

Abstract
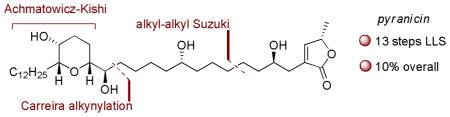
A modular, 13 step, synthesis of the tetrahydropyran-containing annonaceous acetogenin pyranicin is reported. Key features are the use of an Achmatowicz oxidation-Kishi reduction sequence for the assembly of a pyranone from a furan, and the application of Fu’fs alkyl-alkyl Suzuki coupling for subunit union.
The annonaceous acetogenins, a group of polyketides that are generally identified by the presence of one or two tetrahydrofuran rings, now number greater than 400 compounds.1 A small subset of the acetogenins, including mucocin and jimenezin, contain both a tetrahydrofuran ring and a tetrahydropyran ring. As a class, these compounds have attracted substantial scientific attention due to their rich and varied biological activities which include anticancer, antiinfective, immunosuppressive, pesticidal, and antifeedant properties.
Pyranicin (1, Figure 1) was isolated from the stem bark of Goniothalamus giganteus Hook. f. & Thomas (Annonaceae) and is the prototype of acetogenins that contain a single tetrahydropyran ring.2 Despite what could be argued to be substantial structural simplification relative to more complex acetogenins, pyranicin retains impressive biological activity, with <1μg/mL ED50 values against a number of cancer cell lines and particularly noteworthy effects against the PACA-2 (pancreatic cancer) cell line (ED50 = 1.3 ng/mL). As part of our ongoing interest in the synthesis of natural product inhibitors of electron transport,3 we describe in this communication a total synthesis of 1.4
Figure 1.
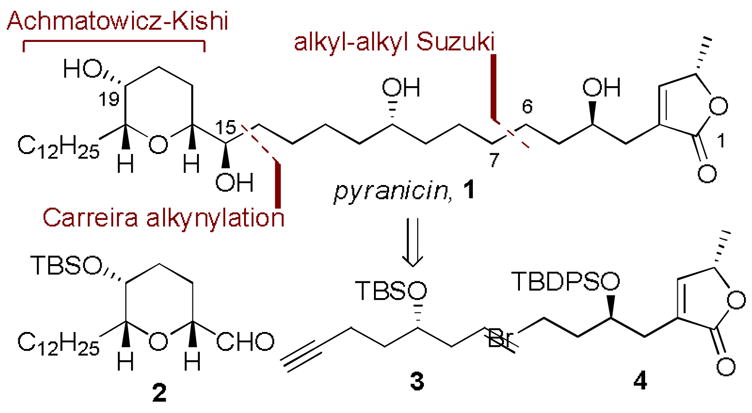
Pyranicin and overall synthesis strategy.
From the outset, an overarching goal of our studies was to develop a concise and modular synthesis that would be amenable to the synthesis of a variety of compounds that could illuminate SAR for pyranicin. With this in mind, we settled on a strategy that called for the preparation of three key subunits: pyran 2, alkynyl olefin 3, and butenolide 4. Pyran 2 would be prepared by our recently described Achamatowicz oxidation-Kishi reduction sequence,5 and our plans for subunit coupling involved the use of Carreira’s asymmetric alkynylation6 and Fu’s recently described alkyl-alkyl Suzuki coupling.7,
The synthesis of the tetahydropyran-containing domain commenced with commercially available furan 58 (Scheme 1). Addition of dodecylmagnesium bromide produced furfuryl alcohol 6, which was subjected to the Sharpless asymmetric kinetic resolution reported by Sato9,10 to yield the intermediate pyranone hemiacetal 7. Immediate reduction with i-Pr3SiH in the presence of BF3·OEt2 gave pyran 8 as a single diastereoisomer (>20:1 by 1H NMR analysis). This three step sequence conveniently provided the desired pyran in 33% overall yield. Hydrogenation of the enone, followed by reduction of the ketone with L-Selectride gave axial alcohol 9 in 86% yield (2 steps). Protection of the secondary alcohol as the TBS ether, removal of the benzyl ether (88% yield, 2 steps), and oxidation with Dess-Martin periodinane led to aldehyde 2 (99% yield). Reaction with known alkyne 311 under Carreira’s asymmetric alkynylation conditions (Et3N, Zn(OTf)2, (−)-NME, PhMe) and subsequent protection of the secondary alcohol as the TBS ether gave 10 (82% yield over 2 steps).
Scheme 1.

Synthesis of the C7–C32 pyran-containing domain.
The synthesis of butenolide 4 began with the alkylation of alkyne 1112 with epoxide 1213 in the presence of BF3·OEt2, followed by immediate protection of the homopropargyl alcohol with TBDSPCl to give 13 in 88% yield (Scheme 2). Careful removal of the TBS ether with ethanolic PPTS (95% yield) and hydroalumination-iodination of the alkyne following Denmark’s procedure14 produced vinyl iodide 14 in 90% yield. In accord with Stille’s original report15 and Hoye’s application in the context of acetogenin synthesis,16 when 14 was subjected to Pd(0) under 50 psi CO, carbonylative lactonization occurred to yield the desired butenolide 15 in 90% yield. Oxidative removal of the PMB ether with DDQ in wet CH2Cl2 (82% yield) unveiled primary alcohol 16. Conversion of the alcohol to the bromide 4 was readily achieved by Hannesian’s NBS-Ph3P protocol17 in 87% yield.
Scheme 2.

