

Abstract
Several neurodegenerative diseases are typified by intraneuronal α-synuclein deposits, synaptic dysfunction, and dementia. While even modest α-synuclein elevations can be pathologic, the precise cascade of events induced by excessive α-synuclein and eventually culminating in synaptotoxicity is unclear. To elucidate this, we developed a quantitative model system to evaluate evolving α-synuclein-induced pathologic events with high spatial and temporal resolution, using cultured neurons from brains of transgenic mice overexpressing fluorescent-human-α-synuclein. Transgenic α-synuclein was pathologically altered over time and overexpressing neurons showed striking neurotransmitter release deficits and enlarged synaptic vesicles; a phenotype reminiscent of previous animal models lacking critical presynaptic proteins. Indeed, several endogenous presynaptic proteins involved in exocytosis and endocytosis were undetectable in a subset of transgenic boutons (“vacant synapses”) with diminished levels in the remainder, suggesting that such diminutions were triggering the overall synaptic pathology. Similar synaptic protein alterations were also retrospectively seen in human pathologic brains, highlighting potential relevance to human disease. Collectively the data suggest a previously unknown cascade of events where pathologic α-synuclein leads to a loss of a number of critical presynaptic proteins, thereby inducing functional synaptic deficits.
Introduction
Pathologic intraneuronal deposits of α-synuclein are seen in diverse neurodegenerative diseases, including Parkinson's disease (PD), dementia with Lewy bodies (DLB), and a subset of Alzheimer's disease (AD), and are often accompanied by dementia (Mukaetova-Ladinska and McKeith, 2006). Under physiologic conditions, α-synuclein localizes to presynaptic terminals, and in pathologic states α-synuclein accumulations are seen in cell bodies, neuronal processes, and synapses (Roy, 2009). Duplications/triplications of the wild-type (WT) human-α-synuclein (h-α-syn) gene are seen in familial forms of PD/DLB (Singleton et al., 2003) (for review, see Cookson and van der Brug, 2008), where a mere twofold increase in WT protein levels leads to profound clinical and pathologic phenotypes (Farrer et al., 2004; Fuchs et al., 2008; Ikeuchi et al., 2008). These data underscore that not only is excessive h-α-syn pathologic, but that even quite modest protein elevations are deleterious to neurons. Synaptic dysfunction and associated neurodegeneration/dementia are established components of α-synuclein-induced pathology as well (McKeith and Mosimann, 2004; Aarsland et al., 2008); collectively indicating that excessive h-α-syn induces a chain of pathologic events that eventually culminate in synaptic dysfunction and dementia.
Though it is evident that excessive α-synuclein has an overall deleterious role in human disease, the precise cascade of early pathologic events resulting from elevations of h-α-syn and culminating in eventual synaptic dysfunction and dementia is unclear (Cookson and van der Brug, 2008). A prevailing hypothesis is that excessive h-α-syn leads to deficits in vesicular transport/trafficking. The initial clues supporting this notion emerged from studies in yeast model systems overexpressing h-α-syn tagged to green fluorescent protein (GFP), which implicated several genes involved in vesicular trafficking pathways (Outeiro and Lindquist, 2003; Gitler et al., 2008; Soper et al., 2008). Subsequent studies in pheochromocytoma (PC12) and chromaffin cell lines overexpressing h-α-syn support the hypothesis, showing that α-synuclein overexpression leads to deficits in vesicular mobilization and release from the cytoplasm of mammalian secretory cells (Larsen et al., 2006).
Though these studies provide important clues into the consequences of h-α-syn overexpression, neuronal correlates of such trafficking defects and their relevance to the synaptic dysfunction seen in human diseases have been difficult to construe. A recent study in cultured neurons overexpressing h-α-syn elegantly demonstrated deficiencies in the presynaptic apparatus (Nemani et al., 2010); however, the pathologic events leading up to these deficits and the relevance of such deficits to human disease are still unclear. Other studies have related losses of endogenous mouse α-synuclein to deficits in vesicular trafficking/exocytosis (Abeliovich et al., 2000; Chandra et al., 2004, 2005); however, the precise pathologic events induced by α-synuclein overexpression within neurons and their relevance to human disease have not been systematically explored.
A comprehension of the sequence of pathologic events induced by excessive h-α-syn is clearly critical to our understanding of the mechanistic basis of these diseases. Here we illustrate a multifaceted approach that we took to address this issue; combining contemporary quantitative cell biology with electrophysiology, ultrastructural studies, and neuropathology. Our studies suggest a surprising cascade of pathologic events that may underlie the h-α-syn-induced synaptic dysfunction seen in these diseases.
Materials and Methods
Cell cultures from transgenic mice.
The PDGF-h-α-syn:GFP mice (C57/B6 background) used in this study have been described previously (Rockenstein et al., 2005). Hippocampal neurons were obtained from brains of heterozygous postnatal (P0–P2) α-synuclein:GFP transgenic pups. Pups were screened using a “GFP flashlight” (Nightsea) that made the GFP+ pups glow. Nontransgenic littermates were used as controls. For all cell biology experiments, dissociated cells were plated at a density of 100,000 cells/cm2 in poly-d-lysine-coated glass-bottom culture dishes (Mattek) and maintained in Neurobasal/B27 media (Invitrogen) supplemented with 0.5 mm glutamine. All animal studies were performed in accordance with University of California guidelines. Immunofluorescence studies were performed as previously described (Roy et al., 2008). Briefly, cultured neurons were fixed with paraformaldehyde/120 mm sucrose, rinsed several times, and stained with the appropriate antibodies. Alexa 488, 594, and 647 dyes (Invitrogen) were used as secondary antibodies.
Antibodies.
Endogenous mouse VAMP, Piccolo, synapsin, and amphiphysin were detected using a mouse monoclonal anti-VAMP-2, a rabbit polyclonal anti-Piccolo, a rabbit polyclonal antibody to amphiphysin (all from Synaptic Systems), and a rabbit polyclonal antibody to synapsin-1 (Invitrogen). Total (mouse + human) synuclein was detected using an in-house guinea-pig α-synuclein antibody (GP-SYN) that was generated in Dr. Virginia Lee's laboratory (University of Pennsylvania, Philadelphia, PA). Human α-synuclein in tissue sections was detected using a rabbit polyclonal antibody (Millipore). Other antibodies used were a mouse monoclonal microtubule-associated protein 2 (MAP2) antibody (gift from Dr. Virginia Lee), a mouse monoclonal anti-PSD-95 antibody (Calbiochem), and the human-specific α-synuclein antibodies (LB509 and syn211, both from Abcam). All chemicals were from Sigma unless otherwise noted.
Microscopy and image analysis.
Images were acquired using an Olympus inverted motorized epifluorescence microscope equipped with a Z-controller (IX81, Olympus) and a motorized X–Y stage controller (Prior Scientific), attached to a ultrastable light source (Exfo exacte) and CCD cameras (Coolsnap HQ2, Photometrics). All images were acquired and processed with MetaMorph software (Molecular Devices). To capture the majority of synaptic profiles in a given field, Z-stack images were obtained using procedures similar to those used in previous studies of synaptic proteins in cultured neurons (Custer et al., 2006). Briefly, a z-series of images was collected at a resolution of 0.2 μm, deconvolved, and saved as a single projection. Subsequent processing for all images was as performed in three steps as described below, largely based on Krueger et al., 2003. (1) Background subtraction: Background fluorescence was calculated as the mean plus two times the SD of 10–20 three-pixel diameter regions of peri-boutonic/axonal fluorescence, and this value was subtracted from respective images. (2) Bouton-size criteria: Background-corrected puncta measuring <3 pixels (0.48 μm) in width and height were not included in the analysis. (3) Colocalization criteria: GFP+ve/α-synuclein+ve puncta were considered to be colocalized with other synaptic proteins if their fluorescence overlapped by >5 pixels in area. For each dataset, a sampling of this analysis was manually counted to confirm the integrity of analysis. Experimental groups were statistically analyzed using a nonparametric Student's t test and expressed as mean ± SEM; all experiments were repeated at least 2–3 times, on separate culture sets. A p value of <0.05 was considered significant.
Electrophysiology.
For these experiments, GFP+ or WT littermate neurons were plated at very low densities (1000 cells/cm2) with an astrocyte feeder layer suspended above them (Kaech and Banker, 2006) to maximize apposition of transgenic boutons to GFP+ somata/dendrites within a coverslip. Under these conditions, 52 ± 3% of the boutons apposed to h-α-syn-GFP+ primary and secondary dendrites were transgenic (10 neurons, 3 coverslips, 546 boutons analyzed). Whole-cell voltage-clamp recordings were performed with an Axopatch 200B amplifier from α-synuclein:GFP-expressing neurons and neurons from wild-type littermates; recordings were obtained after switching to DIC optics. Coverslips were placed into a perfusion chamber (Warner Instruments). The external recording solution was as follows (in mm): 155 NaCl, 3.5 KCl, 2 CaCl2, 1.5 MgSO4, 10 glucose, and 10 HEPES, pH 7.4, 320 mOsm. The pipette solution contained the following (in mm): 140 K-methanesulfonate, 5 KCl, 10 HEPES, 2 MgCl2, 0.5 EGTA, 2 ATP, and 1 GTP, pH 7.3. Miniature EPSCs (mEPSCs/“minis”) were recorded in the presence of 1 μm tetrodotoxin and 50 μm bicuculline at −60 mV holding potential. The solution was allowed to perfuse into cells during whole-cell recording for 3 min, before recording spontaneous postsynaptic currents for 10–20 min. Only cells with stable access resistance were included in the data analysis. The electrode resistance after back-filling was 3–5 MΩ. Recordings were digitized using a Digidata 1320A interface and the pClamp 9 (Molecular Devices) software package. Data for mEPSC analysis were sampled at a rate of 20 kHz and filtered at 3 kHz. Synaptic events were detected and analyzed (amplitude, kinetics, frequency) off-line using a peak detection program (Mini Analysis program, Synaptosoft). The mean frequency (number of events/duration) and amplitude of the synaptic events were computed after automatic detection of a series of at least 500 events from continuous recording stretches lasting 10–20 min.
Proteinase-K and FM4-64 experiments.
