

Abstract
This paper describes a very simple and robust microfluidic device for digitizing samples into an array of discrete volumes. The device is based on an inherent fluidic phenomenon, where an incoming aqueous sample divides itself into an array of chambers that have been primed with an immiscible phase. Self-digitization of sample volumes results from the interplay between fluidic forces, interfacial tension, channel geometry, and the final stability of the digitized samples in the chambers. Here we describe experiments and simulations that were used to characterize these parameters and the conditions under which the self-digitization process occurred. Unlike existing methods used to partition samples into array, our method is able to digitize 100% of a sample into a localized array without any loss of sample volume. The final volume of the discretized sample at each location is defined by the geometry and size of each chamber. Thus, we can form an array of samples with varying but predefined volumes. We exploited this feature to separate the crystal growth of otherwise concomitant polymorphs from a single solution. Additionally, we demonstrated the removal of the digitized samples from the chambers for downstream analysis, as well as the addition of reagents to the digitized samples. We believe this simple method will be useful in a broad range of applications where a large array of discretized samples is required, including digital PCR, single-cell analysis, and cell-based drug screening.
INTRODUCTION
A crucial first step in many chemical and biological experiments, such as in digital PCR or single-cell analysis, is the partitioning of samples into an array of small individual volumes for subsequent assays. Towards this end, several methods exist. Traditional methods of generating small volumes of sample include the use of a nebulizer to create an aerosol and the mixing of a sample with an immiscible phase to create an emulsion1–3. These methods, however, have limitations. The droplets formed by a nebulizer exhibit a large range of sizes. Similarly, mixing two immiscible phases to form an emulsion does not produce monodisperse droplets. The droplets formed by both methods are difficult to manipulate and analyze individually. In addition, neither of these methods would be well suited for the continuous monitoring that is necessary for in situ assays.
For more control over the process and the final volumes that are formed, a common means to generating an array of samples is by dispensing small volumes into microwells4–7. While this approach is successful in creating an array of spatially localized samples, filling the individual wells can be tedious. Human error also can get in the way of dispensing homogeneous volumes. To avoid this, special dispensing equipment, such as robotic pipets, are used. However, specialized dispensing equipment limits the flexibility of these microwell platforms and raises costs. In addition, the dispensing equipment makes it difficult to work with volumes below 0.5μL because of the relatively high dispensing volumes of pipets as well as problems with sample evaporation4, 6, 8.
Given the need to generate and manipulate a large number of samples and the drawbacks of traditional approaches, new methods of sample-volume discretization have emerged over the past decade. These methods include separation of volumes from one another using microfluidic valves9, formation of droplets on a dielectric by electrowetting10, 11, differential wetting of surfaces that have been patterned with self-assembled monolayers12, and generation of individual or a continuous stream of droplets using microfluidics13–20. These newer methods, albeit powerful, still suffer from a number of drawbacks. For example, valves and pumps require complex fluidic control in fabricated devices that tend to be expensive, especially if hundreds to thousands discrete volumes are involved. Steady-state methods to generate a stream of droplets make the subsequent manipulation and analysis of individual droplets difficult. But more importantly, they often result in loss of sample volume because there is some initial waste of the sample as a steady-state flow stream is established before the regular formation of droplets can begin. Additionally, these steady-state methods require accurate control of flow rates which increases the complexity and expense of the final instrument.
A simpler method for creating an array of discretized samples is to drag a fluid along a surface pitted with small wells by a method called discontinuous dewetting21, 22. Another option is to send liquids through paths at the interface between two substrate slabs and then isolating portions of the liquid by moving one slab with respect to the other in a device called the SlipChip23. These methods are appealing in their simplicity but direct fluid contact with the substrate surface could interfere with certain applications and prevents the removal of solutions in their entirety from the wells. Further possibilities include the separation of volumes using capillary valves or in enlarged nodules along a microfluidic channel24, 25. Here, the fluid is not necessarily in contact with the channel walls, yet there is a disadvantage in that it isn’t possible to release a selected digitized volume independent of the other digitized volumes. Therefore, with these two techniques, one could not conduct downstream analysis of identified samples of interest. Additionally, it may be difficult to vary the surface-to-volume ratio of the digitized volumes across a wide range, a capability that is important for certain applications such as screening for polymorphs by varying solvent evaporation rate.
To address the need for digitizing sample volumes into an array format, this paper describes an approach that is simple, robust, does not incur any sample loss during the digitization step, and forms sample volumes at pre-determined locations with pre-defined volumes. Sample formation is not based on a steady state method. The discretized volumes can be manipulated by both the addition of reagents and the removal of samples for further analysis. We believe this easy method will be useful in a broad range of applications, such as digital PCR and cell-based assays. As a demonstration, we applied our device towards the study of polymorphs formed during small-molecule crystallization.
RESULTS AND DISCUSSION
For digitizing samples, we were interested in utilizing designs that contain a main channel with adjacent side cavities (for formation and retention of small volumes) so that once formed, the discretized samples would not impede flow in the main-channel. Such a design enables the main channel to permit access to the side cavities: reagents could be added through the main channel to individual cavities with samples and discretized aliquots could also be removed from the cavities and advanced down the main channel for additional analysis. We investigated a number of designs to see if they met the following requirements: (1) the digitization process had to be simple, robust, and with minimal or no fluidic control, (2) the sample had to be digitized with 100% efficiency without any sample loss, and (3) the digitized sample had to be easy to remove for downstream manipulation and analysis. To satisfy these requirements, we found the channel system had to be primed with an immiscible fluid prior to introducing the aqueous sample phase. We have developed two designs to meet our requirements: one that involved side cavities that were taller than the main channel (Figure 1a-1d) and the other that was shaped like a Greek key (Figure 1e-1h). We found the design with tall side cavities to be particularly simple and useful, and consequently selected this design to present our parametric studies later in the paper.
Figure 1. Schematic and sequence of images showing two microfluidic designs used for digitization of an aqueous sample.

a) Schematic of the digitization platform where the main channel was half the height of the side chambers (shown here as 50 and 100μm, respectively). b) The chip was first primed with an immiscible phase. c) The sample phase was then slowly introduced into the channel. d) The immiscible phase flowed through the channel a second time. In this step, sample phase was displaced from the main channel yet remained in the side chambers, discretizing the sample phase into a volume that was defined by the geometric dimensions of the side cavities. The experimental time sequences (c and d) were made using 100μM Alexa488 as the sample phase and light mineral oil with 0.01% Span 80 as the immiscible phase. The scale bar corresponds to 200μm. e) Schematic of our Greek key-shaped digitization platform. The main channel was the same height as the side chambers. Discretization using this design followed the same two initial steps. f) The channel was primed with immiscible phase, and g) The sample phase was slowly introduced. h) When the sample phase flow was ceased, digitization occured. The scale bar corresponds to 250μm. i) An array containing 810 digitized samples with inset displaying a close-up of 90. The sample consisted of blue food coloring diluted in half with purified water and the immiscible phase was light mineral oil with 0.01% Span 80. Both scale bars correspond to 600 μm.
