes at ≈220 cm-1 are estimated to contribute most to the IR activity followed by the lower-lying symmetric stretch while the highest antisymmetric mode at 290 cm-1 appears to be essentially silent. Thus, the distinct HB donor/acceptor topologies accessible in first solvation shells due to HB fluctuations occurring on the picosecond time scale imprint their signature on the frequencies of vibrational motion involving the first nearest-neighbors and contribute to the broadness of the measured 200 cm-1 resonance in view of their different average frequencies. In addition to the four stretches, the remaining five modes are due to HB bending motion. They all peak significantly between 75–80 cm-1 but they also feature extended tails deep into the 200 cm-1 resonance; permuting the donor/acceptor ordering results in similar spectra. In conjunction with the previous distance-dependent analysis of the IR absorption coefficient, see Fig. 3, we can associate the stretching modes to be responsible for the pronounced maximum at ≈2.7 Å of the 200 cm-1 peak and the bending modes for the first-shell shoulder detected at
at intermediate frequency around 220 cm-1 which involve counterphase donor and acceptor motion; the two modes at ≈220 cm-1 are estimated to contribute most to the IR activity followed by the lower-lying symmetric stretch while the highest antisymmetric mode at 290 cm-1 appears to be essentially silent. Thus, the distinct HB donor/acceptor topologies accessible in first solvation shells due to HB fluctuations occurring on the picosecond time scale imprint their signature on the frequencies of vibrational motion involving the first nearest-neighbors and contribute to the broadness of the measured 200 cm-1 resonance in view of their different average frequencies. In addition to the four stretches, the remaining five modes are due to HB bending motion. They all peak significantly between 75–80 cm-1 but they also feature extended tails deep into the 200 cm-1 resonance; permuting the donor/acceptor ordering results in similar spectra. In conjunction with the previous distance-dependent analysis of the IR absorption coefficient, see Fig. 3, we can associate the stretching modes to be responsible for the pronounced maximum at ≈2.7 Å of the 200 cm-1 peak and the bending modes for the first-shell shoulder detected at 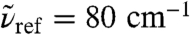 .
.
Fig. 4.

Power spectra 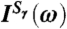 of the nine tetrahedral HB stretching and bending modes and of the antisymmetric umbrella mode as defined in the text; scheme and inset define labeling in Eqs. 8, 9.
of the nine tetrahedral HB stretching and bending modes and of the antisymmetric umbrella mode as defined in the text; scheme and inset define labeling in Eqs. 8, 9.
Still, the major contribution around 80 cm-1 must come from motion involving the second solvation shell according to the 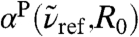 analysis. The antisymmetric umbrella mode, Sau, which involves two HB tetrahedra interconnected via a HB as defined in Eq. 9 and sketched in Fig. 4, accounts for second-shell motion and features a pronounced resonance around 80 cm-1 in the power spectrum. Note that the symmetric umbrella motion, Ssu, leads to a similar resonance, which is however broader with a tail towards higher frequencies due to more pronounced couplings to stretching-like deformations. Cross-correlating the Sau mode with its local dipole demonstrates that its integrated contribution to the total IR activity is indeed very weak, which is consistent with the lack of any resonance around 80 cm-1 in the integrated IR spectrum, see Fig. 1. Combining these findings, we assign the long-range distance-dependent modulation of the IR intensity at 80 cm-1 ≈ 2.4 THz in Fig. 3 to be dominated by relative motion of two hydrogen-bonded water tetrahedra. This finding might explain the rich modulation of absorption intensities uncovered by THz laser spectroscopy of solvated molecules (18, 20) at this particular frequency.
analysis. The antisymmetric umbrella mode, Sau, which involves two HB tetrahedra interconnected via a HB as defined in Eq. 9 and sketched in Fig. 4, accounts for second-shell motion and features a pronounced resonance around 80 cm-1 in the power spectrum. Note that the symmetric umbrella motion, Ssu, leads to a similar resonance, which is however broader with a tail towards higher frequencies due to more pronounced couplings to stretching-like deformations. Cross-correlating the Sau mode with its local dipole demonstrates that its integrated contribution to the total IR activity is indeed very weak, which is consistent with the lack of any resonance around 80 cm-1 in the integrated IR spectrum, see Fig. 1. Combining these findings, we assign the long-range distance-dependent modulation of the IR intensity at 80 cm-1 ≈ 2.4 THz in Fig. 3 to be dominated by relative motion of two hydrogen-bonded water tetrahedra. This finding might explain the rich modulation of absorption intensities uncovered by THz laser spectroscopy of solvated molecules (18, 20) at this particular frequency.
