

Abstract
In order to build more accurate models of cells and tissues, the ability to accurate information on the distributions of proteins (and other macromolecules) will become increasingly important. This review describes current progress towards determining and representing protein subcellular patterns so that the information can be used as part of systems biology efforts. Approaches to decomposing an image of the subcellular pattern of a protein give critical information about the fraction of that protein in each of a number of fundamental patterns (e.g., organelles). Methods for learning generative models from images provide a means of capturing the essential properties, and variation in those properties, of cell shape and organelle patterns. The combination of models of fundamental patterns and vectors specifying the fraction of a protein in each of them provide a much better means of communicating subcellular patterns than the descriptive terms that are currently used. Communicating information about subcellular patterns is important not only for systems biology simulations but also for representing results from microscopy experiments, including high content screening and imaging flow cytometry, in a transportable and generalizable manner.
Introduction
Structured information on subcellular location in protein databases, such as Uniprot (1), currently consists only of terms from a restricted vocabulary. These are usually drawn from the Cellular Component portion of the Gene Ontology (GO) (2). The guiding principle behind this approach is that each protein can be assigned to one, or at most a few, distinct subcellular structures whose names are given in the ontology. Assignments may be based on experimental determinations, unsupported assertions in papers, or predictions based on sequence analysis. Some databases, such as LOCATE (3), provide links to papers describing experimental evidence for a given assignment. Most protein databases have at least some GO terms associated with most proteins.
There are three major considerations that currently limit incorporation of information on protein subcellular distributions into spatially realistic cell simulations. The first is the need for accurate models of the structure (and variation in structure) of each subcellular component. The second is the need for information on the distribution of proteins across different components. The third is the need for information on how protein locations change between different conditions or cell types. This review summarizes progress towards addressing these goals through direct analysis of images. The various approaches are depicted for reference in Figure 1.
Figure 1.

Overview of approaches to communicating subcellular distributions. A) When examples are available of each of the patterns expected to be present, supervised approaches can be used. The top path shows a traditional classification approach: the example images are used to train a classifier and then the image to be analyzed is assigned to one of the classes. The lower path illustrates representing the image by a vector showing the fraction of each of the classes it contains. B) When a large collection is available, unsupervised unmixing can find the fundamental patterns that they contain as well as a vector of pattern fractions for each image. Models of each of the fundamental patterns can then be built for each of the fundamental patterns. Synthetic images can be created using the models and vectors (not shown).
A word on the terminology used here. The terms subcellular component or structure are used below rather than organelle since the latter term usually implies the presence of a limiting membrane and many distinguishable components are not membrane bound. Furthermore, a component is considered to be a collective noun, namely, that it can consist of more than one distinct element or object within a given cell.
The discussion below is intended to capture the current state of a field that is in its infancy and hopefully to stimulate the extensive further work that is needed.
Protein distributions across subcellular components
Boolean vectors: GO terms and classifiers
Assignments of GO terms can be represented as a Boolean vector describing whether or not a particular protein is found in each subcellular component (which we can also think of as fundamental patterns). For example, if only the components plasma membrane, endosome, lysosome, Golgi apparatus, endoplasmic reticulum, cytoplasm and nucleus were being considered, [0,1,0,1,0,0,0] would represent a protein present in both endosomes and the Golgi apparatus.
Given that a number of proteins change their distribution between components, and that such changes can play an important role in cell behavior, simply identifying which components a protein is (or may be) contained in is not sufficient for understanding the characteristics and function of that protein. Of course, the primary reason for using Boolean vectors to represent subcellular location is that in most cases the specific distribution of a protein across components is not known. Boolean vector representations are frequently used in systems that attempt to predict subcellular location from sequence (4,5). In principle, a different vector could be used for each protein for different cell or tissue types, for different conditions, or for different time points within the cell cycle. Very little information on such differences is available, and it is rarely captured in databases.
Fractional distributions: Pattern unmixing
Ideally, the subcellular distribution of a protein could be represented as a vector containing the fraction of that protein that is found in each distinguishable component or fundamental pattern. These fractions must sum to one. Thus, the example Boolean vector given above would be represented as [0, 0.5, 0, 0.5, 0, 0, 0] if the protein were equally distributed between endosomes and Golgi, but [0, 0.2, 0, 0.8, 0, 0, 0] if it was only 20% in endosomes. This approach well represents cases where a protein is in equilibrium between a soluble cytoplasmic form and a form bound to a cytoskeletal structure (like a microtubule) or a membrane compartment (like an endosome). It also can easily represent more complex cases, like a protein that traffics between the endoplasmic reticulum and Golgi via transport vesicles. In this case, transport vesicles would best be considered as a distinct component (especially if the fraction of the protein in them is large), but considering them to be part of the source or destination component may be an acceptable approximation.