1) Operation of the Device
Digitization of Sample Volume
Our chip design with tall side chambers consisted of two inlets feeding into a main channel that was decorated with side cavities roughly twice the height of the main channel (Figure 1a). Unless otherwise specified, the height of the main channel was 50μm and that of side cavities was 100μm. The width of the main channel was 100μm. Rectangular side cavities were 200μm in length along the main channel and 125μm in depth, with an ~10-μm overlap with the main channel. The overlap ensured that the side cavities made contact with the main channel during fabrication.
The second channel design resembling a Greek-key motif also consisted of a main channel decorated with side cavities (Figure 1e). In this design, the main channel and sample cavities were the same height (40μm). The width of the main channel was 25μm. Side cavities extended 300μm away from the main channel and were 350μm long. The side cavity opening was 250μm wide but 60μm into the cavity entrance, it widened to 350μm. We found it was critical that the opening of the side cavity be larger than the cross-sectional area of the main channel in order for flow from the main channel to be redirected into the cavity. A small drain channel (10–20 μm wide) connected each side cavity (from a region furthest to where the sample phase enters) to the main channel and facilitated the removal of immiscible phase from the side cavity as the sample phase entered.
The sample digitization process is commenced by priming the main channel and side cavities with an immiscible phase (Figure 1b and 1f). Next the sample phase is slowly introduced, entering the main channel as well as the side cavities (Figure 1c and 1g; see video in supplementary material). In the Greek key-motif chip (Fig. 1e), there is usually some droplet break-off and recombination upon sample phase filling of the cavity. This phenomenon can be seen in the experimental time sequence of Figure 1g. Drop-wise filling can occur with the tall rectangular side cavities as well (Figure 1a). However, because successive droplets combine as more aqueous sample phase entered the cavity, this phenomenon does not appear to have a negative effect on the digitization function of the device.
In the Greek key-motif chip, digitization occurs when the flow of the sample phase is ceased (Figure 1h). Sample phase in the main channel migrates or retracts to a nearby side cavity. In this design, the main channel was thin enough so that any aqueous sample phase in this channel would have a high interfacial energy. Migration/retraction of the sample phase from the main channel to a sample cavity reduces the interfacial energy of the system.
When using a chip with tall rectangular side chambers (Figure 1a), additional immiscible phase is sent through the main channel in order to initiate digitization (Figure 1d). The immiscible phase moves slowly through the main channel but does not enter the sample cavities. As the front of the immiscible phase advances, sample contained within the side cavities is isolated and becomes partitioned (see video in supplementary material). In this way, large arrays can be formed quickly and easily. Figure 1i shows an array of 810 side chambers.
In both designs, the volume of the final digitized sample is defined by the dimensions of the side cavities. It is therefore possible to create samples of varying volumes simply by changing the size of the side chambers. If the side chambers are constructed out of some gas-permeable material, such as PDMS, an array of digitized samples exhibiting various sizes and thus rates of solvent evaporation can be generated. As we describe later in the paper, we took advantage of this property and used our sample-digitization chip to weigh the role of kinetics in the balance between thermodynamics and kinetics that ultimately determines the polymorphic distribution in a concomitant system26.
Sample Self-Digitization with 100% Efficiency
An attractive feature of the digitization chip is that it can digitize100% of a sample volume without any loss of sample, provided that the volume of the aqueous sample phase is less than the total volume of the side cavities combined. A plug of aqueous sample phase sandwiched between immiscible phase can move through the main channel and fill each subsequent side chamber until the entire aqueous sample had been digitized (Figure 2). This feature is important in applications where the amount of material available is small (e.g. for certain purified biomolecules or cells, or when assaying a drop of blood from a needle prick) or if the loss of sample could translate into uncertainty in the result of the assay (e.g. for digital PCR in clinical diagnostics).
Figure 2. Digitization of 100% of sample volume without loss.
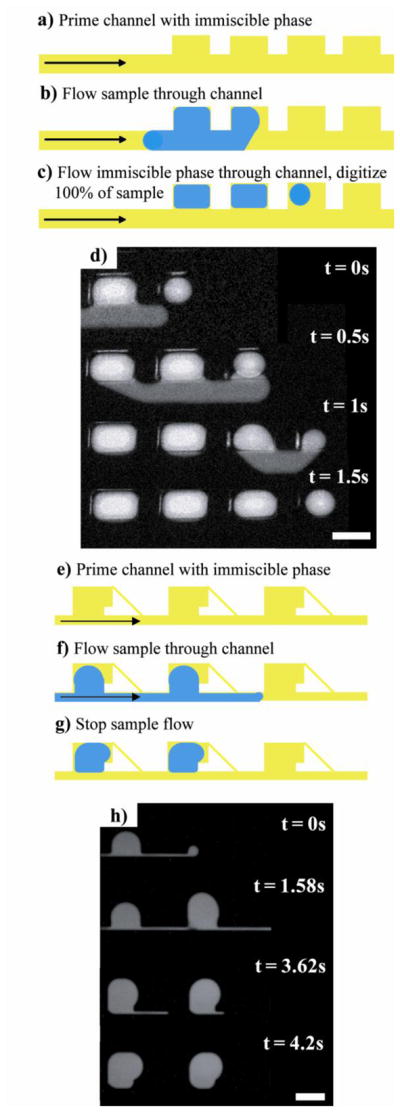
a - c) Schematic depiction of the digitization and retention of 100% of a sample volume using the design with the tall rectangular side cavity. d) An experimental time sequence illustrating this phenomenon. All sample volume moving through the channel continued to fill each subsequent chamber and became discretized as long as the capacity of the chambers (i.e. total volume of the chambers) was larger than the volume of the sample. e - g) Schematic depiction of the digitization and retention of 100% of a sample volume using the Greek key-shaped cavity design. h) An experimental time sequence illustrating this phenomenon. The images show the fluorescence of the sample phase (100μM Alexa488). The immiscible phase was light mineral oil with 0.01% Span 80. The scale bars correspond to 200μm (d) and 250μm (h), respectively.
Manipulation of Digitized Samples
Using our chip it is possible to remove individual samples from the side cavities for downstream analysis either in full (Figure 3a) or in part (Figure 3b). We have also demonstrated sample phase removal from all cavities by simply flowing additional immiscible phase through the main channel at a fast rate (Supplementary Figure 1). We found that connecting small external access channels to the side cavities provided a straightforward means of addressing each individual sample. The external access channels had a small cross-sectional area with a high resistance to flow to ensure that the samples within the chambers would not enter these channels during the digitization process. External access channels were connected to separate fluid inlets and outlets that were independently controlled. If desired, pressure could be applied to the fluid in the external access channels, displacing immiscible phase from the external channel into the side cavity and pushing the digitized sample from the side cavity into the main flow channel. As long as a low flow rate of about 1–2 μL/min was established in the main flow channel, the dislodged sample moved in the direction of flow. By varying the pressure applied to the external access channels and the flow rate in the main channel, we were able to remove entire digitized samples (Figure 3a), partition one sample into multiple aliquots, or remove only a portion of a given digitized sample (Figure 3b). To partition a sample into aliquots or to remove only a portion of a digitized sample, syringe pumps were employed to define a specific ratio of flow rates between the main channel and the access channels that are connected to the side cavities. We were also able to introduce additional reagents to the digitized samples by introducing reagents into the main channel (Supplementary Figure 2). As the immiscible phase was displaced from the main channel by the aqueous reagent phase, the reagent phase fused with aqueous samples in the side cavities. The digitized samples could be later re-partitioned by flowing immiscible phase through the main channel. This phenomenon was observed with all of the aqueous sample phases that were evaluated.