For the proteinase-K (PK) experiments, 21 d in vitro (DIV-21) hippocampal neurons were fixed with paraformaldehyde/120 mm sucrose for 10 min and extensively rinsed with PBS. A 1 μg/ml concentration of proteinase-K was added to the cultures for 2 h, after which the reaction was quenched by adding serum. Images were then processed for immunostaining as described above. All FM4-64 experiments/imaging were performed on living neurons maintained at ∼37°C inside a live-cell environment chamber (Precision Control) mounted on the microscope. Neurons were first rinsed in “live imaging media” containing Hibernate E and supplements (Roy et al., 2008) and the antagonists (10 μm CNQX and 50 μm dl-AP5, Tocris Biosciences). They were then transferred to stimulating media (5 mm HEPES, 31.5 mm NaCl, 1 mm MgCl2, 2 mm CaCl2, 30 mm glucose, and 90 mm KCl) containing 15 μm FM4-64 and antagonists for 2 min. This protocol is known to label mainly the rapidly recycling synaptic vesicles (Deák et al., 2004; Gaffield and Betz, 2006). Immediately thereafter, cells were washed for three times in the “live imaging media” with antagonists and incubated for two 10 min periods (with rinses) to remove excess dye on the cell surface. Living neurons were then imaged as described above. To analyze the temporal kinetics of FM release, transgenic or WT neurons were loaded with FM4-64 as described above and then incubated for an additional 2 min to allow maximal endocytosis (Deák et al., 2004). FM-loaded neurons were washed extensively and destained using the high K+ solution. The temporal release of the dye from boutons was monitored by live imaging and corrected for photobleaching. Decay curves from transgenic (green) and WT (red) boutons were calculated as mean ± SEM F/F 0 values for all boutons analyzed.
Electron microscopy and histology.
For ultrastructural analysis of the synapses expressing α-synuclein:GFP, we performed immuno-EM using a mouse monoclonal anti-GFP antibody (Millipore Bioscience Research Reagents) to unequivocally identify transgenic boutons, and compared them with WT boutons. For these experiments, cultured neurons were fixed with 2.5% glutaraldehyde and 2% paraformaldehyde (Electron Microscopy Sciences) at 4°C for 30 min and postfixed with 1% osmium tetroxide, 0.8% potassium ferrocyanide, and 3 μm calcium chloride in 0.1 m cacodylate buffer, pH 7.4, and embedded in epoxy. Cells were washed and immunostained with a primary antibody to GFP detected using 5 nm gold particles with silver enhancement (Electron Microscopy Sciences). Blocks were sectioned with an Ultracut E ultramicrotome (Leica) and analyzed with a Zeiss EM10 electron microscope (Carl Zeiss). Vesicle sizes were assayed by partitioning each synaptic bouton into four quadrants and then measuring the diameter of all vesicles within a quadrant. Average vesicle sizes were calculated as mean ± SEM for vesicles within each bouton. Average bouton density was calculated as the total number of vesicles within a bouton quadrant divided by the area analyzed. To determine the range in the vesicular sizes within synapses, ∼30 vesicles were randomly selected from each WT or transgenic bouton, and their diameters were measured. These data were then pooled and plotted to compare the variability in the sizes of boutons between the WT and transgenic groups. To analyze human brain tissue, prefrontal cortices of two controls (aged 77 and 85) and two LB dementia cases (aged 79 and 85) were obtained from the Alzheimer's Disease Research Center brain bank at University of California, San Diego. Free-floating vibratome sections were immunolabeled with human α-synuclein and synapsin antibodies as described previously (Rockenstein et al., 2005), and imaged/quantified as noted above.
Results
Model system to evaluate early pathology in α-synuclein-overexpressing neurons
To evaluate the evolution of pathology in a setting of modest α-synuclein overexpression, we used cultured hippocampal neurons from transgenic mice overexpressing h-α-syn tagged at the C terminus to enhanced-GFP (h-α-syn:GFP) (Rockenstein et al., 2005). The h-α-syn in this mouse model is driven by a platelet-derived growth factor (PDGF) promoter. Unlike some other mouse models, the α-synuclein expression is largely restricted to the limbic and cortical areas, and the overall expression levels are much more modest (Rockenstein et al., 2002, 2005). Thus the spatial and quantal distribution of α-synuclein in these mice may more reasonably approximate dementing α-synuclein-related illnesses like DLB. Behavioral testing of 6-month-old PDGF-h-α-syn:GFP mice revealed striking deficits of spatial memory in the Morris water maze test as shown in supplemental Figure 1 (available at www.jneurosci.org as supplemental material), further underlining the suitability of this mouse model in the investigation of α-synuclein-related dementias. Brain homogenates from these transgenic mice show a single band with the expected molecular weight of the h-α-syn:GFP fusion protein (supplemental Fig. 2A, available at www.jneurosci.org as supplemental material), and no proteolytic products were seen, unlike a previous report with transient transfections of C-terminally tagged α-synuclein (McLean et al., 2001). Our results are similar to data from other research groups showing that C terminus tagging of h-α-syn does not appear to alter the biophysical properties of the protein (Nakamura et al., 2008). Moreover, transgenic h-α-syn was robustly and unequivocally enriched at synapses (supplemental Fig. 2B, and see data below) as expected for a presynaptic protein.
The overall experimental strategy is shown in Figure 1 A. First we identified heterozygous GFP-positive pups by optical screening using a “GFP flashlight” (see Materials and Methods) and obtained cultured neurons from the hippocampi of transgenic animals or their WT littermates. The transgenic, presynaptically enriched α-synuclein was easily identifiable within the neurons by their native GFP fluorescence, allowing us to follow the transgenic neurons and rigorously explore the evolving pathology over time. Using these methods, we achieved modest and consistent levels of h-α-syn overexpression (see below) in large numbers of neurons and were able to analyze thousands of overexpressing boutons with relative ease (Fig. 1 B). In these heterozygous cultures, ∼40% of the boutons were GFP positive, and we designed quantitative algorithms to isolate the “green” boutons and analyze the progression of synaptic pathology specifically in these transgenic boutons, including their colocalization with other synaptic elements (see Materials and Methods). In our hands, this strategy was far more reliable than transient transfections (used in all other α-synuclein studies in cultured neurons to date), where cell-to-cell variations and low transfection efficiencies (typically <5%) precluded consistent and reliable inferences from large synapse populations.
Figure 1.

Model system using cultured hippocampal neurons from human-α-synuclein:GFP transgenic mice. A, To develop a model system for α-synuclein overexpression, heterozygous h-α-syn:GFP+ pups (P0–P2) were optically screened (see Materials and Methods) and dissociated hippocampal neurons were plated from these mice (left); neurons from WT littermates were used as controls for all experiments (right). For quantitative studies, GFP+ (α-synuclein-overexpressing) boutons were isolated and colocalization of presynaptic proteins/dyes within the GFP+ve boutons was analyzed (see Materials and Methods for specific details of quantization algorithm). B, Top left, Native GFP fluorescence in a 3-month-old h-α-syn:GFP mouse brain section. Robust GFP expression was seen in the forebrain and hippocampus (boxed), with lower fluorescence levels throughout the neocortex. Low-power scanning representative image of native GFP fluorescence from live DIV-21 hippocampal cultures are shown. Note that numerous h-α-syn-positive punctate, bouton-like varicosities can be readily visualized in these cultures, with diffuse fluorescence in cell-bodies (magnified in inset). These methods allowed us to obtain robust α-synuclein expression in large neuronal populations and examine the evolving pathology over time.
The GFP tag allowed us to precisely identify living h-α-syn-positive cells/boutons and perform functional studies on living neurons. As expected, the vast majority of the overexpressed h-α-syn localized to presynaptic boutons. Figure 2 A shows two representative fields with h-α-syn:GFP boutons apposed to MAP2, a somatodendritic marker, and the postsynaptic density marker PSD-95. Next, we quantitatively determined h-α-syn overexpression levels in our model system. We performed most of our studies at DIV-21, a time when these neurons are mature with well developed presynaptic boutons and dendritic spines. We found that the average α-synuclein fluorescence in transgenic boutons at DIV-21 was only ∼2.5 times higher than that of WT littermates (Fig. 2 B), providing a reasonable approximation of elevated α-synuclein levels in human disease. Importantly, overexpression of h-α-syn in this setting did not result in an overt loss of synapses (Fig. 2 C), allowing us to investigate early/evolving pathologic events, before any obvious synapse/cell loss.
Figure 2.
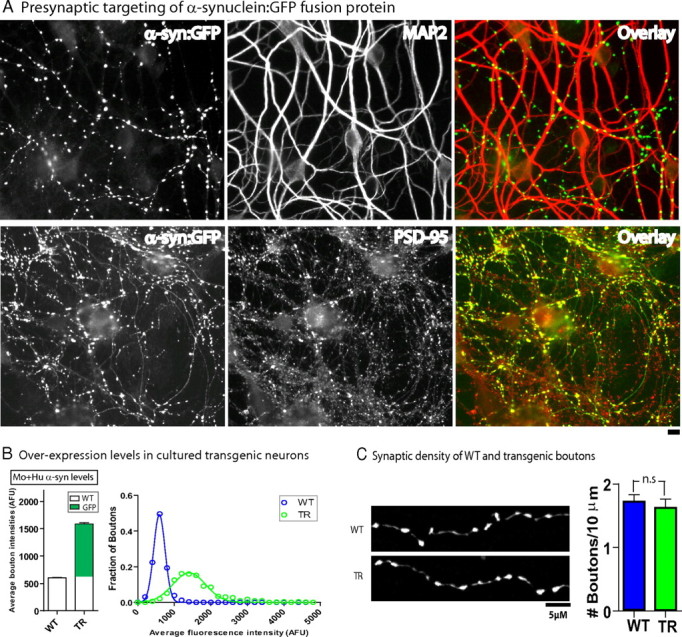
Modest overexpression of human α-synuclein at synapses with no synaptic loss in DIV-21 cultured neurons. A, Transgenic h-α-syn-overexpressing neurons were fixed and stained for a somatodendritic (MAP2) and postsynaptic (PSD-95) marker. Representative images with pseudocolor overlays show apposition of h-α-syn transgenic puncta to these two markers, indicating that the vast majority of visible puncta in these cultures represent presynaptic boutons. B, Transgenic and WT DIV-21 neurons were fixed and immunostained with a pan-synuclein antibody to reveal total (mouse + human) α-synuclein. Quantitative analyses of average α-synuclein fluorescence in WT and transgenic boutons shows ∼2.5 times overexpression; distribution of data is shown as a histogram on right. C, Synaptic density was calculated by counting the number of boutons/axon length. In WT cultures, soluble GFP was transiently transfected and used as an axon/bouton-filler. No differences in presynaptic densities were seen between WT and transgenic neurons (1.7 ± 0.1 and 1.6 ± 0.1 boutons/10 μm, respectively, mean ± SEM, p = 0.63, n = 200–300 boutons analyzed for each group).