As with Boolean vectors, a different fractional distribution could be used for each cell type, condition or cell cycle stage. The important question becomes how these fractional distributions can be determined. Recent work addresses this issue for microscope images of fluorescently-tagged proteins (see Figure 1a, lower path). Only determinations for images of a single cell type and condition are considered in this section.
Supervised Pattern Unmixing
As mentioned above, many, if not most, subcellular components can be viewed as consisting of distinct elements or objects. These objects may all be quite similar to each other (such as is the case for peroxisomes in many cells), or they may be of two or more distinguishable types. (As with any categorization, this distinction may break down if the objects form a continuum spanning two or more types.) We have therefore previously described a three step process for testing how well subcellular components could be represented using objects (6). This consists of first learning the object types from a set of training images, learning to recognize those object types in new (test) images, and then counting the number of each object type present in each test cell. Representing each cell in this way enabled all eight major subcellular components in our 3D HeLa collection to be distinguished with good accuracy (6).
The finding that components can be well represented as a distribution over object types suggested that the distribution of a protein over more than one component can be represented as a linear combination of distributions of object types. More formally, we can define
where fi is a vector of length k containing the number of objects of each type for component i, αi is the fraction of protein in component (fundamental pattern) i, m is the number of components, and y is a vector of length k containing the number of objects of each type in the mixture. Alternatively, fi and y can be defined as the amount of fluorescence in each object type rather than the number of objects.
Given, for each component, a set of images for proteins that are solely found in that component (images of the fundamental patterns), we can directly learn k and fi using the three step process described above and averaging over many images. Then, for any image that might contain a mixture of components, we can measure y and use various estimation methods to learn a vector a containing the αi (for details on unmixing methods see references (6,7)). This approach was initially tested using the 3D HeLa collection by creating synthetic images with various randomly-chosen values of a for eight different components (m=8). An average agreement of 83% was observed between the values estimated for these synthetic images and the values used to synthesize them (6).
While encouraging, this test on synthetic images did not address potential problems that could be encountered with real images of mixed patterns. To test the method on a set of real images required a collection of images containing mixed patterns but where the mixing fractions were at least approximately known. Such as collection was created by Ghislain Bonamy, Sumit Chanda and Dan Rines at the Genomics Institute of the Novartis Foundation using two fluorescent probes that localize primarily to different organelles. Automated microscopy was used to collect a large number of images for a multi-well plate stained with varying combinations of Mitotracker green and Lysotracker green (including wells containing each probe separately). Our group then applied the unmixing method just described to these images (7). An average of 93% correlation was obtained between the fractions estimated by unmixing and those expected based on the ratio of probes added. The method was extended to include tests so that the system could also assign a fraction to an “unknown” component that did not match the ones used for training.
The unmixing method was further validated by applying it to images of cells expressing GFP-tagged microtubule associated protein LC3 in the presence of various concentrations of Bafilomycin A1. Bafilomycin A1 is an inhibitor of the vacuolarATPase that causes trapping of LC2 in autophagosomes. Using images of untreated cells and cells treated with a high drug concentration as the “pure” components, the fraction of protein in the two components was estimated for various drug concentrations (Figure 2). The results confirm the utility of the method.
Figure 2.

Application of pattern unmixing to quantify drug effects on autophagy. Images were collected of cells expressing eGFP-LC3 in the presence or absence of various concentrations of bafilomycin A1 and used to train an unmixing model as described in the text. The fraction of drug treated pattern as a function of concentration of drug was estimated using linear unmixing (squares), multinomial unmixing (circles), and fluorescence fraction unmixing (triangles). eGFP-LC3 showed a gradual relocation between the two patterns as a function of BFA concentration. From reference (7).
Unsupervised Pattern Unmixing
This unmixing method may be described as supervised since it requires the provision in advance of images of each of the fundamental patterns. This may be entirely feasible in many cases, but when considering proteome-wide application of this method it is not at all clear if all fundamental patterns are known. One solution to this problem is to start with what is thought to be a complete set of fundamental patterns but add to that set if unmixing results for a particular protein suggest that it contains a high fraction of unknown pattern. Alternatively, we can attempt an unsupervised approach in which we attempt to learn both the set of fundamental patterns and the unmixing fractions at the same time (8) (see Figure 1b, upper path).