Figure 3. Selective removal of digitized sample volumes.

a) Images showing the complete removal of sample volumes. The sample phase was yellow food coloring and the immiscible phase was light mineral oil with 0.01% Span 80. The flow rate in the main channel was 1μL/min. The external access channels were connected to oil-filled syringes that were operated by hand. b) Images showing the partial removal of a sample volume. Controlled portions of a selected sample could be released by defining the ratio of the flow rate in the main channel to the flow rate in the external access channel. The sample phase was blue food coloring diluted in half with purified water and the immiscible phase was light mineral oil with 0.01% Span 80. The flow rate in the main channel was 1μL/min. The external access channels were connected to oil-filled syringes that were operated by hand. The scale bars correspond to 100μm.
2) The Effect of Varying Physical Parameters Involved in Sample-Volume Digitization
Flow Rate
The volume of the final digitized sample was qualitatively observed to be lower when the sample phase was introduced at a faster flow rate. Closer inspection revealed that this was a reproducible trend (Figure 4). A sample phase moving at faster rates never filled the sample cavity in its entirety (Figure 4 left column) and therefore the volume of the digitized sample decreased with increasing flow rate. We correlated this experimentally observed behavior with simulations using Fluent (Figure 4e). The digitization process was simulated using the design with the tall cavities (Figure 1a) at four different speeds: 0.1, 1, 2.5, 5, and 10mm/s. The results of the simulations mimicked our experimental observations.
Figure 4. Dependence of the digitized sample volume on the aqueous-phase flow rate.

In panels a), b), c), and d) the aqueous phase flow rates were 0.2, 1.1, 6.9 and 8.3 mm/s, respectively. Corresponding images in the column on the right indicate that the volume of the digitized sample was inversely proportional to the aqueous-phase flow rate. The aqueous phase in these images was 100μM 5,6-carboxyflourescein and the oil phase was light mineral oil with 0.01% Span 80. The scale bar corresponds to 200μm. e) The effect of aqueous-phase flow rate was studied using CFD simulations. These images display the final digitized sample volumes generated with different flow rates. As the speed increased from 1 to 10 mm/s, the resultant volume clearly decreased, in agreement with the experimentally observed trend. Blue in schematic denotes aqueous sample phase and yellow denotes immiscible phase.
A bulge of the digitized samples into the main channel was observed in some of our simulations. This bulge could be attributed to the coarse-mesh size used in the simulation. When a narrower mesh size was used, this sample bulge was no longer present (Supplementary Figure 3, RVF > 1 is indicative of a bulge). Although studies with finer mesh sizes would certainly increase the accuracy and direct comparability to the chip experiments, we found them to be impractical, given the large number of simulations needed to characterize the digitization parameters. For example, each simulation depicted in Figure 4e used a mesh size of 5μm and took 6–10 hours to complete. If we use a finer mesh size of 2.5μm, the run time would increase 8-fold to 48–80 hours. The purpose of the simulations was to correlate and help us understand our experimental findings and we were able to do that, and confirm our experimental trends, with a mesh size of 5μm.
Substrate Contact Angle
Prior to using the chips for sample digitization experiments, we baked our freshly sealed PDMS chips in a 115°C oven for 2 days. The baking produced a hydrophobic PDMS surface (≥ 90°) lining the channels and side chambers. To examine the effect of a more hydrophilic surface, we carried out a digitization experiment immediately after sealing the oxidized PDMS chip to glass (Figure 5). Interestingly, the aqueous sample was not able to fully occupy the side cavities as it moved through the chip; more importantly, the aqueous sample was not retained in the side cavities when the immiscible phase subsequently flowed through the channel to digitize the aqueous sample. This result was surprising; we would have expected the aqueous fluid to move easily along the walls of the hydrophilic channel and fill the side cavities.
Figure 5. Dependence of sample-volume digitization on the contact angle (CA) of the substrate.

a), b), and c) are schematic illustrations showing the lack of digitization of a sample phase when the substrate had a hydrophilic surface. Blue in schematic denotes aqueous sample phase and yellow denotes immiscible phase. Below this schematic, b) and c) are experimental time sequences that correspond to the processes depicted in the illustration. Digitization did not occur in a freshly oxidized PDMS channel. The sample phase introduced into the device in panel b) was not retained during subsequent immiscible phase flow shown in c). The experimental images were obtained using a sample phase consisting of 100μM 5,6-carboxyfluorescein; the immiscible phase was light mineral oil with 0.01% Span 80. The scale bar corresponds to 200μm. d) Images of resultant digitized samples from CFD simulations with three different substrate contact angles. Here the contact angle was given its usual definition, that is, the angle measured between the substrate surface and the tangent line to the upper surface of a water droplet placed on top of the substrate. The digitized sample volume increased with contact angle of the substrate. At or below a contact angle of ~120°, digitization of sample volume did not occur, in agreement with our experimental observations.
One possible explanation for the observed behavior is that it was difficult for the oil to exit the sample chamber because the aqueous sample phase was coating the walls of the channel. In other words, aqueous phase coating of the channel walls restricted immiscible phase flow out of the chamber. If this were true, it would imply that a thin layer of hydrophobic immiscible phase must exist between the aqueous sample phase and the channel walls in order for the sample phase to replace immiscible phase inside of the side cavity27,28, 29. To confirm this hypothesis, we simulated the effect of changing the substrate contact angle on the digitization process (Figure 5d). Indeed, we found that sample discretization did not occur at low contact angles, in agreement with our experimental observation. Similar to the experiments, the sample phase never fully entered the side chamber when the substrate had a low contact angle. Furthermore, the volume of the discretized sample was seen to increase as the substrate contact angle was raised.
Interfacial Tension
Interfacial tension is expected to be an important parameter in determining the digitization dynamics. Therefore, we conducted a series of simulations in which the interfacial tension between the two phases was varied while keeping other physical parameters, such as density and viscosity of the immiscible phase, constant (Figure 6a). As a reference, 49.255 mN/m was an estimate of the interfacial energy between an aqueous sample phase and an immiscible phase of light mineral oil without surfactant30.
Figure 6. Dependence of sample-volume digitization on the interfacial tension and the viscosity of the immiscible phase.

Images of the final digitized sample volumes were generated from CFD simulations. a) Light mineral oil (δ = 862.0 kg m−3, μ = 0.03 kg m−1 s−1) was used as the immiscible phase and water was used as the sample phase. Interfacial tension was the only condition to vary between each successive simulation. b) Volume of digitized sample decreases with increasing viscosity of the oil phase. Water was used as the sample phase.