Summary, Conclusions, and Outlook
THz spectroscopy of aqueous solutions has been shown of late to provide a stimulating experimental perspective on the dynamics of interfacial water molecules, for instance hydration water around biomolecules. At variance with traditional IR spectroscopy, which draws on the strength of well defined intramolecular (normal or local) modes and thus harmonic analysis, e.g., in the fingerprint region, low-frequency bands in the THz domain are typically extremely broad and void of features due to rather complex intermolecular motion dominated by anharmonicity. Based on AIMD simulations of liquid water, H2O, we provide insights into the underlying nature of the absorption of bulk water at THz frequencies. Our results offer a firm microscopic understanding of the ability of THz spectroscopy to probe the collective dynamics of water molecules on a spatial scale spanning several solvation shells. The study also shows that the traditional IR spectra at frequencies above 1,000 cm-1 and THz spectra are governed by distinctly different mechanisms. While the underlying vibrations of the former show no significant correlation in particle motions beyond the first solvation shell, but a pronounced coupling of electronic origin, strongly correlated particle motion dominates at THz frequencies. This peculiar behavior can point towards the significance of solvent dynamics in the THz regime for the influence on large amplitude fluctuations of proteins.
Space-resolved analyses of spectra and vibrational motion confirm at high frequencies pronounced correlations mainly within the first solvation shell that are dominated by intramolecular motion and induced electronic polarization of close-by neighbors. In stark contrast, the correlation length is shown to extend beyond nearest-neighbors in the THz domain, thus involving the second and even third solvation shell. Decomposition of particle motion in terms of symmetry-adapted coordinates inspired by a tetrahedral coordination model of water shows that the four HB stretching modes contribute to the resonance around 200 cm-1 at three distinct frequencies depending on the particular HB donor/acceptor pattern. The remaining five tetrahedral HB bending modes all yield pronounced maxima at ≈80 cm-1 whereas their tails extend deep into the 200 cm-1 peak.
The introduced distance-dependent absorption cross section is found to be characterized by strong intensity modulations that depend crucially on the probe frequency. Indeed, the strongest contribution to the 200 cm-1 peak is shown to come from the first solvation shell in agreement with the assignment to intermolecular HB stretching modes. At variance, the main contribution to the intensity around 80 cm-1 is shown to stem from motion within the second solvation shell; the data even indicate the presence of nonnegligible third shell effects. This dynamics is well captured by umbrella-like displacements of two HB tetrahedra with respect to their interconnecting HB axis, thus involving concerted motion of second-nearest-neighbors. Most importantly, although these modes around 80 cm-1 ≈ 2.4 THz neither lead to a peak nor to a significant shoulder in the IR spectrum and have therefore initially been underestimated in their importance, they do induce a very characteristic modulation of the absorption intensity in space, i.e., a spatial response. One is tempted to speculate that this distinct modulation could be the mechanism that allows THz spectroscopy to sensitively detect changes of the solvation behavior from small molecules to proteins undergoing folding, which is the topic of future research. The analysis approach established here for liquid water can be generalized to probe the hydration shell dynamics around solvated molecules including small biomolecules.
Methods
The AIMD simulations (33) have been carried out using the CP2k package with Born-Oppenheimer propagation generated via the Quickstep electronic structure module (41). The Perdew-Burke-Ernzerhof (PBE) functional has been employed together with dual-space norm-conserving pseudo potentials (42) in conjunction with plane waves up to 400 Rydberg and a TZV2P basis set (41) to expand density and orbitals, respectively. A sample of 128 H2O molecules in a cubic ≈15.6 Å supercell corresponding to 1 g/cm3 has been used. The system was equilibrated at ambient conditions using massive Nosé-Hoover chain thermostatting (33) at a rescaled temperature of 400 K to approximately counterbalance the systematic underestimation (43) of temperature by about 20%–30% when using PBE. Sixteen statistically independent initial conditions were then sampled from a 90 ps canonical simulation to initialize microcanonical trajectories of 20 ps length using a time step of 0.5 fs. Every 2 fs the maximally localized Wannier functions have been computed (see Sec. 7.2.4 in ref. 33) and molecular dipole moments μI(t) (assigned to the center-of-mass RI(t)) were constructed using the Wannier centers (see Sec. 7.2.6 in ref. 33). To obtain spatially-resolved IR absorption spectra and vibrational density of states, the local dipole and velocity densities according to Eq. 3 have been computed on a cubic grid with a spacing of 0.489 Å using a regularization parameter σ of 0.4 Å. The corresponding data shown in Fig. 2 have been smoothened in the frequency domain via convolution with a Gaussian filter with  . The sharpness parameter was set to D = 0.25 Å in order to compute the distance-dependent absorption according to Eq. 7. All data shown have been averaged over all NVE trajectories thus yielding canonical averages.