We have used the Lysotracker/Mitotracker images to evaluate two approaches to this task (out of many possible). In both cases, the entire collection of images was provided to the algorithm without specifying the combination of probes that they received (but specifying which images were from the same well). The first approach was a basis pursuit method that assumes the same linear mixing model described above. The idea is to search for two or more wells whose object type distributions can be combined to generate all of the other wells. The second uses a Latent Dirichlet Allocation (LDA) method in which a process for generating mixtures is created and then the parameters corresponding to a real mixture are estimated from random trials. The methods must estimate the number of components as well as their composition. Good results were obtained with both methods, with the LDA method performing a bit better. The basis pursuit method found two components that corresponded closely to the pure Lysotracker and Mitotracker patterns. The LDA method found three components, two of which corresponded well to the pure patterns and one of which was a minor pattern consisting of new object types created by overlaps between objects from the two pure patterns. A correlation of 91% was observed for the LDA method, and a comparison of estimate and expected mixing fractions is shown in Figure 3. This correlation is even better than for the supervised methods.
Figure 3.

Results from unsupervised pattern unmixing. The estimated fraction of each probe in a given component is plotted as a function of the expected fraction (points for both the lysosomal and mitochondrial components are shown together). The dotted line shows agreement between the two fractions. After reference (8).
Limitations
Note that to be strictly accurate, this approach assumes that all molecules of a given protein that are in a given structure are indistinguishable (that is, that they are randomly distributed within that structure). This is rarely the case, since proteins may prefer to bind to specific regions on an organelle, or a protein in equilibrium between a bound and soluble cytoplasmic state may be in higher concentration near its binding partner. The unmixing methods described above do not capture such distinctions, but one can readily imagine extensions to them that would.
Models of subcellular organization
Given methods for learning the fundamental patterns and estimating how much of a given protein is associated with each of them, we now turn to the question of how to communicate the nature of each of those patterns. At a conceptual level, the most complete model of subcellular organization is probably the GO Cellular Component ontology. We can imagine easily associating GO terms with most (but perhaps not all) fundamental patterns by checking which organelle markers are assigned to each. However, such a conceptual model does not provide a spatially accurate representation of each fundamental pattern (including how that pattern varies from cell to cell).
To be useful for spatially realistic modeling, ontology terms must be associated with a representation of things like the number of objects in a component and their structure and distribution within cells. Currently, such representations are abstract and implicit rather than concrete and they often leave unspecified how the organelle would look in different cell types. For example, the abstract concept of a mitochondrion is well-understood by biologists but most would be hard pressed to describe how mitochondria vary in number, size, shape and distribution from cell type to cell type or organism to organism.
Of course, one approach is simply to represent each pattern with an example image containing it. This can be extended by representing a pattern by all (or a subsampling) of its images. This leaves open the question of how to integrate the information into other systems, especially when it is desirable to know how large numbers of patterns would look in the same cell. We have proposed that the learning of generative models of each pattern is a solution to this problem (9) (see Figure 1b, lower path). In this context, we define a model as generative if it can produce synthetic images that are by some specified criteria statistically indistinguishable from real images used to train it.
A key issue in building generative models of cells is that they need to contain pieces that depend on each other. For example, synthesizing an image showing the distribution of lysosomes is dependent on having a cell boundary within which to place them, and the position of the cell boundary and the nuclear boundary must be dependent on each other so that the nucleus is inside the cell. We have chosen to address the latter issue by starting with a model of nuclear shape and making the cell shape model dependent (conditional) on it, but the opposite approach is also feasible.
Nuclear models
Another important issue in building models is choosing an appropriate level of complexity with which to represent instances (examples) of the model. For example, we can consider everything from modeling all nuclei as ellipsoids (10,11) to making a detailed tracing or mesh representation of the surface of each nucleus. For building models of 2D subcellular patterns, we considered a compromise in which eleven parameters were used to describe a medial axis representation of each nucleus. This process is illustrated in Figure 4. The advantage is that the model is compact but still captures much of the variation in length, width, and curvature. The disadvantage is that it cannot represent forked shapes, which were not observed in the images of unperturbed HeLa cells used in our initial work, but can be observed under other conditions.