As the interfacial tension was increased, the volume of the discretized sample increased as well. One explanation for this trend is that in a system with a high interfacial energy, the overall energy of the system is somewhat reduced when the sample phase enters the two-layer side cavities. Since the cavity is taller than the main channel, the sample phase surrounded by the immiscible phase can take on a more spherical shape than in the main channel, thereby minimizing the surface area and thus surface energy. When the interfacial tension was high, the sample phase was quite stable in the cavity as it would have to deform and increase its surface area to exit the cavity. When the interfacial tension was low (2 mN/m), deformation of the interface was facile and would likely require comparatively little energy for sample phase to be pulled out from the side cavity.
It is also possible that the stability of the discretized sample once formed contributed to the observed trend. A sample of smaller volume, as was observed in the 20mN/m simulation, has a higher surface area to volume ratio and therefore would be more stable in this lower interfacial tension system than in one of higher interfacial tension systems, such as the 60 mN/m one.
We experimentally observed that the addition of a small amount of surfactant (0.01 – 0.05% w/w) facilitated sample digitization and retention in the side cavities. Without a surfactant, we found many of the cavities were left empty or partially filled. Even though sample phase entered and filled the cavities, it had difficulty partitioning from the bulk sample volume upon flowing the immiscible phase into the main channel. Adding surfactant beyond 0.01 – 0.05% w/w resulted in an increased occurrence of the drop-wise filling of the side cavities that was described in the —Digitization of Sample Volumes section.
Identity and Viscosity of the Immiscible Phase
Different immiscible phases can be used in the digitization chip. We successfully digitized aqueous fluids using light mineral oil with 0.01% (and 0.05%) Span 80 (30cSt), or 0.01% octaethylene glycol monodecyl ether, or 0.01% tetraethylene glycol monodecyl ether, as well as Fluorinert FC-40, AS-4 (6cSt), AS-4 with 0.05% Span 80, AR-20, 50cSt Silicone Oil with 0.05% Span 80, and 100cSt Silicone Oil with 0.05% Span 80. Digitization behavior did not seem to depend greatly on the identity of the immiscible phase as long as a small amount of surfactant was present.
We also investigated the effect of viscosity of the immiscible phase in simulations where we imported parameters for light mineral oil and light white mineral oil, as well as 50 and 75 cSt PDMS oils (Figure 6b). Interestingly, the higher viscosity oils resulted in smaller discretized sample volumes. When the viscosity of the PDMS oil was 75 cSt, no sample phase was left in the cavity. If viscosity is increased while interfacial tension and flow rate are held constant, two immiscible fluids will continue to flow alongside one another as they move downstream. It is only when the interfacial energy is high enough with respect to the viscosity that a reduction in the interfacial area between the two fluids will sufficiently lower the energy of the system to overcome the viscous forces that drive the sample/immiscible phase interface downstream31. Therefore, digitization was not expected to occur when the immiscible phase viscosity was very high relative to the interfacial tension.
Capillary Number Captures the Effect of Flow Rate, Interfacial Tension, and Viscosity
The formation of droplets is often dependent on the capillary number (Ca), a dimensionless quantity that represents the ratio of the viscous to the interfacial forces:
where U (m s−1) is the flow velocity, μ (kg m−1 s−1) the dynamic viscosity of the immiscible phase, and γ (kg s−2) the interfacial tension between the sample and immiscible phases.
To better understand the effect of the different physical parameters described above on the digitization behavior of sample in our device, we plotted the retained volume fraction (RVF; this is the volume of the digitized sample divided by the volume of the side cavity) as a function of Ca for three different substrate contact angles: 120, 150, and 175° (Figure 7). We generated this data using simulations. As anticipated from the dependence of digitization on contact angle, we didn’t see any sample digitization when the contact angle was 120° (Figure 7a). When the contact angle was 150°, samples were only digitized at Ca of 0.009 and below (Figure 7b). The threshold at Ca ~ 0.009 was quite sharp. It has been reported previously that Ca under 0.01 favor the partitioning of nanoliter-sized plugs of an aqueous solution in an immiscible phase using a microfluidic T-junction32. Inverse proportionality of sample volume to Ca has also been observed previously33. This variation was even more pronounced when the contact angle was 175° (Figure 7c) where a large range of sample volumes was observed between Ca = 0.0008 to Ca = 0.0125. Therefore, given a fixed geometry for the side cavities and channel in a sufficiently hydrophobic chip, digitization dynamics is governed largely by the capillary number.
Figure 7. Dependence of digitized sample volume on the capillary number (Ca) at three different contact angles of the substrate.
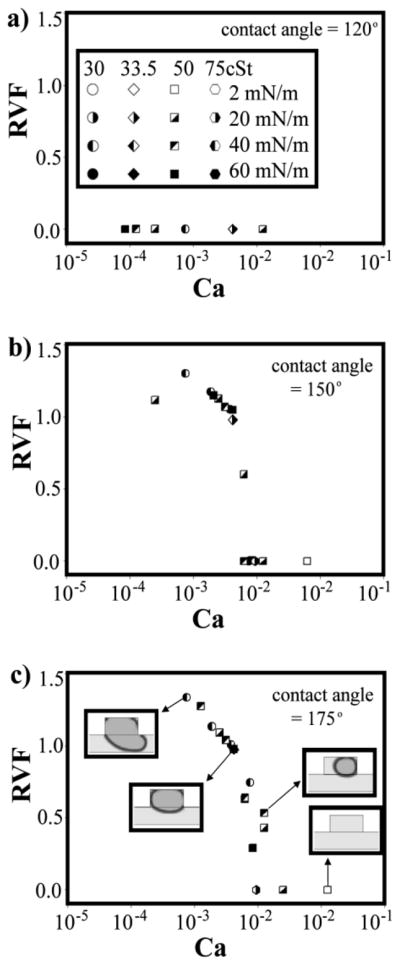
Digitized sample volume divided by the volume of the side chamber, defined as the RVF, plotted as a function of Ca at contact angles of a) 120°, b) 150° and c) 175°. The contact angle here was given its usual definition, that is, the angle measured between the substrate surface and the tangent line to the upper surface of a water droplet placed on top of the substrate. Note the effect of contact angle on sample-volume retention where the surface needs to be sufficiently hydrophobic (α ≫ 90°) to retain the sample volume inside the side cavity. Also note the abrupt decrease in RVF (Vdrop/Vchamber) — above a certain Ca, sample digitization no longer occurs. Sample-volume digitization is a low-Ca phenomenon. The shape and shading of each data point corresponds to the interfacial tension and viscosity of the immiscible phase.
Geometry of the Sample Cavity and Main Channel
In addition to capillary number—which summarizes the effect of flow rate, interfacial tension, and viscosity—we found digitization dynamics were also affected by the dimensions of the sample chamber and the main channel. In particular, we found two ratios to be important: (1) the aspect ratio between the cross-sectional area of the chamber opening and the cross-sectional area of the main channel, and (2) the aspect ratio between the total surface area of the chamber and the cross-sectional area of the main channel.
We investigated sample digitization in a series of forty different side chambers with various dimensions (Figure 8; see Supplementary Figure 4 for a list). For these experiments, the sample phase was 1nM phosphate buffer and the immiscible phase was light mineral oil with 0.01% Span 80. The dimensions varied were d2, d3 and h2 (Figure 8a). Because the exact volume of the sample phase in the side cavity was experimentally difficult to measure, we counted the fraction of filled cavities for each geometric variant. In a few cases where partial filling was observed, the volume of the discretized sample was estimated from optical micrographs and rounded to the nearest tenth of a completely filled sample cavity.