. The sharpness parameter was set to D = 0.25 Å in order to compute the distance-dependent absorption according to Eq. 7. All data shown have been averaged over all NVE trajectories thus yielding canonical averages.
The THz laser spectroscopy experiments have been performed as described earlier (20), whereas the Fourier transform far IR measurements have been carried out using the Bruker VERTEX 80v FTIR spectrometer with diamond window sample cells and a sample thickness of 44 μm. The data evaluation has been performed as described in detail in ref. 44; the refractive indices  of H2O and D2O are tabulated in ref. 22. For each H2O/D2O concentration, three independent samples were prepared followed by three independent measurements each at a constant temperature of (20 ± 1) °C.
of H2O and D2O are tabulated in ref. 22. For each H2O/D2O concentration, three independent samples were prepared followed by three independent measurements each at a constant temperature of (20 ± 1) °C.
Acknowledgments.
We thank David Bosquet (ENS Paris) for helping with preliminary analyses at an early stage of the project. Diedrich Schmidt and Gerhard Schwaab are thanked for valuable discussions and comments. We acknowledge financial support from the Studienstiftung des Deutschen Volkes (Fellowship to M. Heyden), Alexander von Humboldt Stiftung (Fellowship to J.S.), Grant BMBF 05KS7PC2 (to M. Havenith), Volkswagenstiftung (to M. Havenith), as well as Research Department Interfacial Systems Chemistry and Fonds der Chemischen Industrie (to M. Havenith and D.M.). The calculations were carried out at HLRB II München, Bovilab @ RUB, and Rechnerverbund-NRW.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
References
- 1.Franks F. Water: a matrix of life. 2nd Ed. Cambridge: Royal Society of Chemistry; 2000. [Google Scholar]
- 2.Ball P. Water as an active constituent in cell biology. Chem Rev. 2008;108:74–108. doi: 10.1021/cr068037a. [DOI] [PubMed] [Google Scholar]
- 3.Stillinger FH. Water revisited. Science. 1980;209:451–457. doi: 10.1126/science.209.4455.451. [DOI] [PubMed] [Google Scholar]
- 4.Cheung MS, Garcia AE, Onuchic JN. Protein folding mediated by solvation: water expulsion and formation of the hydrophobic core occur after the structural collapse. Proc Natl Acad Sci USA. 2002;99:685–690. doi: 10.1073/pnas.022387699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rhee YM, Sorin EJ, Jayachandran G, Lindahl E, Pande VS. Simulations of the role of water in the protein folding mechanism. Proc Natl Acad Sci USA. 2004;101:6456–6461. doi: 10.1073/pnas.0307898101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Levy Y, Onuchic JN. Water mediation in protein folding and molecular recognition. Annu Rev Biophys Biomol Struct. 2006;35:389–415. doi: 10.1146/annurev.biophys.35.040405.102134. [DOI] [PubMed] [Google Scholar]
- 7.Chaplin M. Do we underestimate the importance of water in cell biology? Nat Rev Mol Cell Biol. 2006;7:861–866. doi: 10.1038/nrm2021. [DOI] [PubMed] [Google Scholar]
- 8.Pal SK, Zewail AH. Dynamics of water in biological recognition. Chem Rev. 2004;104:2099–2123. doi: 10.1021/cr020689l. [DOI] [PubMed] [Google Scholar]
- 9.Fenimore PW, Frauenfelder H, McMahon BH, Parak FG. Slaving: Solvent fluctuations dominate protein dynamics and functions. Proc Natl Acad Sci USA. 2002;99:16047–16051. doi: 10.1073/pnas.212637899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smith JD, Saykally RJ, Geissler PL. The effects of dissolved halide anions on hydrogen bonding in liquid water. J Am Chem Soc. 2007;129:13847–13856. doi: 10.1021/ja071933z. [DOI] [PubMed] [Google Scholar]