Figure 4.

Illustration of medial axis model fitting for nuclear shape. The original nuclear image (a) was processed into a binarized image (b), in which the nuclear object consists of the white pixels. The nuclear object was rotated so that its major axis is vertical (c) and converted into the medial axis representation (d). The horizontal positions of the medial axis as a function of the fractional distance along it are shown by the symbols in (e), along with a B-spline fit (solid curve). The width as a function of fractional distance is shown by the symbols in (f), along with the corresponding fit (solid curve). Scale bar, 5 μm. From reference (9).
To address this, we have developed an alternative, diffeomorphic approach to describing and modeling nuclear shape (12–14). A related approach was first described by Yang et al (15) for the purpose of registering nuclear images. The principle is that the variation in shape among a population of nuclei can be represented by a measure of the pairwise differences in their shapes (12). This measure is found by determining how much work must be done to morph one of them into the other. The result is a square, symmetric matrix of dimensions corresponding to the number of nuclei. Using multidimensional scaling, this matrix can be converted such that each nucleus is represented by a vector in some Euclidean space. The higher the dimension of that space, the closer the approach comes to perfectly capturing the original distance matrix (it is perfect to within numerical accuracy when the dimension equals the number of nuclei). Figure 5 shows reducing the dimension to just two. Remarkably, variation along the first dimension corresponds to nuclear elongation, and variation along the second dimension corresponds to bending. This gives a very compact representation of the shape variation in the nuclear population (12,13).
Figure 5.

Plot of the first two components of the low-dimensional representation of the nuclear shape computed by the diffeomorphic method discussed in the text. Each small circle corresponds to one nuclear image. Images associated with specific data points are shown on the left (diamonds) or across the bottom (squares). Each dark square corresponds, in order, to each image shown in the horizontal bottom series of images. Likewise, each light triangle corresponds to each image stacked vertically. Note that the method separates different modes of shape variation (bending and elongation) into separate coordinates. From reference (12).
However, this approach does not directly give a means of generating new shapes. This limitation was overcome by recursively interpolating shapes at points in the shape space chosen according to a probability density function estimated from the original nuclei (14). This permits the diffeomorphic approach to be used in a generative model, but requires that the original nuclear shapes be saved along with the reduced shape space coordinates of each and the probability density function. The amount of storage required can be reduced by saving a smaller number of examples (e.g., just examples at peaks in the probability density function).
We can now consider a single generative framework for storing models of nuclei and other cell components in which the first “slot” of the framework specifies which type of nuclear model to use as well as the parameters for that model. In the case of the diffeomorphic model, the parameters are very extensive. Other types of generative models of nuclear shape can be used (16,17), although our overall philosophy is to prefer models whose parameters are automatically learned from images.
Cell shape models
The next “slot” of the generative framework is filled by a cell shape model. While approaches that model cell shape alone have been described (18–20), we focus on building a cell shape model that is learned directly from images and conditional on the nuclear shape. This is in order to ensure that the two shapes are compatible with each other (e.g., that the nucleus is inside the cell!) and that any relative orientation of the two is captured. For this we use a simple approach in which a cell to be modeled is first rotated so that its major axis is pointing in a defined direction and flipped (if necessary) so that the side (relative to the major axis) with the larger area is also matched. The ratio between the distance from the center of the nucleus to the nearest point on the plasma membrane and the distance from the center to the nearest point on the nuclear membrane is then measured at angles from 0° to 360° relative to the major axis. This set of relative coordinates is reduced to a small number (10) of principal components. New cell shapes can then be synthesized (after synthesizing a nuclear shape) by randomly choosing values for the principal components and using the synthesized ratios to mark out the cell boundary. Conditional, diffeomorphic models of cell shape can also be made.
Models of subcellular components
We now turn to the most difficult part of building cell models, representing subcellular components. Much work remains to be done in this area. Two distinct but preliminary approaches for representing a subset of protein patterns are described here.