Figure 8. Dependence of sample-volume digitization on the geometry of the side chamber with respect to the main channel.

a) Definition of the device dimensions. b) Dependence of sample digitization on the aspect ratio between the cross-sectional area of the chamber opening and the cross-sectional area of the main channel. When the ratio of these two areas was equal, different fractions of filled cavities were observed. At a ratio of greater than 1, sample digitization generally occurred successfully; at a ratio of less than 1, sample-volume digitization was absent. c) Dependence of sample digitization on the aspect ratio between the total surface area of the chamber and the cross-sectional area of the main channel. As the surface area of the chamber increased, a clear trend towards fully filled cavities was observed. Exceptions to the trend depicted in b) and c) were very shallow side chambers with a large opening area; at this extreme, the presence of the side chamber was similar to a slight widening of the main channel, and thus no sample-volume digitization occurred. Images of digitized samples have been highlighted in bold.
The fraction of filled cavities was plotted against the cross-sectional area of the chamber opening divided by the cross-sectional area of the main channel (Figure 8b); the fraction of filled cavities was also plotted versus the total surface area of the chamber divided by the cross-sectional area of the main channel (Figure 8c). The cross-sectional area of the main channel was held constant (4000 μm2; 100 μm wide by 40 μm tall). Both plots resemble a sharp sigmoid function. There was an abrupt increase in the fraction of filled cavities where the area of the cavity opening was equal to the cross-sectional area of the channel (Figure 8b); digitization did not occur if this ratio fell below 1. Above a ratio of 1, digitization was successful with the exception of one design where d2 = 30 μm and d3 = 300 μm. In this case, the area of the cavity opening (12,000 μm2) was much larger than the cross sectional area of the channel (4000 μm2); however, the cavity depth (< 30 μm) was very shallow. From the perspective of the fluids, the presence of a side chamber of these dimensions appeared to be similar to a slightly wider main channel. Therefore, the depth of the cavity must be sufficiently large in order to retain and digitize the sample phase.
In Figure 8c, all three distances defining the size of the cavity were taken into account instead of just d3. The fraction of filled cavities increased with increasing cavity surface area. Data points that deviated from this trend were d2 = 125μm with d3 = 50μm, and d2 = 30μm with d3 = 300μm (again). When d3 = 50, the area of the chamber opening was half of the cross-sectional area of the channel. Therefore, the ratio of the area of the chamber opening to the main-channel cross section was less than 1, and based on observations from Figure 8b, this design was not expected to result in digitization of the sample phase. For the design where d2 = 30μm with d3 = 300μm, it failed to digitize samples for the same reason as described previously in Figure 8b. The smallest value for cavity depth (d2) in which filling was observed was 62.5μm (here the area of the chamber opening was equal to the cross-sectional area of the main channel). Only a fraction of cavities were filled in this case. At a cavity depth d2 = 125μm, every cavity was filled as long as the area of the chamber opening was larger than the cross-sectional area of the chamber.
In summary, the geometric criteria for successful sample digitization were: 1) the chamber opening must have an area that is equal to or larger than the cross-sectional area of the main channel, and 2) the chamber must have sufficient depth (approximately 2/3 the width of the main channel). The first criterion stems from the need for the aqueous sample phase to enter the chamber; if the chamber opening was too small in comparison with the cross-section of the main channel, the aqueous sample did not efficiently enter the chamber. The second criterion is based on the retention and break-off of the sample phase in the chamber: if the chamber was not sufficiently deep, the aqueous sample phase simply could not be retained within it.
3) Separation of the Concomitant Polymorphs of 2,5-Dihydroxybenzoic Acid
Polymorphism, the ability of a solid material to exist in more than one form or crystal structure, is a fascinating phenomenon in which the same molecule can have different physical properties depending on molecular packing and crystal habit or morphology34. Variations in the solid-state properties of a material due to form can have significant implications in the function of crystalline products, such as pharmaceutical dissolution rates and ease of forming tablets35. For this reason, all possible polymorphs of a crystalline product must be identified and characterized for the formulation of drugs. We are interested in the utility of our digitization chip in these efforts.
Of particular interest to us is concomitant polymorphism26, where two polymorphs grow at the same time under the same conditions. The resultant material is a mixture of the two forms. Previously we have studied the concomitant polymorphs of 2,5-dihydroxybenzoic acid (2,5-DHB)36. We observed that crystallization from a saturated 50% aqueous ethanol solution, with 10−3M of the pH indicator dye methyl red, produced a mixture of two polymorphs that could be identified as red needles and purple plates (Forms I and II, respectively). We were curious whether we could separate the growth of these two crystals with our digitization chip. Because the chip is constructed out of PDMS, a gas-permeable material, aliquots of different volumes and surface areas will evaporate at different rates, resulting in an inherent change in the timescale of the crystallization event. During the growth of concomitant polymorphs, there is competition between a kinetic and a thermodynamic product. Therefore, it seemed likely that we could produce different results from digitized samples that formed in side cavities of different size.
The digitization chip is advantageous in that we could easily screen a large number of different sample volumes and surface areas simply using pre-defined side chambers with a range of sizes and shapes. For this experiment, we used the same forty cavities of differing dimensions as we did for the experiments described in Figure 8 (Supplementary Figure 4). We filled our digitization chip of different sized chambers using a saturated solution of 1 × 10−2 M 2,5-DHB and 1 × 10−5 M methyl red in 1:1 EtOH:H2O as the sample phase. The immiscible phase was light mineral oil with 0.05% Span 80. After being kept at room temperature for 2–3 days (all tubing was removed following digitization and the chip was placed on a shelf), a solid mass was observed in each chamber and no liquid remained. The solid material was found to be crystalline when viewed between crossed polarizers. In some cases, a single crystal was observed (Figure 9b and 9c). Upon close inspection, these crystals resembled the two known polymorphs of 2,5-DHB36, 37. The crystals that had grown from the larger-volume side cavities were red, and their interfacial angles and overall morphology resembled that of Form I (Figure 9b). Purple crystals in the smaller chambers displayed a distinct apex that is expressed by Form II at the intersection of the (1 22̄) and (1̄ 2 2) faces (Figure 9c). The purple color of these crystals was consistent with the previously reported color of methyl red inside of Form II 2,5-DHB crystals; a red color of the dye has been reported in Form I36.
Figure 9. Separation of otherwise concomitant polymorphs of 2,5-dihydroxybenzoic acid (2,5-DHB).

a) Images of crystals that were grown by slow evaporation in a sample self-digitization chip; solvent evaporation rates were adjusted by varying the volume and surface area of the sample compartments. The sample was a 50% aqueous ethanol solution containing 10 mM 2,5-DHB and 10 μM methyl red; immiscible phase was light mineral oil with 0.05% Span 80. Crystals grew in the chip at room temperature over 2–3 days. The scale bar corresponds to 100μm. b) and c) are representative polymorphic crystals grown in the chip. d) Raman spectra of 2,5-DHB. Although there was some fluorescence, spectra of crystals grown inside of the self-digitization device can be clearly correlated to the reference spectra for the two forms.