Pathologic alterations of transgenic α-synuclein in overexpressing neurons
We next asked whether there were any pathologic modifications of the transgenic h-α-syn in DIV-21 neurons over time, as seen in human diseases. A well recognized pathologic alteration of α-synuclein in human pathologic brains and animal models is the accumulation of PK-resistant α-synuclein. In human DLB brains, PK-resistant α-synuclein has been shown to accumulate at presynaptic terminals (Neumann et al., 2002; Kramer and Schulz-Schaeffer, 2007), in addition to the more obvious somatic α-synuclein accumulations. Synaptic PK-resistant α-synuclein aggregates are also seen in animal α-synuclein overexpression models (Neumann et al., 2002; Periquet et al., 2007) and are thought to be pathologic correlates of synaptic dysfunction and dementia in DLB (Kramer and Schulz-Schaeffer, 2007).
To test whether PK-resistant α-synuclein accumulated in our transgenic synapses, hippocampal neurons from h-α-syn:GFP transgenic mice (or their WT littermates) were cultured for 3 weeks (DIV-21), treated with 1 μg/ml proteinase-K as previously described (Periquet et al., 2007) (see Materials and Methods for specifics), and stained with an antibody to total (mouse + human) α-synuclein (GP-SYN) as well as a human-specific α-synuclein antibody (LB509) to reveal transgenic boutons [note that it is not appropriate to use the GFP as a readout in these experiments as the fluorophore is digested by PK treatment (our unpublished observations)]. We first qualitatively compared total (mouse + human) α-synuclein levels in WT and transgenic neurons. We noticed that while the α-synuclein staining in WT neurons was uniformly diminished, focal areas within transgenic boutons retained high levels of α-synuclein as shown in the panels in Figure 3. This suggests that while the PK treatment was sufficient to digest the vast majority of the mouse α-synuclein, a subset of transgenic boutons (perhaps the highest overexpressors) were resistant to PK treatment. Indeed, quantification of human α-synuclein levels in transgenic boutons with/without PK extraction showed that a larger fraction of higher-α-synuclein-overexpressing boutons are present in the post-PK dataset; suggesting preferential retention of h-α-syn in those boutons.
Figure 3.
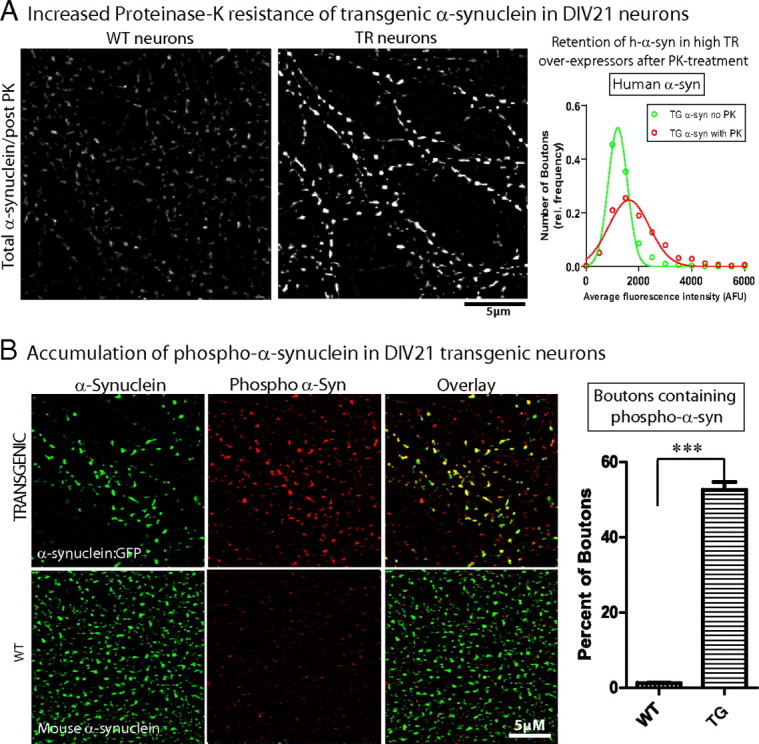
Pathologic alterations in DIV-21 human α-synuclein-overexpressing neurons. A, DIV-21 cultured hippocampal neurons from h-α-syn:GFP mice or their WT littermates were treated with 1 μg/ml PK as described in Materials and Methods and total (mouse + human) and human α-synuclein levels within boutons was analyzed. Focal retention of total α-synuclein (detected with GP-SYN) was seen in transgenic boutons after PK treatment as shown in the panels. Histograms of average human α-synuclein fluorescence (detected with antibody LB509) within transgenic boutons with and without PK treatment show that a larger fraction of high-α-synuclein-overexpressing boutons are present in the post-PK dataset, suggesting that boutons expressing higher h-α-syn levels are more resistant to PK treatment (average bouton intensity of human α-synuclein = 1317 ± 21.83 AFU and 1900 ± 52.20 AFU in non-PK- and PK-treated boutons, respectively, mean ± SEM; n = 700–1950 boutons). B, DIV-21 cultured hippocampal neurons from h-α-syn:GFP mice were stained with an antibody specific for the pathology-associated Ser-129 phosphorylation site of α-synuclein. While ∼53% of the transgenic boutons contained Ser-129 phosphorylated α-synuclein (top panels), negligible levels of phospho-α-synuclein was seen in WT boutons; data quantified on right (n = 500–750 boutons, ***p < 0.0001). Scale bars, lower right of images.
Next we asked whether the overexpressed h-α-syn in our DIV-21 neurons acquired disease-associated posttranslational modifications, namely phosphorylation at Ser-129 residue. Ser-129 phosphorylation is an established pathologic component of α-synuclein inclusions in human disease, as well as animal models of the disease (Fujiwara et al., 2002; Kahle et al., 2002; Chen and Feany, 2005; Anderson et al., 2006; Waxman and Giasson, 2008), and it is generally recognized that such phosphorylation contributes to the pathology in these diseases, though the exact role of such modifications is unclear. In these experiments, we stained DIV-21 h-α-syn:GFP-overexpressing boutons with an antibody that specifically recognizes α-synuclein when it is phosphorylated at the Ser-129 residue (pSer-129/81A antibody) (Waxman and Giasson, 2008) and compared them to boutons from WT littermates expressing endogenous mouse α-synuclein (Fig. 3 B). We found that while approximately half of the transgenic boutons also contained phospho-α-synuclein, it was virtually absent in WT boutons (Fig. 3 B, right). Collectively the proteinase-K resistance and the phospho-h-α-syn accumulation indicate that the transgenically expressed h-α-syn in DIV-21 neurons is pathologically altered.
Functional presynaptic deficits in α-synuclein-overexpressing boutons
Next we examined the functionality of h-α-syn-overexpressing boutons. To do this, we analyzed synaptic transmission in DIV-21 h-α-syn-overexpressing neurons using electrophysiology. Live GFP-positive neurons plated at low densities were identified (see Materials and Methods), and miniature excitatory currents (“minis”) were recorded from perikarya receiving inputs from h-α-syn-overexpressing boutons. Cultured neurons from WT littermates were used as controls. All analyses were performed in parallel transgenic and WT culture sets, to eliminate any possible culture-specific artifacts. Mini frequencies correspond to spontaneous glutamate release from presynaptic boutons and are a classical measure of neurotransmitter exocytosis. There was a striking decrease in the frequencies of the minis in h-α-syn-overexpressing neurons, as shown in the representative tracings (Fig. 4 A). There was no significant change in the amplitudes or the time constants of decay in these experiments (Fig. 4 B), suggesting that the functionality of the postsynaptic receptors is not significantly altered by h-α-syn overexpression. These data are quantified in Figure 4 C. In the absence of overt synapse loss (see Fig. 2 C), these data indicate that the overall synaptic release apparatus is operating at a lower efficiency in h-α-syn-overexpressing boutons.
Figure 4.

Diminished spontaneous synaptic responses in DIV-21 human α-synuclein-overexpressing neurons. DIV-21 transgenic neurons were identified by their native GFP fluorescence and electrophysiologic recordings were obtained from live neurons. For controls, WT neurons from parallel littermate cultures were analyzed. A, Representative tracings (“minis”) of spontaneous synaptic responses from hippocampal neurons overexpressing h-α-syn compared to controls. B, Individual tracings of 15 randomly selected minis from control neurons (left tracings) or h-α-syn-overexpressing neurons (middle tracings), with normalized and overlapped averages of the same events (right tracings). C, Quantitative analyses of spontaneous synaptic events. Frequencies of spontaneous events in WT and h-α-syn-overexpressing neurons were 5.20 ± 1.68 Hz and 1.15 ± 0.46 Hz, respectively, mean ± SEM, p < 0.02; n = 12–15 for each group, 500–2000 events/experiment. The amplitudes of synaptic events averaged 9.95 ± 0.8 pA and 11.47 ± 1.18 pA, and the tau decay was 6.34 ± 0.54 ms and 6.1 ± 0.68 ms in these WT and α synuclein-overexpressing neurons, respectively (mean ± SEM; *p > 0.5 for both parameters). These experiments were repeated on three separate occasions using neurons obtained from three separate litters (for each WT/transgenic group), and the data were pooled.
To further examine exocytosis and endocytosis in individual presynaptic boutons overexpressing α-synuclein, we used FM4-64—a red styryl dye that is selectively endocytosed into rapidly recycling synaptic vesicles and widely used to evaluate synaptic function. Upon brief stimulation in the presence of FM dyes, synapses rapidly endocytose the dye, which can be then released upon a second stimulation (Gaffield and Betz, 2006). Thus the endocytosis/exocytosis of the FM dye is a measure of the functional status of the synaptic apparatus. We first asked whether there was a subset of boutons in our transgenic cultures that were “presynaptically silent,” a possibility given our electrophysiology data. Such “silent synapses” should presumably fail to uptake the FM dye completely, as the recycling machinery would be inoperable. Indeed, we found that the FM dye was undetectable over background in ∼25% of the h-α-syn-overexpressing boutons, indicating that these boutons failed to endocytose the FM4-64, compared to WT boutons (Fig. 5 A, left). When we examined the transgenic boutons that did endocytose the FM dye, we found that the overall average fluorescence intensity of FM in these boutons was diminished by ∼35%. Next we examined the capacity of the FM+ve transgenic boutons to exocytose the FM dye. For these experiments, neurons were loaded with the FM dye, rinsed, and then unloaded by high K+ stimulation as described in Materials and Methods. As shown in Figure 5 B, the temporal kinetics of FM release is significantly slower in TR boutons compared to their WT littermates, further implicating a failure of the presynaptic exocytic machinery in these α-synuclein-overexpressing boutons.