- 11.Kropman MF, Bakker HJ. Dynamics of water molecules in aqueous solvation shells. Science. 2001;291:2118–2120. doi: 10.1126/science.1058190. [DOI] [PubMed] [Google Scholar]
- 12.Wood K, et al. Coupling of protein and hydration-water dynamics in biological membranes. Proc Natl Acad Sci USA. 2007;104:18049–18054. doi: 10.1073/pnas.0706566104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pal SK, Peon J, Bagchi B, Zewail AH. Biological water: femtosecond dynamics of macromolecular hydration. J Phys Chem B. 2002;106:12376–12395. [Google Scholar]
- 14.Laage D, Hynes JT. Reorientional dynamics of water molecules in anionic hydration shells. Proc Natl Am Soc USA. 2007;104:1167–11172. doi: 10.1073/pnas.0701699104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tarek M, Tobias DJ. Single-particle and collective dynamics of protein hydration water: a molecular dynamics study. Phys Rev Lett. 2002;89:275501. doi: 10.1103/PhysRevLett.89.275501. [DOI] [PubMed] [Google Scholar]
- 16.Schmuttenmaer CA. Exploring dynamics in the far-infrared with terahertz spectroscopy. Chem Rev. 2004;104:1759–1779. doi: 10.1021/cr020685g. [DOI] [PubMed] [Google Scholar]
- 17.Plusquellic DF, Siegrist K, Heilweil EJ, Esenturk O. Applications of terahertz spectroscopy in biosystems. ChemPhysChem. 2007;8:2412–2431. doi: 10.1002/cphc.200700332. [DOI] [PubMed] [Google Scholar]
- 18.Heugen U, et al. Solute-induced retardation of water dynamics probed directly by terahertz spectroscopy. Proc Natl Acad Sci USA. 2006;103:12301–12306. doi: 10.1073/pnas.0604897103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang C, Durbin SM. Hydration-induced far-infrared absorption increase in myoglobin. J Phys Chem B. 2006;110:23607–23613. doi: 10.1021/jp063545+. [DOI] [PubMed] [Google Scholar]
- 20.Ebbinghaus S, et al. An extended dynamical hydration shell around proteins. Proc Natl Acad Sci USA. 2007;104:20749–20752. doi: 10.1073/pnas.0709207104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim SJ, Born B, Havenith M, Gruebele M. Real-time detection of protein-water dynamics upon protein folding by terahertz absorption spectroscopy. Angew Chem Int Ed. 2008;47:6486–6489. doi: 10.1002/anie.200802281. [DOI] [PubMed] [Google Scholar]
- 22.Zelsmann HR. Temperature dependence of the optical constants for liquid H2O and D2O in the far IR region. J Mol Struct. 1995;350:95–114. [Google Scholar]
- 23.Bertie JE, Lan Z. Infrared intensities of liquids: The intensity of the OH stretching band of liquid water revisited, and the best current values of the optical constants of H2O(l) at 25 °C between 15,000 and 1 cm-1. Appl Spectrosc. 1996;50:1047–1057. [Google Scholar]
- 24.Walrafen GE. Raman spectrum of water: Transverse and longitudinal acoustic modes below ≈300 cm-1 and optic modes above ≈300 cm-1. J Phys Chem. 1990;94:2237–2239. [Google Scholar]
- 25.Ohmine I, Saito S. Water dynamics: Fluctuation, relaxation, and chemical reactions in hydrogen bond network rearrangement. Acc Chem Res. 1999;32:741–749. [Google Scholar]
- 26.Madden PA, Impey RW. On the infrared and Raman spectra of water in the region 5–250 cm-1. Chem Phys Lett. 1986;123:502–506. [Google Scholar]
- 27.Harder E, Eaves JD, Tokmakoff A, Berne BJ. Polarizable molecules in the vibrational spectroscopy of water. Proc Natl Acad Sci USA. 2005;102:11611–11616. doi: 10.1073/pnas.0505206102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fanourgakis GS, Xantheas S. Development of transferable interaction potentials for water. V. Extension of the flexible, polarizable, Thole-type model potential (TTM3-F, v. 3.0) to describe the vibrational spectra of water clusters and liquid water. J Chem Phys. 2008;128:074506. doi: 10.1063/1.2837299. [DOI] [PubMed] [Google Scholar]