To confirm our visual observations, we collected Raman spectra of the crystals inside the chambers and compared them to reference spectra for each polymorph (Figure 9d). The Form I reference spectrum in green exhibits characteristic peaks at 1327 cm−1 from phenol CO stretching or OH in-plane deformations and at 287 cm−1 that is likely a skeletal vibration. Form II (in red) has distinct features at 1665 cm−1 from an intra-molecular hydrogen-bonded carboxylic acid and at 1199 cm−1 due to either phenol CO stretching or OH in-plane deformations. Raman spectra of crystals inside the chip were collected by sending the Raman laser through the glass coverslip (130 – 170μm thick). Although there was some fluorescence (evidenced by the elevated baselines), we were able to collect spectra and correlate them to references for either form.
Form I was found in the larger chambers, confirming our visual observations. Out of six crystallization experiments, Form I was found in 11.4 and 10.17-nL capacity cavities four times, in 9.9 and 9.12-nL cavities twice, and always in the case of the fused 2-nL chambers that had a total capacity of approximately 0.104μL38. Raman spectra consistent with Form II were collected in all smaller volume chambers, except for some cavities with a volume below 2nL where the crystals melted during the Raman experiment and produced spectra of poor quality. Single crystals in these smaller compartments visibly resembled Form II. Figure 9d shows representative Raman spectra of crystals found in the larger and smaller cavities. We believe the observed variation in crossover volume between the occurrence of one crystal form over the other was due to differences in the evaporation rate caused by either variations in our laboratory’s temperature and humidity, inconsistencies in the thickness of the PDMS, or a combination of both of these effects.
Form I, melting between 214.85 and 216.85 °C, is the more stable polymorph (Form II melts between 207.85 and 209.85 °C). Therefore, it was not surprising to find Form II in the smaller cavities where faster evaporation rates gave rise to high super-saturation quickly resulting in growth of the less stable polymorph to be energetically favored. Rapid evaporation from small volumes has previously been observed to yield less stable crystal forms39. There has even been some evidence that a less stable crystal actually becomes more stable in a confined environment40.
CONCLUSIONS
We described a robust and versatile approach to digitize 100% of a sample volume that relies on a persistent fluidic behavior rather than complex fluidic controls. The procedure was simple: prime chip with oil, introduce aqueous sample, then follow with oil again. The main channel acted as the principal conduit for the chip, making it possible to select and remove the digitized aqueous samples for further manipulations or analysis. We characterized the effect of different physical parameters on the digitization behavior in detail and demonstrated how these physical parameters could be adjusted to fine-tune the volume of a digitized sample.
We highlighted the ease of generating digitized samples of different volumes to separate the growth of otherwise concomitant polymorphs. In doing so, we added to a growing body of evidence that crystallization in confined environments can produce the same results as crystallization from a variety of solvents41, 42. This observation has the potential to simplify the way in which a typical polymorph screen is conducted35, as well as significantly reduce the amount of material required to do so. We anticipate our chip for sample self-digitization will be useful in polymorph screens that must be conducted in industries where the final product is a crystalline material.
MATERIALS AND METHODS
Chip Design and Fabrication
Microfluidic chips were generated by replicating poly(dimethylsiloxane) (PDMS, (Sylgard 184) bought from Dow Corning Co. (Midland, MI).) from a SU-8-on-silicon master fabricated using two-layer photolithography. This procedure has been extensively described elsewhere43. Briefly, channel designs were drawn in Autocad 2007 (Autodesk, San Rafael, CA) and high-resolution photomasks bearing these patterns were printed by a photoplotter. (Fineline Imaging, Colorado Springs, CO). Prior to photolithography, SU-8-3050 photoresist (Microchem, Newton, MA) was spin-coated onto a silicon wafer at 40- or 50-μm thickness to form the first layer of features on the silicon wafer (the main channel in Figure 1a-d and the entire design in Figure 1e-h). The photomask was then placed over the SU-8 coated wafer and exposed to UV light with a mask aligner (Newport, Irvine, CA). Following exposure, curing, and development of these features, SU-8-3050 was spin-coated onto the same wafer again at 100-μm thickness (unless otherwise specified), and exposed using a second photomask that defined the taller features of the SU-8-on-silicon master. In Figure 1a-d the second layer corresponds to the sample cavities and in Figure 1e-h a second layer does not exist. The second photomask and features of the first layer of photoresist were aligned and exposed to UV light. After curing and development, the finished two-layer master was coated with (tridecafluoro-1,1,2,2-tectrhydrooctyl)trichlorosilane (Gelest, Morrisville, PA) by gas-phase deposition to prevent PDMS from sticking during molding and peel-off. The molded PDMS replica was punched with access holes (16 G) for fluid inlets and outlets, and then the PDMS mold was permanently sealed to glass coverslips that were purchased from Electron Microscopy Sciences (Hatfield, PA). The PDMS-glass chips were exposed to oxygen plasma. To ensure thorough curing, the coated PDMS layer was left in a 115°C oven for at least 1day.
Materials Used in the Self-Digitization Chip
The self-digitization chip was primed with immiscible phases that were composed of light mineral oil and light white mineral oil (Mineral Oil, Embryo Tested, Sterile Filtered, Sigma-Aldrich, St. Louis, MO), Fluorinert (FC-40, 3-M, St. Paul, MN), AS-4 (Fluka, Buchs, Switzerland), AR-20 (Fluka), 50cSt PDMS oil, or 100cSt PDMS oil (Poly(dimethylsiloxane) 200 fluid, Sigma-Aldrich). Where specified, the immiscible phase was mixed with SPAN 80 (sorbitan mono-oleate purchased from Sigma-Aldrich), octaethylene glycol monodecyl ether (Sigma-Aldrich), and tetraethylene glycol monodecyl ether(Sigma-Aldrich). Unless otherwise noted, the sample phase consisted of 1nM phosphate buffer. Other sample phases were 100μM Alexa488 (Invitrogen, Carlsbad, CA), 100μM 5,6-carboxyflourescein (Molecular Probes, Eugene, OR), yellow food coloring, blue food coloring (Kroger, Cincinnati, OH) diluted by half, and 1 × 10−2 M 2,5-dihydroxybenzoic acid (Sigma-Aldrich) and 1 × 10−5 M methyl red (Color Index #13020, Sigma-Aldrich) in 1:1 EtOH:H2O.
Experimental Setup
To reach the microchannels, access holes to the channels were punched with a 16-gauge needle prior to sealing the chip. After removal from the 115°C oven, polyethylene tubing (PE 100, Becton Dickinson, Franklin Lakes, NJ) was inserted into the access holes. The tubing leading to one of the inlets was attached to a 3-mL plastic syringe (Becton Dickinson) that was filled with the immiscible phase. Pressure was applied until all of the sample cavities were filled and no air bubbles remained. After 30 to 60 minutes, sample phase was introduced using a 50-μL glass syringe (Hamilton, Reno, NV) with a micrometer connected to the plunger. The syringe and the tubing were filled with mineral oil, except for the very tip of the tubing containing the sample phase. This setup enabled us to work with and dispense very small volumes (less than or equal to the sum of volumes for all side cavities connected to an individual main channel). In a typical experiment, we were able to introduce volumes as small as 7nL to the digitization chip (see Figure 2). The tubing connected to the sample phase inlet during filling was removed and the tubing connected to glass syringe was inserted in its place, taking care not to introduce any air bubbles into the system. The sample phase was pushed forward with fine control using the micrometer.