Figure 5.
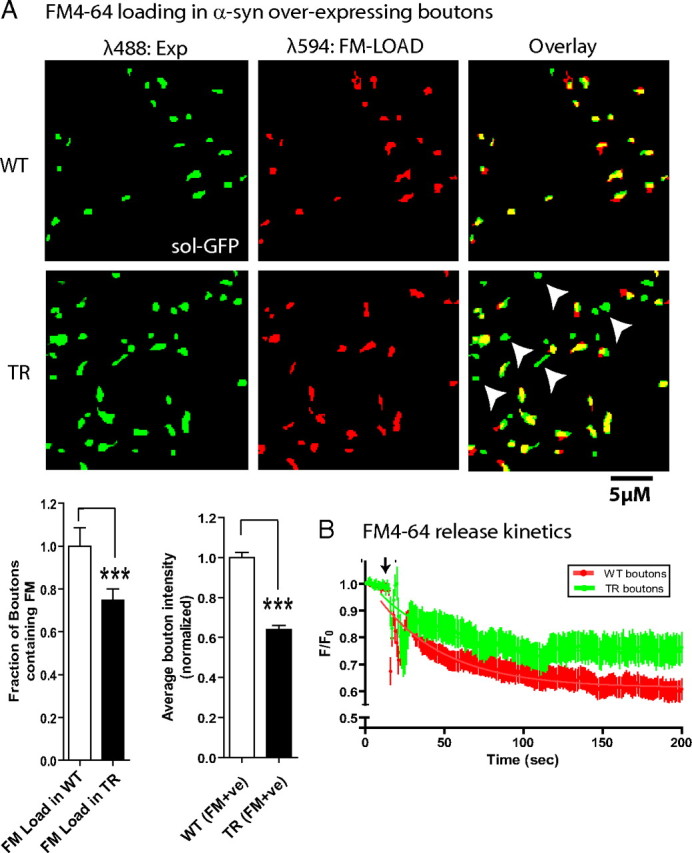
FM4-64 loading experiments. A, WT neurons transfected with soluble GFP, or h-α-syn:GFP-overexpressing neurons were loaded with 15 μm FM4-64 to label synapses, rinsed extensively, and imaged as described in Materials and Methods. First, the fraction of FM+ve boutons in the WT or transgenic group was analyzed. Left, GFP-positive boutons (green) and their corresponding FM images (red) along with the pseudocolor overlay. Note the presence of several GFP+/FM− boutons in the pseudocolor overlay from boutons overexpressing h-α-syn (some marked with arrowheads). Left graph, The fraction of transgenic boutons uptaking the FM dye relative to WT boutons was 0.748 ± 0.052 (mean ± SEM, p = 0.0115; n = 300–700 boutons), indicating that ∼25% of the boutons failed to endocytose any detectable FM dye. Right graph, Second, the average FM intensities in the boutons that did endocytose the FM dye (FM+ve) were analyzed. Average bouton intensity (in AFU) in transgenic boutons relative to WT boutons was 0.641 ± 0.021 (mean ± SEM, p < 0.0001; n = 300–500 boutons). B, Temporal kinetics of FM release. Transgenic or WT neurons were incubated with FM4-64 (see Materials and Methods), FM-loaded neurons were washed extensively, destained with high K+, and the temporal release of the dye (decay of fluorescence after stimulation) from boutons was monitored by live imaging. Decay curves from transgenic (green) and WT (red) boutons are shown; arrow marks the time point when the neurons were stimulated. F/F 0 represents the ratio of the bouton fluorescence at any given time to the initial fluorescence. The symbols and error bars are mean ± SEM, and the solid lines represent the best single-phase decay fit to the data. Note that there was a significant inhibition of FM exocytosis in the transgenic boutons, compared to boutons from their WT littermates (n = 25–40 boutons; p < 0.0001).
Ultrastructural abnormalities in α-synuclein-overexpressing boutons
To gain further insights into the morphologic changes induced by α-synuclein overexpression, we examined the ultrastructure of h-α-syn-overexpressing boutons in cultured neurons by electron microscopy (EM). To unequivocally identify the subset of overexpressing boutons in our α-synuclein transgenic (+/−) cultures (typically ∼40%), we performed immuno-EM using an anti-GFP antibody, and then compared immunopositive, transgenic synapses to WT littermate boutons. Control labeling in WT (GFP-negative) cultures was nonspecific (data not shown). Though synaptic vesicles were seen in both WT and transgenic boutons, we noticed striking variations in the sizes of the vesicles located within transgenic synapses. While control synapses were populated with small synaptic vesicles of relatively uniform diameters as expected, vesicles in transgenic boutons were highly variable in size and occasionally enormous, as shown in Figure 6 A (bottom right). Overall quantitative analyses showed that there was no change in the overall size of boutons, but there was a reduction in the density of vesicles at synapses (Fig. 6 A). Quantization of the size of vesicles within synapses showed an increase in the number of vesicular structures over 50 nm in transgenic synapses, with a corresponding decrease in vesicles that were <50 nm (Fig. 6 B, top). The overall variation in the sizes of vesicles at α-synuclein transgenic boutons, as well as the presence of enlarged vesicles, is also evident in vesicle-diameter scatter plots from transgenic and WT boutons (Fig. 6 B, bottom). Though we recognize that calling such enormous vesicles “synaptic” is questionable, we use this term to indicate their location within boutons (which is unequivocal), as well as to highlight this curious morphologic alteration in our α-synuclein-overexpressing boutons.
Figure 6.
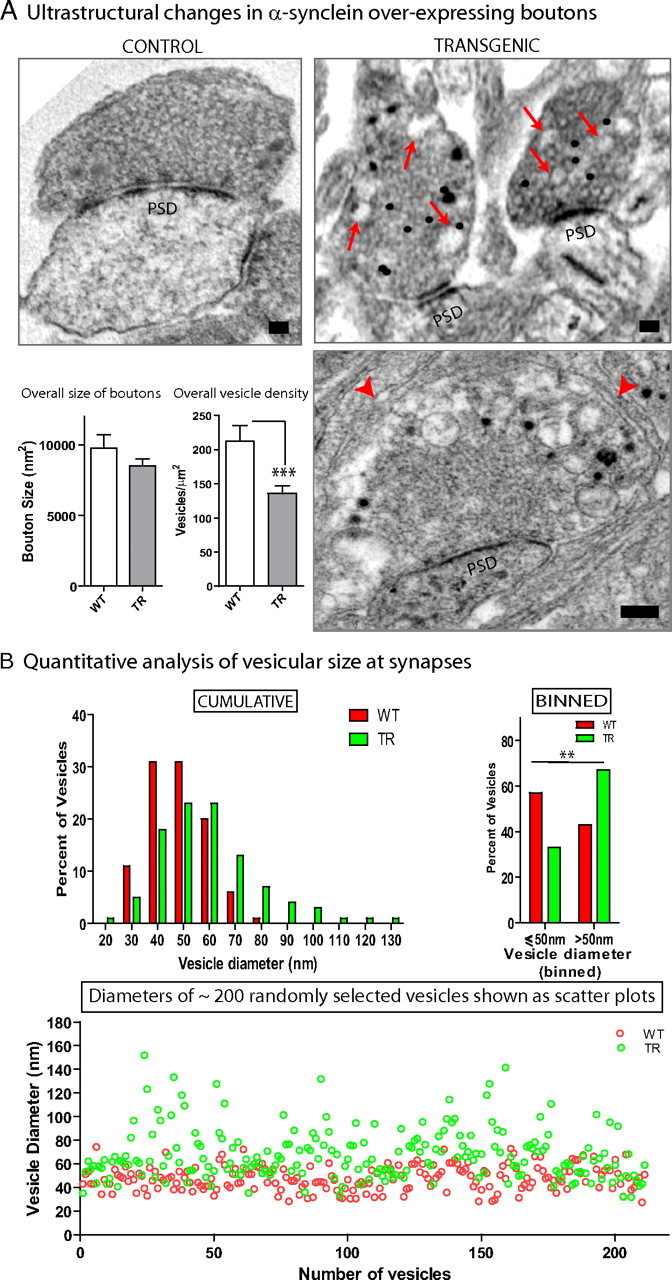
Ultrastructural alterations in DIV-21 human α-synuclein-overexpressing boutons. A, Boutons overexpressing h-α-syn:GFP were identified by immuno-EM using an antibody to GFP (immunogold signals appear as black dots) and compared to boutons from WT littermates. Top left, A typical control bouton with synaptic vesicles that are largely of uniform diameter (PSD: postsynaptic density). Top right, Two representative transgenic boutons with abnormal large vesicular profiles intermingled within synaptic vesicles of relatively normal diameters (red arrows). Bottom right, An extreme example of enlarged vesicles in transgenic synapses. Note the large vacuolar accumulations between the red arrowheads. Quantitative analyses showed that while the overall size of the boutons was unchanged, there was a significant reduction in vesicle densities within synapses (p = 0.0052; n = 5–14 boutons). Scale bar, 100 nm. B, Histograms of vesicle diameters (10 nm bins) from WT (red bars) and transgenic (green bars) highlight the variations in the overall distributions of vesicular sizes between the two vesicular populations. When the data were binned into two groups (<50 nm and >50 nm), a Fisher's exact test showed a significant increase in vesicles >50 nm in the transgenic group with a corresponding decrease in the other group (<50 nm group: 57% and 33% for WT and TR; >50 nm group: 43% and 67%, respectively; n = 200–400 vesicles). The variation in vesicular sizes within transgenic boutons is also readily visualized in the scatter plots (bottom), where vesicle diameters of ∼200 randomly selected vesicles from WT and transgenic boutons were plotted. Note that while most vesicles are ∼40–50 nm, abnormally large vesicles are far more common in the transgenic group.