Object-based models: Direct learning
This first is building object-based models (6). This approach is most suited to organelles such as endosomes, lysosomes, and peroxisomes that largely exist as discrete vesicles. As a first approximation, these can be modeled as Gaussian objects, that is, as circles (or spheres in 3D) whose intensity decreases with distance from its center (as expected if its intensity in a given pixel was proportional to the volume that underlies that pixel). Since cell images often have two or more vesicles touching or overlapping, we estimate the number and sizes of the vesicles that are most likely to have given rise to a particular image using non-linear fitting. Doing this for many cells allows distributions to be learned for the number of objects per cell and their size variation in each cell. The position of each object relative to the nearest point on the nucleus and the nearest point on the cell membrane is then calculated and used to create a 2D (or 3D) position probability density function. The synthesis of new patterns is then quite simple. For each cell, a number of objects is drawn from the number distribution, and a size and position are sampled from the size distribution and the position probability density function, respectively. These are used to place the objects into the nuclear and cell shape model described above. An example of a synthesized image showing a lysosomal pattern is shown in Figure 6.
Figure 6.
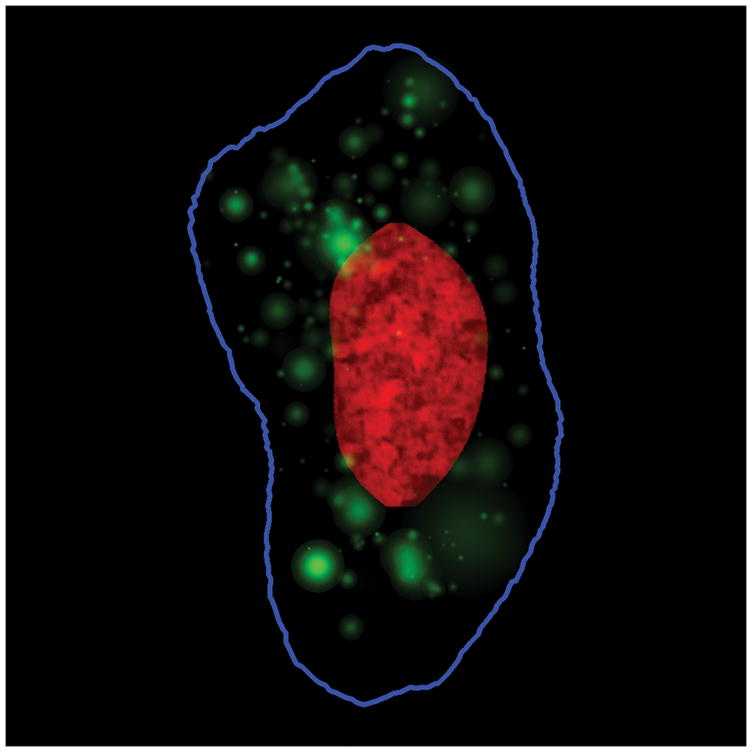
Example synthetic image generated by a model learned from images of the endosomal protein transferrin. The DNA distribution is shown in red, the cell outline in blue, and transferrin-containing objects in green.
Network models: Inverse Modeling
The second approach is designed for network distributions, such as the tubulin cytoskeleton, that are not appropriately modeled as objects. Since elements of such networks frequently cross and pile up near the center of the cell, it is difficult to estimate parameters of a model from conventional microscope images. One solution is to use specialized microscopic methods, such as speckle microscopy, that image only a portion of the network at a time (21). Excellent models of actin polymerization in the leading edge of a crawling cell have been obtained by this approach (22). Speckle microscopy requires suitable polymerization and depolymerization rates and may not be appropriate for all network proteins. An alternative for extracting model parameters from wide-field microscope images is to use inverse modeling. The principle is that the parameters that describe the state of a network in a real image can be estimated using a model that can synthesize images for many parameters values and a comparator that finds the synthetic image whose appearance is closest to the real one. One of the earliest uses of this approach was to estimate spindle dynamics (23). We have recently described a simple but justifiable model of microtubule polymerization in interphase cells and demonstrated that it can be used to make reasonable estimates of the number, length distribution and degree of growth direction persistence of HeLa cells (24).
Combining component models: independent or conditional
A major goal of the model building described here is to be able to create cell models containing spatially realistic distributions for many different proteins. Since the number of different proteins that can be measured in the same living cell is currently less than ten (although the number in fixed cells is at least one hundred (25)), it is difficult to imagine using multi-color images directly for this purpose. An alternative is to combine subcellular models learned from separate sets of images. This can be done by constructing a single nuclear and cell shape and then adding objects or networks in turn for each additional component. This assumes that these distributions are independent of each other. If this is not the case, the placement of one component can be made conditional on that of another. For example, endosomal positions can be preferentially placed along microtubules.