Discretization experiments were visualized using the AZ-100 zoom microscope (Nikon Instruments, Melville, NY). The light source was an Intensilight C-HGFI (Nikon). The Brightline FTIC-3540B-000 filter set from Semrock (Rochester, NY) was used when imaging the fluorescence of either Alexa488 or 5,6-carboxyfluorescein. A Prosilica (Vancouver, BC) GC640 CMOS fast camera controlled with a program written in LabView (National Instruments, Austin, TX) collected bright-field images and time sequences. A Prosilica GC660 CCD camera was used for the fluorescence experiments.
To remove samples from the side cavities, a syringe pump (KD Scientific, Holliston, MA) established a low flow rate of 1 – 2 μL/min of immiscible phase through the main channel. External access channels were connected to syringes filled with immiscible phase. These syringes were operated either by hand or controlled using additional syringe pumps. Sample phase flow rates were calculated by measuring the physical advancement of the immiscible/sample phase interface between individual frames of movies collected using the GC640 camera. The flow rate was calculated when the interface was moving in between side cavities, not during the process of filling a cavity.
Raman spectra were collected using a Renishaw inVia Raman Microscope equipped with a 785-nm diode laser (Wotton-under-Edge, Gloucestershire, UK). The reference spectra belonged to large single crystals that had been indexed to confirm polymorphic identity. Crystals in chambers were analyzed using a 100× objective at 100% or 10% power with a 10s or 60s accumulation time, respectively. Spectra were analyzed using the Renishaw WiRE 2.0 software.
Computational Fluid Dynamics (CFD)
Computational analyses of the various process parameters (interfacial tension, contact angle, linear flow velocities) that are important for self-digitization of sample volume were conducted using a commercial computational fluid dynamics (CFD) package (Fluent, Version 6.3.26; Fluent Inc.; Ann Arbor, MI). The filling was simulated using a side chamber with a volume of 0.25 nl (length × width × height: 100 μm × 50 μm × 50 μm) adjacent to a main channel (width × height: 50 μm × 25 μm) that was filled with a plug of aqueous disperse phase that had a volume of at least twice the volume of the side chamber. The channel and side cavity geometry were converted into finite elements using a hexahedral meshing strategy that produced 10,533 nodes and 8,100 cells (mesh size of 5.0 μm between node points). Geometric designs that were a 1:1 replica of the on-chip design were studied as well.
The different oils used in the experiments were applied in the CFD analysis. These oils were: (1) light mineral oil (Sigma M8410; δ = 862.0 kg m−3, μ = 0.03 kg m−1 s−1), (2) light white mineral oil (Sigma 330779; δ = 838.0 kg m−3, μ = 0.0335 kg m−1 s−1), (3) silicone oil (Sigma 378356; δ = 980.0 kg m−3, μ = 0.05 kg m−1 s−1), and (4) an artificial oil with higher viscosity (δ = 980.0 kg m−3, μ = 0.075 kg m−1 s−1). Water (δ = 998.0 kg m−3, μ = 1.003 × 10−3 kg m−1 s−1) was used in all simulations as the disperse or sample phase. The model solver was defined as pressure-based, with a 3-dimensional and implicit formulation, unsteady with non-iterative time advancement, with an absolute velocity formulation, and with first-order implicit unsteady formulation. The multiphase model was set to a Volume of Fluid (VOF) solver with two phases, explicit VOF scheme, and a Courant number of 0.25. The phase interaction was defined with interfacial tension values ranging from 2.0 to 60 mN m−1. Subsequently, depending on the parameter to be investigated, the boundary conditions were set as follows: the inlet was defined by a flat velocity profile with a linear flow velocity ranging from 0.1 to 10.0 mm s−1; the outlet was defined as outflow with a constant pressure (POutlet = Patm = 101,325 Pa); the walls enclosing the channel and side cavity were set to have contact angles ranging from 120° to 175°. A water plug was defined through the Fluent —adapt -tool, while all other nodes of the mesh were defined with an initial water volume fraction of 0. The fluid flow profile and the volume fractions were then simulated with fractional steps for the pressure-velocity coupling. The spatial discretization was implemented by a first order upwind scheme for the momentum, a PREssure STagering Option (PRESTO!) set for the pressure, and the volume fraction was discretized using the Geo-Reconstruct option. All residual tolerances were set to 0.001.
Supplementary Material
Acknowledgments
We are grateful to NSF (CHE-0844688) and NIH (EB005197) for support of this work. Part of this work was conducted at the University of Washington NanoTech User Facility, a member of the NSF National Nanotechnology Infrastructure Network (NNIN).