Absence of endogenous mouse presynaptic proteins in a subset of α-synuclein-overexpressing boutons
The above data show that prolonged h-α-syn overexpression in neurons leads to defects in the synaptic vesicular release apparatus, as well as heterogeneity and enlargement in the size of vesicles at synapses. We were struck by the similarity of this constellation of synaptic alterations to previous animal models lacking synaptic vesicular proteins, including soluble NSF-attachment protein receptors (SNAREs) and active-zone components as well as endocytic proteins that are critical to the synaptic release machinery. Specifically, reduced spontaneous synaptic responses and deficits in neurotransmitter release have been demonstrated in mice (Schoch et al., 2001), Caenorhabditis elegans (Nonet et al., 1998), and Drosophila (Deitcher et al., 1998) deleted for a critical vesicular (v-SNARE) protein—synaptobrevin/vesicle-associated membrane protein (VAMP or its homologues). Pharmacologic disruption of SNAREs in other studies also led to deficits in neurotransmitter release (Neale et al., 1999; Verderio et al., 1999), and similar alterations have been reported in animal models lacking active-zone proteins as well (Schoch and Gundelfinger, 2006). Neurons lacking other major presynaptic proteins like synapsin also show defects in neurotransmitter release (Gitler et al., 2004). Intriguingly, EM studies of cultured hippocampal neurons from mice lacking VAMP-2 and dynamin, as well as neurons from flies and worms lacking synaptotagmin and clathrin (various synaptic proteins involved in recycling/endocytosis), all show heterogeneity and increases in the size of synaptic vesicles (Zhang et al., 1998; Nonet et al., 1999; Poskanzer et al., 2006; Ferguson et al., 2007), somewhat similar to our results.
Thus we wondered whether the aggregate of synaptic deficits in our h-α-syn-overexpressing neurons were simply a consequence of a depletion of critical mouse presynaptic proteins at h-α-syn-overexpressing synapses. Accordingly, we quantitatively evaluated levels of the following endogenous mouse presynaptic proteins in h-α-syn-overexpressing boutons: a vesicular-SNARE protein (VAMP-2), an active-zone protein (Piccolo), a versatile synaptic vesicle-associated protein thought to play a role in vesicular mobilization and fusion at synapses (synapsin), and amphiphysin—a presynaptically enriched protein that is thought to recruit dynamin to sites of clathrin-mediated endocytosis. For these experiments, we fixed DIV-21 h-α-syn:GFP-overexpressing neurons, retrospectively immunostained them for the respective endogenous mouse presynaptic proteins (using red secondary antibodies), then quantified colocalization of GFP+ve transgenic boutons to the respective (red fluorescent) bouton containing mouse presynaptic proteins. WT littermate boutons containing endogenous mouse α-synuclein and the respective presynaptic protein under consideration were used as controls.
Surprisingly, we found that all four mouse presynaptic proteins were undetectable over background in subsets of h-α-syn-overexpressing boutons (Fig. 7 A,B), and we started calling them “vacant synapses.” We retain this term here to recognize this subset of boutons in our studies. The appearance of such vacant synapses was gradual, increasing in numbers from DIV-7 to DIV-21 (Fig. 7 C). These temporal changes were likely due to sustained h-α-syn overexpression, and not a result of a sudden escalation in α-synuclein from DIV-7 to 21, as the average levels of overexpressed GFP fluorescence within boutons was practically unchanged during this period (1126 ± 34.56 AFU and 1117 ± 31.27 AFU in DIV-7 and DIV-21 neurons, respectively, n = 300–600 boutons). Similar diminutions in endogenous presynaptic proteins was also seen in hippocampal and cortical in vivo tissue sections from h-α-syn:GFP transgenic mice (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). We also considered the possibility (albeit remote) that these “vacant synapses” could represent GABAergic boutons. Though neurons in hippocampal cultures like ours are predominantly glutamatergic, a small fraction of GABAergic neurons are present. However, this is unlikely to be the case here, as in a set of experiments only 4% of our transgenic boutons were opposed to GABAergic postsynaptic terminals (out of 1229 α-synuclein transgenic boutons, only 54 were opposed to postsynaptic terminals immunopositive for the GABAA receptor). Thus the vast majority of “vacant synapses” in our system are likely glutamatergic.
Figure 7.
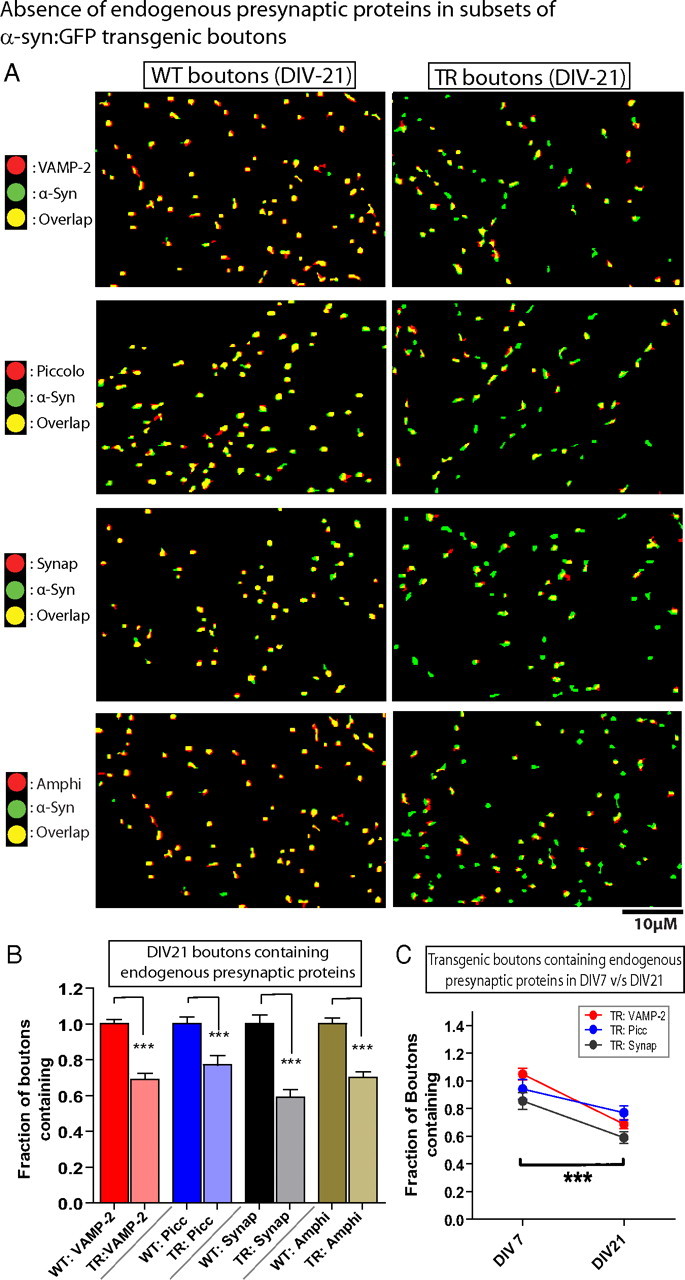
Undetectable endogenous presynaptic proteins in subsets of h-α-syn-overexpressing boutons (“vacant synapses”). A, DIV-21 boutons overexpressing h-α-syn:GFP (green) were fixed and stained with antibodies recognizing endogenous mouse VAMP-2/piccolo/synapsin-1/amphiphysin (red). For controls, neurons from WT littermates were fixed and stained for mouse α-synuclein (green) as well as endogenous mouse VAMP-2/piccolo/synapsin-1/amphiphysin (red). The panels are pseudocolor overlays of the WT mouse (left) or the transgenic human (right) α-synucleins to the corresponding mouse endogenous proteins; colocalized boutons appear yellow. Note that while there is an almost complete colocalization of the four mouse presynaptic proteins with mouse α-synuclein in WT boutons as expected (yellow in left panels), many h-α-syn:GFP transgenic boutons did not contain any detectable presynaptic proteins (green boutons in right panels), as shown in these representative images. B, Quantification of the colocalization data. The fraction of transgenic boutons containing VAMP-2, piccolo, synapsin-1, and amphiphysin relative to WT boutons was 0.6884 ± 0.034, 0.7702 ± 0.051, 0.5902 ± 0.043, and 0.7015 ± 0.031, respectively (mean ± SEM, p < 0.0001 for all groups; n = 1000–1600 boutons/group). C, The number of boutons lacking endogenous mouse proteins increased over time, with a significantly larger numbers of boutons lacking the synaptic proteins at DIV-21, compared to DIV-7, suggesting a gradual evolution of the synaptic pathology (p < 0.0001, two-way ANOVA between groups; n = 400–700 boutons).
Diminished levels of mouse presynaptic proteins in transgenic boutons containing mouse presynaptic proteins (“nonvacant synapses”)
We wondered whether the “vacant synapses” in our system represented a unique subset of transgenic boutons that lacked endogenous presynaptic proteins, or whether these vacant boutons were simply on the extreme end of a continuum where α-synuclein overexpression diminished the physiologic presynaptic targeting of these endogenous proteins. To address this, we quantitatively evaluated average fluorescence levels of the four endogenous presynaptic proteins, focusing only on the transgenic boutons that did contain these presynaptic proteins (i.e., the “nonvacant synapses”). Levels of all four proteins were diminished in “nonvacant” boutons, as shown in representative pseudocolor (heat-map) panels in Figure 8 A and the corresponding quantitative analyses in Figure 8 B. Note that the extent of quantitative diminution of a specific protein generally mirrored the fraction of “vacant synapses” within the given group (compare Fig. 8 B to Fig. 7 B), further suggesting that these changes represent a continuum. Collectively, these data show that α-synuclein overexpression in our system leads to global diminutions in the levels of several presynaptic proteins involved in exocytosis and endocytosis, and that subsets of such overexpressing boutons do not contain any detectable amounts of these proteins.
Figure 8.

Diminished levels of presynaptic proteins in “non-vacant” transgenic boutons. Neurons were processed the same way as in Figure 7, and the analysis was focused on the transgenic boutons that did contain VAMP/Piccolo/synapsin-1/amphiphysin. A, Images on left show pseudocolor (heat maps) indicating levels of proteins within boutons, where red/white indicate higher fluorescence levels and blue/purple indicate lower (key: upper right). Note that in general, average fluorescence levels of these four proteins are higher within WT boutons, compared to transgenics. B, Average fluorescence intensities of the four mouse presynaptic proteins in transgenic boutons in transgenic boutons relative to WT boutons were 0.6992 ± 0.023, 0.8504 ± 0.026, 0.4920 ± 0.013, and 0.8250 ± 0.016, respectively (mean ± SEM, ***p < 0.001 for all groups, n = 1000–1600 boutons/each group). Scale bars, lower right of images.