Use of models for testing algorithms
Much of the work from other groups described above for constructing models of cells and nuclei has been motivated not by a desire to learn parameters from real images but rather to create synthetic images whose underlying cell or nuclear shape is known so that they can be used to test analysis software. When used this way, models can be considered as digital “phantoms” by analogy to the test samples of known properties frequently used to test imaging hardware. All of the methods described in this article can be used for this purpose, especially methods than can generate combinations of patterns.
Future directions
While the focus of this article has been on building models of static images, cell behaviors are clearly dynamic. One of the first next steps therefore, is to extend these methods to time series images. The goal would be to have a model that can generate not only an object or network distribution, but also how it changes over time. Similarly, it will be important to model how patterns vary between cell types and conditions, especially with the goal of being able to construct a general model for a given component that can then be instantiated in a given cell type. Lastly, the possibility of differences in subcellular location for different splicing isoforms from the same gene has been given relatively little attention and will need more. While much work remains to be done, it is hoped that the approaches reviewed here suggest that all of these goals are attainable.
Acknowledgments
I would like to express my thanks for the work done by Michael Boland, Meel Velliste, Ting Zhao, Tao Peng, Luis Coelho, Wei Wang and Gustavo Rohde and for many helpful discussions with them and with Joel Stiles, Eric Xing, Geoffrey Gordon, Russell Schwartz, Klaus Palme, Olaf Ronneberger, Hagit Shatkay, Leslie Loew, Ion Moraru, Karl Rohr, Ghislain Bonamy, Sumit Chanda, and Daniel Rines. The original work from my group reviewed here was supported in part by National Institutes of Health grants R01 GM068845 and R01 GM075205 and National Science Foundation grant EF-0331657, and part of the discussions and writing of this article were supported by a Research Award from the Alexander von Humboldt Foundation and a Senior External Research Fellowship from the Freiburg Institute of Advanced Studies.
Literature Cited
- 1.The Universal Protein Resource (UniProt) in 2010. Nucleic Acids Res. 38(Database issue):D142–8. doi: 10.1093/nar/gkp846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium Nat Genet. 2000;25(1):25–9. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sprenger J, Lynn Fink J, Karunaratne S, Hanson K, Hamilton NA, Teasdale RD. LOCATE: a mammalian protein subcellular localization database. Nucleic Acids Res. 2008;36(Database issue):D230–3. doi: 10.1093/nar/gkm950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chou KC, Shen HB. Recent progress in protein subcellular location prediction. Anal Biochem. 2007;370(1):1–16. doi: 10.1016/j.ab.2007.07.006. [DOI] [PubMed] [Google Scholar]
- 5.Shatkay H, Hoglund A, Brady S, Blum T, Donnes P, Kohlbacher O. SherLoc: high-accuracy prediction of protein subcellular localization by integrating text and protein sequence data. Bioinformatics. 2007;23(11):1410–7. doi: 10.1093/bioinformatics/btm115. [DOI] [PubMed] [Google Scholar]
- 6.Zhao T, Velliste M, Boland MV, Murphy RF. Object type recognition for automated analysis of protein subcellular location. IEEE Trans Image Process. 2005;14(9):1351–9. doi: 10.1109/tip.2005.852456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peng T, Bonamy GM, Glory-Afshar E, Rines DR, Chanda SK, Murphy RF. Determining the distribution of probes between different subcellular locations through automated unmixing of subcellular patterns. Proc Natl Acad Sci U S A. 2010;107(7):2944–9. doi: 10.1073/pnas.0912090107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coelho LP, Peng T, Murphy RF. Quantifying the distribution of probes between subcellular locations using unsupervised pattern unmixing. Bioinformatics. 2010;26 doi: 10.1093/bioinformatics/btq220. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao T, Murphy RF. Automated learning of generative models for subcellular location: building blocks for systems biology. Cytometry A. 2007;71(12):978–90. doi: 10.1002/cyto.a.20487. [DOI] [PubMed] [Google Scholar]