Footnotes
SUPPORTING INFORMATION AVAILABLE: (1) Videos of experiments showing the self-digitization of sample volumes (CavityFilling.avi and Digitization.avi); (2) A sequence of experimental images showing the drop-wise removal of digitized sample volumes using elevated flow rate in the main channel; (3) A sequence of experimental images showing the addition of a reagent to digitized samples and subsequent re-digitization; (4) CFD characterizations showing the effect of mesh size (and simulation time) on the final digitized sample volume; (5) A list of all side-chamber dimensions that were evaluated for filling efficiency. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Tawfik DS, Griffiths AD. Nat Biotechnol. 1998;16:652–656. doi: 10.1038/nbt0798-652. [DOI] [PubMed] [Google Scholar]
- 2.Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen ZT, Dewell SB, Du L, Fierro JM, Gomes XV, Godwin BC, He W, Helgesen S, Ho CH, Irzyk GP, Jando SC, Alenquer MLI, Jarvie TP, Jirage KB, Kim JB, Knight JR, Lanza JR, Leamon JH, Lefkowitz SM, Lei M, Li J, Lohman KL, Lu H, Makhijani VB, McDade KE, McKenna MP, Myers EW, Nickerson E, Nobile JR, Plant R, Puc BP, Ronan MT, Roth GT, Sarkis GJ, Simons JF, Simpson JW, Srinivasan M, Tartaro KR, Tomasz A, Vogt KA, Volkmer GA, Wang SH, Wang Y, Weiner MP, Yu PG, Begley RF, Rothberg JM. Nature. 2005;437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shendure J, Porreca GJ, Reppas NB, Lin XX, McCutcheon JP, Rosenbaum AM, Wang MD, Zhang K, Mitra RD, Church GM. Science. 2005;309:1728–1732. doi: 10.1126/science.1117389. [DOI] [PubMed] [Google Scholar]
- 4.Rose D. Drug Discov Today. 1999;4:411–419. doi: 10.1016/s1359-6446(99)01388-4. [DOI] [PubMed] [Google Scholar]
- 5.Morrison T, Hurley J, Garcia J, Yoder K, Katz A, Roberts D, Cho J, Kanigan T, Ilyin SE, Horowitz D, Dixon JM, Brenan CJH. Nucleic Acids Res. 2006;34:9. doi: 10.1093/nar/gkl639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Litborn E, Emmer A, Roeraade J. Electrophoresis. 2000;21:91–99. doi: 10.1002/(SICI)1522-2683(20000101)21:1<91::AID-ELPS91>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 7.Clark RA, Hietpas PB, Ewing AG. Anal Chem. 1997;69:259–263. doi: 10.1021/ac970070x. [DOI] [PubMed] [Google Scholar]
- 8.Lynn NS, Henry CS, Dandy DS. Lab on a Chip. 2009;9:1780–1788. doi: 10.1039/b900556k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Unger MA, Chou HP, Thorsen T, Scherer A, Quake SR. Science. 2000;288:113–116. doi: 10.1126/science.288.5463.113. [DOI] [PubMed] [Google Scholar]
- 10.Pollack MG, Fair RB, Shenderov AD. Appl Phys Lett. 2000;77:1725–1726. [Google Scholar]
- 11.Wheeler AR. Science. 2008;322:539–540. doi: 10.1126/science.1165719. [DOI] [PubMed] [Google Scholar]
- 12.Biebuyck HA, Whitesides GM. Langmuir. 1994;10:2790–2793. [Google Scholar]
- 13.Sgro AE, Allen PB, Chiu DT. Anal Chem. 2007;79:4845–4851. doi: 10.1021/ac062458a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lorenz RM, Edgar JS, Jeffries GDM, Chiu DT. Anal Chem. 2006;78:6433–6439. doi: 10.1021/ac060748l. [DOI] [PubMed] [Google Scholar]
- 15.Shim JU, Cristobal G, Link DR, Thorsen T, Jia YW, Piattelli K, Fraden S. J Am Chem Soc. 2007;129:8825–8835. doi: 10.1021/ja071820f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Christopher GF, Anna SL. J Phys D-Appl Phys. 2007;40:R319–R336. [Google Scholar]
- 17.Shah RK, Shum HC, Rowat AC, Lee D, Agresti JJ, Utada AS, Chu LY, Kim JW, Fernandez-Nieves A, Martinez CJ, Weitz DA. Mater Today. 2008;11:18–27. [Google Scholar]
- 18.Garstecki P, Fuerstman MJ, Stone HA, Whitesides GM. Lab on a Chip. 2006;6:437–446. doi: 10.1039/b510841a. [DOI] [PubMed] [Google Scholar]
- 19.Fidalgo LM, Abell C, Huck WTS. Lab on a Chip. 2007;7:984–986. doi: 10.1039/b708091c. [DOI] [PubMed] [Google Scholar]
- 20.Huebner A, Sharma S, Srisa-Art M, Hollfelder F, Edel JB, Demello AJ. Lab on a Chip. 2008;8:1244–1254. doi: 10.1039/b806405a. [DOI] [PubMed] [Google Scholar]
- 21.Jackman RJ, Duffy DC, Ostuni E, Willmore ND, Whitesides GM. Anal Chem. 1998;70:2280–2287. doi: 10.1021/ac971295a. [DOI] [PubMed] [Google Scholar]
- 22.Shen H, Fang Q, Fang ZL. Lab on a Chip. 2006;6:1387–1389. doi: 10.1039/b605332g. [DOI] [PubMed] [Google Scholar]
- 23.Du W, Li L, Nichols KP, Ismagilov RF. Lab on a Chip. 2009;9:2286–2292. doi: 10.1039/b908978k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boukellal H, Selimovic S, Jia YW, Cristobal G, Fraden S. Lab on a Chip. 2009;9:331–338. doi: 10.1039/b808579j. [DOI] [PubMed] [Google Scholar]
- 25.Wu L, Li GP, Xu W, Bachman M. Appl Phys Lett. 2006;89:3. [Google Scholar]
- 26.Bernstein J, Davey RJ, Henck JO. Angew Chem-Int Edit. 1999;38:3441–3461. doi: 10.1002/(sici)1521-3773(19991203)38:23<3440::aid-anie3440>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 27.When the chip is hydrophobic, the immiscible phase preferentially wets the channel walls and therefore there is expected to be a thin layer of immiscible phase in between the aqueous sample phase and the channel wall.
- 28.Tice JD, Song H, Lyon AD, Ismagilov RF. Langmuir. 2003;19:9127–9133. [Google Scholar]
- 29.Bico J, Quere D. J Fluid Mech. 2002;467:101–127. [Google Scholar]
- 30.Stan CA, Tang SKY, Whitesides GM. Anal Chem. 2009;81:2399–2402. doi: 10.1021/ac8026542. [DOI] [PubMed] [Google Scholar]
- 31.Squires TM, Quake SR. Rev Mod Phys. 2005;77:977–1026. [Google Scholar]
- 32.Tice JD, Lyon AD, Ismagilov RF. Anal Chim Acta. 2004;507:73–77. [Google Scholar]
- 33.Christopher GF, Noharuddin NN, Taylor JA, Anna SL. Phys Rev E. 2008;78:12. doi: 10.1103/PhysRevE.78.036317. [DOI] [PubMed] [Google Scholar]
- 34.Bernstein J. Polymorphism in Molecular Crystals. Claredon Press; Oxford, U. K: 2002. [Google Scholar]
- 35.Hilfiker R. Polymorphism: in the Pharmaceutical Industry. Wiley-VCH; Weinheim, Germany: 2006. [Google Scholar]
- 36.Cohen DE, Benedict JB, Morlan B, Chiu DT, Kahr B. Cryst Growth Des. 2007;7:492–495. [Google Scholar]
- 37.Haisa M, Kashino S, Hanada SI, Tanaka K, Okazaki S, Shibagaki M. Acta Crystallogr Sect B-Struct Commun. 1982;38:1480–1485. [Google Scholar]
- 38.Footnote – We attempted to fabricate a digitization chip in which d2 = 125μm, d3 = 200μm, h2 = 122μm, and d4 = 20μm. We were not able to resolve two features that were 122μm high and only 20μm apart and thus our resultant master consisted of 52 fused side cavities. Side cavity filling and sample digitization was successful in this chip.
- 39.Lee AY, Lee IS, Dettet SS, Boerner J, Myerson AS. J Am Chem Soc. 2005;127:14982–14983. doi: 10.1021/ja055416x. [DOI] [PubMed] [Google Scholar]
- 40.Hamilton BD, Hillmyer MA, Ward MD. Cryst Growth Des. 2008;8:3368–3375. [Google Scholar]
- 41.Lee IS, Lee AY, Myerson AS. Pharm Res. 2008;25:960–968. doi: 10.1007/s11095-007-9424-z. [DOI] [PubMed] [Google Scholar]
- 42.Ha JM, Hamilton BD, Hillmyer MA, Ward MD. Cryst Growth Des. 2009;9:4766–4777. [Google Scholar]
- 43.Duffy DC, McDonald JC, Schueller OJA, Whitesides GM. Anal Chem. 1998;70:4974–4984. doi: 10.1021/ac980656z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.