Pathologic synaptic alterations in human synucleinopathy brains
The above data in cultured neurons and intact mouse brain support a disease model where excessive h-α-syn is pathologically altered in a subset of presynaptic boutons, inducing depletion of various endogenous presynaptic proteins at these boutons and subsequent functional deficits resulting from the loss of critical synaptic components. In DLB, the prototypical human disease with α-synuclein-related dementia, presynaptic aggregates of proteinase-K-resistant α-synuclein are also seen in frontal cortical sections (Kramer and Schulz-Schaeffer, 2007), prompting the question of whether the mechanistic events professed by our single-cell experiments also operate in human disease as well. If so, one would expect that h-α-syn-containing synapses in these diseased brains would have lesser amounts of other presynaptic proteins. To evaluate this, we immunostained frontal cortical sections from autopsy brains of patients with DLB (or age-matched controls) for h-α-syn and synapsin (the protein showing the greatest diminution in our culture studies), and evaluated synaptic protein colocalization. Many h-α-syn-positive synapses in DLB brains had undetectable synapsin levels as well (Fig. 9), suggesting that h-α-syn-induced depletion of presynaptic proteins may play a role in α-synuclein-induced human disease as well. We noticed that the colocalization of different presynaptic proteins in tissue sections (even in control brains) was not complete, unlike the situation in WT cultured neurons, where such colocalization consistently approached 100%. The reasons for this are unclear, but may be related to the complex neuronal architecture in brain slices compared to the relatively simple morphology in cultured neurons, where individual boutons can be resolved precisely by light microscopy.
Figure 9.

Undetectable levels of a presynaptic protein in human brains with α-synuclein related pathology. Prefrontal cortices of autopsy human brains from controls and patients with DLB were immunostained with antibodies to h-α-syn and synapsin, and colocalization of the two antigens was evaluated. Representative images from control and LB dementia brains are shown with quantization. Note that while extensive colocalization (yellow) is evident in the control brain (left), many α-synuclein-positive boutons (green) do not contain any detectable synapsin-1 in the DLB brain (right). The area within the box was magnified (inset); arrowheads denote α-synuclein+ boutons that do not contain synapsin. The fraction of α-synuclein+ve boutons that also contained synapsin in DLB brains relative to controls was 0.6690 ± 0.046 (mean ± SEM, ***p < 0.0001; n = 2000 boutons/each group). Note that in WT brain sections, the colocalization of the various synaptic antigens was not complete (unlike cultured neurons), and there were occasional α-synuclein+ boutons in control brains that did not contain synapsin (one marked with an asterisk). Scale bars, lower right of images.
Discussion
To investigate evolving pathologic events induced by excessive α-synuclein, we developed a model system reliably overexpressing modest amounts of fluorescent α-synuclein in large neuronal populations and used quantitative cell-biological tools to evaluate the evolving α-synuclein-induced pathology in live and fixed neurons before the onset of overt synaptic losses. We found that in mature (DIV-21) neurons, the expressed α-synuclein was PK resistant and also acquired disease-associated posttranslational modifications. At a functional level, there were profound deficits in neurotransmitter release, providing a basis for synaptotoxicity. Ultrastructural examination revealed a peculiar enlargement of synaptic vesicles in transgenic boutons. This constellation of functional and morphologic changes resembled previous animal models lacking presynaptic proteins, prompting us to quantitatively evaluate endogenous mouse presynaptic proteins in our h-α-syn-overexpressing boutons. Indeed, we found that several critical exocytic and endocytic endogenous presynaptic proteins were absent in transgenic boutons (“vacant synapses”); along with striking diminutions in the levels of such mouse proteins in the remainder. Importantly, these changes may be disease relevant as diminutions in presynaptic protein levels are also visible in human autopsy brains with α-synuclein-related dementias.
Collectively, the data point to a previously unknown mechanistic cascade of events induced by elevated h-α-syn levels that may underlie the early synaptic dysfunction seen in diseased states. Specifically, increased intracellular α-synuclein leads to attenuated levels of several proteins critical to the physiologic operation of the synaptic vesicle machinery; such attenuations lead to functional impairments at synapses manifested by an inhibition of neurotransmitter release; and this toxic chain of events eventually leads to synaptic dysfunction and dementia. Though we have not looked at the entire gamut of synaptic proteins in h-α-syn-overexpressing boutons, we rationally selected a few proteins critically important in exocytosis and endocytosis, and surprisingly, they were all depleted/diminished in transgenic boutons.
What is the specific defect induced by excessive α-synuclein at the synapse? The available evidence strongly suggests that inhibition of neurotransmitter release is the overall pathologic mechanism induced by excessive α-synuclein, as shown here and in a recent study (Nemani et al., 2010). However, the specific synaptic defect(s) induced by excessive α-synuclein is less obvious. Previous unbiased studies of genetic modifiers of α-synuclein toxicity clearly implicate multiple vesicular trafficking pathways in the pathogenesis of α-synuclein toxicity (Gitler et al., 2008; Soper et al., 2008), and a similar approach in C. elegans α-synuclein models implicated genes involved in the endocytosis, in addition to other vesicular trafficking pathways (Kuwahara et al., 2008; van Ham et al., 2008). Other groups have implicated SNARE proteins in α-synuclein pathogenesis (Chandra et al., 2005). Recently, Larsen et al. (2006) suggested that excessive α-synuclein may impair vesicular release machinery by inhibiting “priming”—the step immediately preceding vesicular docking to presynaptic membranes. Most recently, Nemani et al. (2010) rigorously demonstrated that excessive α-synuclein inhibits neurotransmission, reduces the recycling pool of vesicles, and impairs the activity-dependent mobility of synaptic vesicles, suggesting that excessive α-synuclein induces a specific defect in the synaptic vesicle recycling pathway by preventing the reclustering of vesicles after endocytosis.
The focus of our studies was to elucidate the evolution of pathologic events resulting from α-synuclein overexpression and culminating in synaptic dysfunction. Though we are agnostic about the precise defect(s) induced by α-synuclein at synapses, we favor the view that the pathologic effects are pleiotropic and not restricted to a specific and exclusive impairment of a singular step in the synaptic machinery. First, as previously noted, unbiased screens of α-synuclein toxicity modifiers suggest involvement of multiple vesicular trafficking pathways, including those involved in exocytosis and endocytosis. Second, our FM data show that ∼25% of the transgenic boutons fail to uptake any dye (Fig. 5 A, lower left graph). Though a diminution in the recycling pool as proposed by Nemani et al. (2010) should technically reduce FM levels within boutons as well, a complete absence of FM loading may reflect accompanying endocytic defects. Third, abnormally enlarged synaptic vesicles, as seen in our EM studies, have been repeatedly observed in animal models deficient in proteins associated with endocytosis (Zhang et al., 1998; Nonet et al., 1999; Deák et al., 2004; Poskanzer et al., 2006; Ferguson et al., 2007), suggesting that such morphologic changes may be due (at least in part) to abnormalities in endocytosis. Finally, we show that α-synuclein-overexpressing synapses also have clear diminutions of amphiphysin—a protein involved in endocytosis, further implicating the involvement of multiple exocytic and endocytic pathways in α-synuclein pathogenesis.
In contrast to the prevalent viewpoint, our data suggest that the eventual pathology in α-synuclein-overexpressing synapses is likely to be a complex phenotype induced by the absence of an assortment of presynaptic proteins, including SNARE and vesicle-associated proteins involved in recycling and mobilization of synaptic vesicles (VAMP-2 and synapsin-1), active-zone proteins that act as scaffolds at synapses and are thought to play a role in exocytosis and endocytosis (piccolo), and proteins critical for the endocytosis (amphiphysin). The absence/diminution of these assorted proteins would likely induce defects in recycling, priming as well as endocytosis, perhaps reconciling evidence from existing studies. We note that Nemani et al. (2010) also showed changes in some protein levels from brains of α-synuclein transgenic mice, but it is difficult to compare their biochemical data from brain homogenates to our examination of single-boutons that unequivocally contain α-synuclein and can be analyzed with precision.
The enlarged vesicles seen in our EM studies also resemble the h-α-syn-positive vesicular clusters that we and others reported in the yeast model system overexpressing h-α-syn:GFP (Gitler et al., 2008; Soper et al., 2008). Though the yeast data were somewhat puzzling at the time, in retrospect, perhaps the vesicular clusters represent failed cycles of vesicular fusion/fission and/or endocytosis in yeast, in the presence of excessive h-α-syn. Regarding neurons, though the exact reason for the presence of enlarged vesicles is unclear, given the existing EM evidence from endocytic mutants (cited above), it seems plausible that this synaptic phenotype is simply due to the lack of proteins involved in endocytosis. Alternatively/additionally, α-synuclein binding to synaptic vesicles may directly cause vesicular fusion abnormalities as well (Kamp and Beyer, 2006).
Studies in α-synuclein-null mice suggest a physiologic role of α-synuclein in regulating the release of neurotransmitters (Abeliovich et al., 2000), and also in the maintenance of the SNARE machinery itself (Chandra et al., 2004, 2005). Thus it is conceivable that in diseased states, an imbalance of α-synuclein levels at synapses interferes with its physiologic role of releasing neurotransmitters, eventually leading the exquisite synaptic machinery to go awry. Such mechanisms can be imagined as being analogous to the role of amyloid-β (Aβ) in Alzheimer's disease, where emerging evidence implicates a concentration-dependent role for Aβ oligomers in both normal synaptic functioning as well as synaptic damage (Giuffrida et al., 2009; Parodi et al., 2010).
Though our studies are focused on h-α-syn-induced synaptotoxicity, our proposed mechanistic cascade leading to “vacant synapses” may have more general implications for neurodegeneration as well. In a recent report, prolonged treatment of hippocampal neurons with oligomeric Aβ diminished spontaneous synaptic responses in cultured hippocampal neurons, along with a diminution of various presynaptic proteins (Parodi et al., 2010), somewhat similar to our results. Presynaptic protein depletion and synaptic dysfunction was also reported in a model of amyotrophic lateral sclerosis associated with mutant superoxide dismutase as well (SOD1) (Wang et al., 2009), suggesting further commonalities between apparently diverse neurodegenerative diseases.