- 10.Lockett SJ, Sudar D, Thompson CT, Pinkel D, Gray JW. Efficient, interactive, and three-dimensional segmentation of cell nuclei in thick tissue sections. Cytometry. 1998;31(4):275–86. doi: 10.1002/(sici)1097-0320(19980401)31:4<275::aid-cyto7>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 11.Ortiz de Solorzano C, Garcia Rodriguez E, Jones A, Pinkel D, Gray JW, Sudar D, Lockett SJ. Segmentation of confocal microscope images of cell nuclei in thick tissue sections. J Microsc. 1999;193(Pt 3):212–26. doi: 10.1046/j.1365-2818.1999.00463.x. [DOI] [PubMed] [Google Scholar]
- 12.Rohde GK, Ribeiro AJ, Dahl KN, Murphy RF. Deformation-based nuclear morphometry: capturing nuclear shape variation in HeLa cells. Cytometry A. 2008;73(4):341–50. doi: 10.1002/cyto.a.20506. [DOI] [PubMed] [Google Scholar]
- 13.Rohde GK, Wang W, Peng T, Murphy RF. Deformation-based nonlinear dimension reduction: Applications to nuclear morphometry. Proc 2008 Intl Symp Biomed Imaging. 2008:500–503. [Google Scholar]
- 14.Peng T, Wang W, Rohde GK, Murphy RF. Instance-based generative biological shape modeling. Proc 2009 Intl Symp Biomed Imaging. 2009:690–693. doi: 10.1109/ISBI.2009.5193141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang S, Kohler D, Teller K, Cremer T, Le Baccon P, Heard E, Eils R, Rohr K. Non-rigid registration of 3D multi-channel microscopy images of cell nuclei. Med Image Comput Comput Assist Interv. 2006;9(Pt 1):907–14. doi: 10.1007/11866565_111. [DOI] [PubMed] [Google Scholar]
- 16.Svoboda D, Kasik M, Maska M, Hubeny J, Stejskal S, Zimmermann M. On simulating 3d fluorescent microscope images. Lect Notes Comp Sci. 2007;4673:309–316. [Google Scholar]
- 17.Svoboda D, Kozubek M, Stejskal S. Generation of digital phantoms of cell nuclei and simulation of image formation in 3D image cytometry. Cytometry A. 2009;75(6):494–509. doi: 10.1002/cyto.a.20714. [DOI] [PubMed] [Google Scholar]
- 18.Dufour A, Shinin V, Tajbakhsh S, Guillen-Aghion N, Olivo-Marin JC, Zimmer C. Segmenting and tracking fluorescent cells in dynamic 3-D microscopy with coupled active surfaces. IEEE Trans Image Process. 2005;14(9):1396–410. doi: 10.1109/tip.2005.852790. [DOI] [PubMed] [Google Scholar]
- 19.Lehmussola A, Selinummi J, Ruusuvuori P, Niemisto A, Yli-Harja O. Simulating fluorescent microscope images of cell populations. Conf Proc IEEE Eng Med Biol Soc. 2005;3:3153–6. doi: 10.1109/IEMBS.2005.1617144. [DOI] [PubMed] [Google Scholar]
- 20.Lehmussola A, Ruusuvuori P, Selinummi J, Huttunen H, Yli-Harja O. Computational framework for simulating fluorescence microscope images with cell populations. IEEE Trans Med Imaging. 2007;26(7):1010–6. doi: 10.1109/TMI.2007.896925. [DOI] [PubMed] [Google Scholar]
- 21.Danuser G, Waterman-Storer CM. Quantitative fluorescent speckle microscopy of cytoskeleton dynamics. Annu Rev Biophys Biomol Struct. 2006;35:361–87. doi: 10.1146/annurev.biophys.35.040405.102114. [DOI] [PubMed] [Google Scholar]
- 22.Ponti A, Machacek M, Gupton SL, Waterman-Storer CM, Danuser G. Two distinct actin networks drive the protrusion of migrating cells. Science. 2004;305(5691):1782–6. doi: 10.1126/science.1100533. [DOI] [PubMed] [Google Scholar]
- 23.Sprague BL, Pearson CG, Maddox PS, Bloom KS, Salmon ED, Odde DJ. Mechanisms of microtubule-based kinetochore positioning in the yeast metaphase spindle. Biophys J. 2003;84(6):3529–46. doi: 10.1016/S0006-3495(03)75087-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shariff A, Murphy RF, Rohde GK. A generative model of microtubule distributions, and indirect estimation of its parameters from fluorescence microscopy images. Cytometry A. 2010;77(5):457–66. doi: 10.1002/cyto.a.20854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schubert W. A three-symbol code for organized proteomes based on cyclical imaging of protein locations. Cytometry A. 2007;71(6):352–60. doi: 10.1002/cyto.a.20281. [DOI] [PubMed] [Google Scholar]