- 29.Silvestrelli PL, Bernasconi M, Parrinello M. Ab initio infrared spectrum of liquid water. Chem Phys Lett. 1997;277:478–482. [Google Scholar]
- 30.Sharma M, Resta R, Car R. Intermolecular dynamical charge fluctuations in water: A signature of the H-bond network. Phys Rev Lett. 2005;95:187401. doi: 10.1103/PhysRevLett.95.187401. Erratum: Phys Rev Lett 100:019901 (2008) [DOI] [PubMed] [Google Scholar]
- 31.Chen W, Sharma M, Resta R, Galli G, Car R. Role of dipolar correlations in the infrared spectra of water and ice. Phys Rev B. 2008;77:245114. [Google Scholar]
- 32.Ramírez R, López-Ciudad T, Kumar P, Marx D. Quantum corrections to classical time-correlation functions: Hydrogen bonding and anharmonic floppy modes. J Chem Phys. 2004;121:3973–3983. doi: 10.1063/1.1774986. [DOI] [PubMed] [Google Scholar]
- 33.Marx D, Hutter J. Ab initio molecular dynamics: basic theory and advanced methods. Cambridge: Cambridge Univ Press; 2009. [Google Scholar]
- 34.Gaigeot MP, Sprik M. Ab initio molecular dynamics computation of the infrared spectrum of aqueous uracil. J Phys Chem B. 2003;107:10344–10358. [Google Scholar]
- 35.Iftimie R, Tuckerman ME. Decomposing total IR spectra of aqueous systems into solute and solvent contributions: A computational approach using maximally localized Wannier orbitals. J Chem Phys. 2005;122:214508. doi: 10.1063/1.1908950. [DOI] [PubMed] [Google Scholar]
- 36.Marx D. Throwing tetrahedral dice. Science. 2004;303:634–636. doi: 10.1126/science.1094001. [DOI] [PubMed] [Google Scholar]
- 37.Marx D. Proton transfer 200 years after von Grotthuss: Insights from ab initio simulations. ChemPhysChem. 2006;7:1848–1870. doi: 10.1002/cphc.200600128. Addendum: ChemPhysChem 8, 209–210 (2007) [DOI] [PubMed] [Google Scholar]
- 38.Chatzidimitriou-Dreismann CA, Krieger UK, Möller A, Stern M. Evidence of quantum correlation effects of protons and deuterons in Raman spectra of liquid H2O-D2O. Phys Rev Lett. 1995;75:3008–3011. doi: 10.1103/PhysRevLett.75.3008. Torii H. Phys Rev Lett 84:5236 (2000). Reply: Chatzidimitriou-Dreismann CA, Abdul-Redah T, Kolaric B, Juranic I. Phys Rev Lett 84:5237 (2000) [DOI] [PubMed] [Google Scholar]
- 39.Torii H. Time-domain calculations of the polarized Raman spectra, the transient infrared absorption anisotropy, and the extent of delocalization of the OH stretching mode of liquid water. J Phys Chem A. 2006;110:9469–9477. doi: 10.1021/jp062033s. [DOI] [PubMed] [Google Scholar]
- 40.Auer BM, Skinner JL. IR and Raman spectra of liquid water: Theory and interpretation. J Chem Phys. 2008;128:224511. doi: 10.1063/1.2925258. [DOI] [PubMed] [Google Scholar]
- 41.VandeVondele J, et al. Quickstep: Fast and accurate density functional calculations using a mixed Gaussian and plane waves approach. Comput Phys Commun. 2005;167:103–128. the CP2k Developers Group, http://cp2k.berlios.de/ [Google Scholar]
- 42.Goedecker S, Teter M, Hutter J. Separable dual-space Gaussian pseudopotentials. Phys Rev B. 1996;54:1703–1710. doi: 10.1103/physrevb.54.1703. [DOI] [PubMed] [Google Scholar]
- 43.Schwegler E, Grossman JC, Gygi F, Galli G. Towards an assessment of the accuracy of density functional theory for first principles simulations of water. II. J Chem Phys. 2004;121:5400–5409. doi: 10.1063/1.1782074. [DOI] [PubMed] [Google Scholar]
- 44.Schmidt DA, et al. Rattling in the cage: Ions as probes of sub-picosecond water network dynamics. J Am Chem Soc. 2010;131:18512–18517. doi: 10.1021/ja9083545. [DOI] [PubMed] [Google Scholar]