How does excessive α-synuclein lead to a diminution of other presynaptic proteins? One possibility is that excessive α-synuclein may inhibit the axonal transport and/or presynaptic targeting of other proteins. The continuum of protein loss in transgenic boutons as seen in our studies, ranging from a total absence in some (Fig. 7), to ∼20–50% diminutions in the remainder (Fig. 8), supports this idea. In human DLB brains, widespread varicosities containing phospho-α-synuclein are commonly seen (Saito et al., 2003), and may represent axonal transport defects. In this scenario, as α-synuclein itself is conveyed in slow axonal transport with other proteins (Roy et al., 2007, 2008), misfolding/aggregation of the protein may alter its mobility, and also the movements of other cotransported cargoes as they navigate along elongated axons. Indeed, we see the greatest diminutions in synapsin, the only protein examined that moves in slow axonal transport. Recent studies showing that mutant α-synuclein (A53T) diminished levels of various motor proteins in neurons (Chung et al., 2009) and that axonal transport abnormalities are also seen in squid axons treated with 1-methyl-4-phenylpyridinium—a model of experimentally induced Parkinsonism (Morfini et al., 2007)—also implicate axonal transport defects. Alternatively, excessive amounts of misfolded α-synuclein may aggregate at synapses (Kramer and Schulz-Schaeffer, 2007), physically preventing the targeting of other presynaptic proteins; or alter the biophysical properties/turnover of synaptic vesicles/proteins. Another possibility is that excessive α-synuclein may alter the structural properties of synapses over time, preventing the robust association of synaptic proteins to these sites. Indeed, previous studies suggest that actin or other synaptic scaffolding proteins may be involved in α-synuclein pathogenesis (Sousa et al., 2009); Ihara et al., 2007). Future studies will focus on distinguishing between these possibilities.
Footnotes
This work was supported by grants to S.R. from the Hillblom Foundation, the American Parkinson's Disease Association, the Alzheimer's Association (NIRG-08-90769), the March of Dimes Foundation (Basil O'Connor Award), and the National Institute for Aging (2P50AG005131-P2); a generous gift from Darlene and Donald Shiley to the University of California, San Diego Alzheimer's Center; and a National Institutes of Health Grant to I.T. (NS060799). We thank Margarita Trejo and Anthony Adame for technical assistance with electron microscopy and histology. D.S. and S.R. designed and performed the imaging experiments, developed quantitative algorithms, analyzed the data, and wrote the manuscript. Electrophysiological studies/analyses were done by I.T.; Y.T. contributed to immunofluorescence experiments. E.M. and A.C. contributed to the ultrastructural studies and human tissue histology.
References
- Aarsland D, Beyer MK, Kurz MW. Dementia in Parkinson's disease. Curr Opin Neurol. 2008;21:676–682. doi: 10.1097/WCO.0b013e3283168df0. [DOI] [PubMed] [Google Scholar]
- Abeliovich A, Schmitz Y, Fariñas I, Choi-Lundberg D, Ho WH, Castillo PE, Shinsky N, Verdugo JM, Armanini M, Ryan A, Hynes M, Phillips H, Sulzer D, Rosenthal A. Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron. 2000;25:239–252. doi: 10.1016/s0896-6273(00)80886-7. [DOI] [PubMed] [Google Scholar]
- Anderson JP, Walker DE, Goldstein JM, de Laat R, Banducci K, Caccavello RJ, Barbour R, Huang J, Kling K, Lee M, Diep L, Keim PS, Shen X, Chataway T, Schlossmacher MG, Seubert P, Schenk D, Sinha S, Gai WP, Chilcote TJ. Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. J Biol Chem. 2006;281:29739–29752. doi: 10.1074/jbc.M600933200. [DOI] [PubMed] [Google Scholar]
- Chandra S, Fornai F, Kwon HB, Yazdani U, Atasoy D, Liu X, Hammer RE, Battaglia G, German DC, Castillo PE, Südhof TC. Double-knockout mice for alpha- and beta-synucleins: effect on synaptic functions. Proc Natl Acad Sci U S A. 2004;101:14966–14971. doi: 10.1073/pnas.0406283101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra S, Gallardo G, Fernández-Chacón R, Schlüter OM, Südhof TC. Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell. 2005;123:383–396. doi: 10.1016/j.cell.2005.09.028. [DOI] [PubMed] [Google Scholar]
- Chen L, Feany MB. Alpha-synuclein phosphorylation controls neurotoxicity and inclusion formation in a Drosophila model of Parkinson disease. Nat Neurosci. 2005;8:657–663. doi: 10.1038/nn1443. [DOI] [PubMed] [Google Scholar]
- Chung CY, Koprich JB, Siddiqi H, Isacson O. Dynamic changes in presynaptic and axonal transport proteins combined with striatal neuroinflammation precede dopaminergic neuronal loss in a rat model of AAV α-synucleinopathy. J Neurosci. 2009;29:3365–3373. doi: 10.1523/JNEUROSCI.5427-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cookson MR, van der Brug M. Cell systems and the toxic mechanism(s) of alpha-synuclein. Exp Neurol. 2008;209:5–11. doi: 10.1016/j.expneurol.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Custer KL, Austin NS, Sullivan JM, Bajjalieh SM. Synaptic vesicle protein 2 enhances release probability at quiescent synapses. J Neurosci. 2006;26:1303–1313. doi: 10.1523/JNEUROSCI.2699-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deák F, Schoch S, Liu X, Südhof TC, Kavalali ET. Synaptobrevin is essential for fast synaptic-vesicle endocytosis. Nat Cell Biol. 2004;6:1102–1108. doi: 10.1038/ncb1185. [DOI] [PubMed] [Google Scholar]
- Deitcher DL, Ueda A, Stewart BA, Burgess RW, Kidokoro Y, Schwarz TL. Distinct requirements for evoked and spontaneous release of neurotransmitter are revealed by mutations in the Drosophila gene neuronal-synaptobrevin. J Neurosci. 1998;18:2028–2039. doi: 10.1523/JNEUROSCI.18-06-02028.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrer M, Kachergus J, Forno L, Lincoln S, Wang DS, Hulihan M, Maraganore D, Gwinn-Hardy K, Wszolek Z, Dickson D, Langston JW. Comparison of kindreds with parkinsonism and alpha-synuclein genomic multiplications. Ann Neurol. 2004;55:174–179. doi: 10.1002/ana.10846. [DOI] [PubMed] [Google Scholar]
- Ferguson SM, Brasnjo G, Hayashi M, Wölfel M, Collesi C, Giovedi S, Raimondi A, Gong LW, Ariel P, Paradise S, O'Toole E, Flavell R, Cremona O, Miesenböck G, Ryan TA, De Camilli P. A selective activity-dependent requirement for dynamin 1 in synaptic vesicle endocytosis. Science. 2007;316:570–574. doi: 10.1126/science.1140621. [DOI] [PubMed] [Google Scholar]
- Fuchs J, Tichopad A, Golub Y, Munz M, Schweitzer KJ, Wolf B, Berg D, Mueller JC, Gasser T. Genetic variability in the SNCA gene influences alpha-synuclein levels in the blood and brain. FASEB J. 2008;22:1327–1334. doi: 10.1096/fj.07-9348com. [DOI] [PubMed] [Google Scholar]
- Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, Shen J, Takio K, Iwatsubo T. alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol. 2002;4:160–164. doi: 10.1038/ncb748. [DOI] [PubMed] [Google Scholar]
- Gaffield MA, Betz WJ. Imaging synaptic vesicle exocytosis and endocytosis with FM dyes. Nat Protoc. 2006;1:2916–2921. doi: 10.1038/nprot.2006.476. [DOI] [PubMed] [Google Scholar]
- Gitler AD, Bevis BJ, Shorter J, Strathearn KE, Hamamichi S, Su LJ, Caldwell KA, Caldwell GA, Rochet JC, McCaffery JM, Barlowe C, Lindquist S. The Parkinson's disease protein alpha-synuclein disrupts cellular Rab homeostasis. Proc Natl Acad Sci U S A. 2008;105:145–150. doi: 10.1073/pnas.0710685105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitler D, Takagishi Y, Feng J, Ren Y, Rodriguiz RM, Wetsel WC, Greengard P, Augustine GJ. Different presynaptic roles of synapsins at excitatory and inhibitory synapses. J Neurosci. 2004;24:11368–11380. doi: 10.1523/JNEUROSCI.3795-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giuffrida ML, Caraci F, Pignataro B, Cataldo S, De Bona P, Bruno V, Molinaro G, Pappalardo G, Messina A, Palmigiano A, Garozzo D, Nicoletti F, Rizzarelli E, Copani A. β-Amyloid monomers are neuroprotective. J Neurosci. 2009;29:10582–10587. doi: 10.1523/JNEUROSCI.1736-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihara M, Yamasaki N, Hagiwara A, Tanigaki A, Kitano A, Hikawa R, Tomimoto H, Noda M, Takanashi M, Mori H, Hattori N, Miyakawa T, Kinoshita M. Sept4, a component of presynaptic scaffold and Lewy bodies, is required for the suppression of alpha-synuclein neurotoxicity. Neuron. 2007;53:519–533. doi: 10.1016/j.neuron.2007.01.019. [DOI] [PubMed] [Google Scholar]
- Ikeuchi T, Kakita A, Shiga A, Kasuga K, Kaneko H, Tan CF, Idezuka J, Wakabayashi K, Onodera O, Iwatsubo T, Nishizawa M, Takahashi H, Ishikawa A. Patients homozygous and heterozygous for SNCA duplication in a family with parkinsonism and dementia. Arch Neurol. 2008;65:514–519. doi: 10.1001/archneur.65.4.514. [DOI] [PubMed] [Google Scholar]
- Kaech S, Banker G. Culturing hippocampal neurons. Nat Protoc. 2006;1:2406–2415. doi: 10.1038/nprot.2006.356. [DOI] [PubMed] [Google Scholar]
- Kahle PJ, Neumann M, Ozmen L, Muller V, Jacobsen H, Spooren W, Fuss B, Mallon B, Macklin WB, Fujiwara H, Hasegawa M, Iwatsubo T, Kretzschmar HA, Haass C. Hyperphosphorylation and insolubility of alpha-synuclein in transgenic mouse oligodendrocytes. EMBO Rep. 2002;3:583–588. doi: 10.1093/embo-reports/kvf109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamp F, Beyer K. Binding of alpha-synuclein affects the lipid packing in bilayers of small vesicles. J Biol Chem. 2006;281:9251–9259. doi: 10.1074/jbc.M512292200. [DOI] [PubMed] [Google Scholar]
- Kramer ML, Schulz-Schaeffer WJ. Presynaptic α-synuclein aggregates, not Lewy bodies, cause neurodegeneration in dementia with Lewy bodies. J Neurosci. 2007;27:1405–1410. doi: 10.1523/JNEUROSCI.4564-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger SR, Kolar A, Fitzsimonds RM. The presynaptic release apparatus is functional in the absence of dendritic contact and highly mobile within isolated axons. Neuron. 2003;40:945–957. doi: 10.1016/s0896-6273(03)00729-3. [DOI] [PubMed] [Google Scholar]
- Kuwahara T, Koyama A, Koyama S, Yoshina S, Ren CH, Kato T, Mitani S, Iwatsubo T. A systematic RNAi screen reveals involvement of endocytic pathway in neuronal dysfunction in alpha-synuclein transgenic C. elegans . Hum Mol Genet. 2008;17:2997–3009. doi: 10.1093/hmg/ddn198. [DOI] [PubMed] [Google Scholar]
- Larsen KE, Schmitz Y, Troyer MD, Mosharov E, Dietrich P, Quazi AZ, Savalle M, Nemani V, Chaudhry FA, Edwards RH, Stefanis L, Sulzer D. α-Synuclein overexpression in PC12 and chromaffin cells impairs catecholamine release by interfering with a late step in exocytosis. J Neurosci. 2006;26:11915–11922. doi: 10.1523/JNEUROSCI.3821-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeith IG, Mosimann UP. Dementia with Lewy bodies and Parkinson's disease. Parkinsonism Relat Disord. 2004;10(Suppl 1):S15–S18. doi: 10.1016/j.parkreldis.2003.12.005. [DOI] [PubMed] [Google Scholar]
- McLean PJ, Kawamata H, Hyman BT. Alpha-synuclein-enhanced green fluorescent protein fusion proteins form proteasome sensitive inclusions in primary neurons. Neuroscience. 2001;104:901–912. doi: 10.1016/s0306-4522(01)00113-0. [DOI] [PubMed] [Google Scholar]
- Morfini G, Pigino G, Opalach K, Serulle Y, Moreira JE, Sugimori M, Llinás RR, Brady ST. 1-Methyl-4-phenylpyridinium affects fast axonal transport by activation of caspase and protein kinase C. Proc Natl Acad Sci U S A. 2007;104:2442–2447. doi: 10.1073/pnas.0611231104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukaetova-Ladinska EB, McKeith IG. Pathophysiology of synuclein aggregation in Lewy body disease. Mech Ageing Dev. 2006;127:188–202. doi: 10.1016/j.mad.2005.09.014. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Nemani VM, Wallender EK, Kaehlcke K, Ott M, Edwards RH. Optical reporters for the conformation of α-synuclein reveal a specific interaction with mitochondria. J Neurosci. 2008;28:12305–12317. doi: 10.1523/JNEUROSCI.3088-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale EA, Bowers LM, Jia M, Bateman KE, Williamson LC. Botulinum neurotoxin A blocks synaptic vesicle exocytosis but not endocytosis at the nerve terminal. J Cell Biol. 1999;147:1249–1260. doi: 10.1083/jcb.147.6.1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemani VM, Lu W, Berge V, Nakamura K, Onoa B, Lee MK, Chaudhry FA, Nicoll RA, Edwards RH. Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron. 2010;65:66–79. doi: 10.1016/j.neuron.2009.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M, Kahle PJ, Giasson BI, Ozmen L, Borroni E, Spooren W, Müller V, Odoy S, Fujiwara H, Hasegawa M, Iwatsubo T, Trojanowski JQ, Kretzschmar HA, Haass C. Misfolded proteinase K-resistant hyperphosphorylated alpha-synuclein in aged transgenic mice with locomotor deterioration and in human alpha-synucleinopathies. J Clin Invest. 2002;110:1429–1439. doi: 10.1172/JCI15777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonet ML, Saifee O, Zhao H, Rand JB, Wei L. Synaptic transmission deficits in Caenorhabditis elegans synaptobrevin mutants. J Neurosci. 1998;18:70–80. doi: 10.1523/JNEUROSCI.18-01-00070.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonet ML, Holgado AM, Brewer F, Serpe CJ, Norbeck BA, Holleran J, Wei L, Hartwieg E, Jorgensen EM, Alfonso A. UNC-11, a Caenorhabditis elegans AP180 homologue, regulates the size and protein composition of synaptic vesicles. Mol Biol Cell. 1999;10:2343–2360. doi: 10.1091/mbc.10.7.2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Outeiro TF, Lindquist S. Yeast cells provide insight into alpha-synuclein biology and pathobiology. Science. 2003;302:1772–1775. doi: 10.1126/science.1090439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parodi J, Sepúlveda FJ, Roa J, Opazo C, Inestrosa NC, Aguayo LG. β-Amyloid causes depletion of synaptic vesicles leading to neurotransmission failure. J Biol Chem. 2010;285:2506–2514. doi: 10.1074/jbc.M109.030023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Periquet M, Fulga T, Myllykangas L, Schlossmacher MG, Feany MB. Aggregated α-synuclein mediates dopaminergic neurotoxicity in vivo . J Neurosci. 2007;27:3338–3346. doi: 10.1523/JNEUROSCI.0285-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poskanzer KE, Fetter RD, Davis GW. Discrete residues in the c(2)b domain of synaptotagmin I independently specify endocytic rate and synaptic vesicle size. Neuron. 2006;50:49–62. doi: 10.1016/j.neuron.2006.02.021. [DOI] [PubMed] [Google Scholar]
- Rockenstein E, Mallory M, Hashimoto M, Song D, Shults CW, Lang I, Masliah E. Differential neuropathological alterations in transgenic mice expressing alpha-synuclein from the platelet-derived growth factor and Thy-1 promoters. J Neurosci Res. 2002;68:568–578. doi: 10.1002/jnr.10231. [DOI] [PubMed] [Google Scholar]
- Rockenstein E, Schwach G, Ingolic E, Adame A, Crews L, Mante M, Pfragner R, Schreiner E, Windisch M, Masliah E. Lysosomal pathology associated with alpha-synuclein accumulation in transgenic models using an eGFP fusion protein. J Neurosci Res. 2005;80:247–259. doi: 10.1002/jnr.20446. [DOI] [PubMed] [Google Scholar]
- Roy S. The paradoxical cell biology of α-synuclein. Results Probl Cell Differ. 2009;48:159–172. doi: 10.1007/400_2009_23. [DOI] [PubMed] [Google Scholar]
- Roy S, Winton MJ, Black MM, Trojanowski JQ, Lee VM. Rapid and intermittent cotransport of slow component-b proteins. J Neurosci. 2007;27:3131–3138. doi: 10.1523/JNEUROSCI.4999-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy S, Winton MJ, Black MM, Trojanowski JQ, Lee VM. Cytoskeletal requirements in axonal transport of slow component-b. J Neurosci. 2008;28:5248–5256. doi: 10.1523/JNEUROSCI.0309-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito Y, Kawashima A, Ruberu NN, Fujiwara H, Koyama S, Sawabe M, Arai T, Nagura H, Yamanouchi H, Hasegawa M, Iwatsubo T, Murayama S. Accumulation of phosphorylated alpha-synuclein in aging human brain. J Neuropathol Exp Neurol. 2003;62:644–654. doi: 10.1093/jnen/62.6.644. [DOI] [PubMed] [Google Scholar]
- Schoch S, Gundelfinger ED. Molecular organization of the presynaptic active zone. Cell Tissue Res. 2006;326:379–391. doi: 10.1007/s00441-006-0244-y. [DOI] [PubMed] [Google Scholar]
- Schoch S, Deák F, Königstorfer A, Mozhayeva M, Sara Y, Südhof TC, Kavalali ET. SNARE function analyzed in synaptobrevin/VAMP knockout mice. Science. 2001;294:1117–1122. doi: 10.1126/science.1064335. [DOI] [PubMed] [Google Scholar]
- Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, et al. alpha-Synuclein locus triplication causes Parkinson's disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- Soper JH, Roy S, Stieber A, Lee E, Wilson RB, Trojanowski JQ, Burd CG, Lee VM. {alpha}-Synuclein-induced aggregation of cytoplasmic vesicles in Saccharomyces cerevisiae . Mol Biol Cell. 2008;19:1093–1103. doi: 10.1091/mbc.E07-08-0827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa VL, Bellani S, Giannandrea M, Yousuf M, Valtorta F, Meldolesi J, Chieregatti E. {alpha}-Synuclein and its A30P mutant affect actin cytoskeletal structure and dynamics. Mol Biol Cell. 2009;20:3725–3739. doi: 10.1091/mbc.E08-03-0302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Ham TJ, Thijssen KL, Breitling R, Hofstra RM, Plasterk RH, Nollen EA. C. elegans model identifies genetic modifiers of alpha-synuclein inclusion formation during aging. PLoS Genet. 2008;4:e1000027. doi: 10.1371/journal.pgen.1000027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verderio C, Coco S, Bacci A, Rossetto O, De Camilli P, Montecucco C, Matteoli M. Tetanus toxin blocks the exocytosis of synaptic vesicles clustered at synapses but not of synaptic vesicles in isolated axons. J Neurosci. 1999;19:6723–6732. doi: 10.1523/JNEUROSCI.19-16-06723.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Farr GW, Hall DH, Li F, Furtak K, Dreier L, Horwich AL. An ALS-linked mutant SOD1 produces a locomotor defect associated with aggregation and synaptic dysfunction when expressed in neurons of Caenorhabditis elegans . PLoS Genet. 2009;5:e1000350. doi: 10.1371/journal.pgen.1000350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman EA, Giasson BI. Specificity and regulation of casein kinase-mediated phosphorylation of alpha-synuclein. J Neuropathol Exp Neurol. 2008;67:402–416. doi: 10.1097/NEN.0b013e3186fc995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Koh YH, Beckstead RB, Budnik V, Ganetzky B, Bellen HJ. Synaptic vesicle size and number are regulated by a clathrin adaptor protein required for endocytosis. Neuron. 1998;21:1465–1475. doi: 10.1016/s0896-6273(00)80664-9. [DOI] [PubMed] [Google Scholar]