Synthesis of the butenolide domain and completion of the synthesis.
With routes to 10 and 4 secured we were positioned to examine the proposed Fu-type Suzuki coupling for subunit union (Scheme 2).18 To this end, when 10 was treated with 1.1 equivalents of freshly prepared 9-BBN in THF at room temperature, hydroboration was selective for the terminal alkene over the internal alkyne19 to yield intermediate borane 17. When this borane was exposed to bromide 4, Pd(PCy3)2 and K3PO4.H2O smooth cross-coupling ensued to yield the desired compound 18 in 60% yield. In our hands, this coupling was robust with 20 mol% Pd(PCy3)2 catalyst loading and 1.2 equivalents of bromide 4. In the current example, deviation from these conditions resulted in decreased and variable yields of the desired compound. The synthesis was then completed in a straightforward fashion by removal of the silyl protecting groups with aqueous HF in acetonitrile (92% yield), and reduction of the alkyne to the hydrocarbon using Wilkinson’s catalyst in benzene-ethanol to produce pyranicin, 1 in 89% yield. The physical characteristics of synthetic material (1H and 13C NMR, HRMS) matched those reported by McLaughlin.2 Optical rotation data was in accord with that reported by Rein and Takahashi.20,21
In conclusion, we have described a concise (13 steps longest linear sequence) and efficient (10% overall yield) synthesis of pyranicin. Beyond these metrics, the synthesis is modular and will prove amenable to the synthesis of a variety of analogs. This work also highlights the utility of the Achmatowicz oxidation-Kishi reduction sequence for the assembly of pyrans and the strategic potential of alkyl-alkyl Suzuki couplings in a complex setting.
Supplementary Material
Acknowledgments
We thank Eli Lilly and Company (via the Lilly Grantee Program), the AP Sloan Foundation, and the National Cancer Institute (CA 110246) for support of this research.
Footnotes
Supporting Information Available: Procedures for the synthesis of new compounds, along with characterization data, and spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For a review see: Bermejo A, Figadere B, Zafra-Polo M-C, Barrachina I, Estornell E, Cortes D. Nat Prod Rep. 2005;22:269. doi: 10.1039/b500186m. and references cited therein.
- 2.Alali FQ, Rogers L, Zhang Y, McLaughlin JL. Tetrahedron. 1998;54:5833. [Google Scholar]
- 3.Keaton KA, Phillips AJ. J Am Chem Soc. 2006;128:408. doi: 10.1021/ja057434k. [DOI] [PubMed] [Google Scholar]
- 4.For other total syntheses see: Strand D, Norrby PO, Rein T. J Org Chem. 2006;71:1879. doi: 10.1021/jo052233k.Strand D, Rein T. Org Lett. 2005;7:199. doi: 10.1021/ol0479242.Takahashi S, Kubota A, Nakata T. Org Lett. 2003;5:1353. doi: 10.1021/ol034323m.Hattori Y, Furuhata S-i, Okajima M, Konno H, Abe M, Miyoshi H, Goto T, Makabe H. Org Lett. 2008;10:717. doi: 10.1021/ol702902w.Furuhata S-i, Hattori Y, Okajima M, Konno H, Abe M, Miyoshi H, Goto T, Makabe H. Tetrahedron. 2008;64:7695. doi: 10.1021/ol702902w.
- 5.Henderson JA, Jackson KL, Phillips AJ. Org Lett. 2007;9:5299. doi: 10.1021/ol702559e.Um JM, Houk KN, Phillips AJ. Org Lett. 2008;10:3769. doi: 10.1021/ol801421n.See also Nicolaou KC, Frederick MO, Burtoloso ACB, Denton RM, Rivas F, Cole KP, Aversa RJ, Gibe R, Umezawa T, Suzuki TJ. Am Chem Soc. 2008;130:7466. doi: 10.1021/ja801139f.
- 6.Anand NK, Carreira EM. J Am Chem Soc. 2001;123:9687. doi: 10.1021/ja016378u. [DOI] [PubMed] [Google Scholar]
- 7.Netherton MR, Dai C, Neuschutz K, Fu GC. J Am Chem Soc. 2001;123:10099. doi: 10.1021/ja011306o. [DOI] [PubMed] [Google Scholar]
- 8.This furan is also readily prepared by benzylation of (hydroxymethyl) furfural. See Cottier L, Descotes G, Eymard L, Rapp K. Synthesis. 1995:303.
- 9.Kobayashi Y, Kusakabe M, Kitano Y, Sato F. J Org Chem. 1988;53:1586. [Google Scholar]
- 10.For the CaH2/SiO2 modified conditions employed here see: Yang ZC, Zhou WS. Tetrahedron Lett. 1995;36:5617.Zhou WS, Lu Z, Wang Z. Tetrahedron Lett. 1991;32:1467.Wang Z, Zhou W, Lin G. Tetrahedron Lett. 1985;26:6221.
- 11.Garcia-Fandino R, Aldegunde MJ, Codesido EM, Castedo L, Granja JR. J Org Chem. 2005;70:8281. doi: 10.1021/jo050568w. [DOI] [PubMed] [Google Scholar]
- 12.Brimble MA, Bryant CJ. Org Biomol Chem. 2007;5:2858. doi: 10.1039/b709932k. [DOI] [PubMed] [Google Scholar]
- 13.Marshall JA, Schaaf G, Nolting A. Org Lett. 2005;7:5331. doi: 10.1021/ol0523493. [DOI] [PubMed] [Google Scholar]
- 14.(a) Chan K, Cohen N, DeNoble JP, Specian AC, Saucy G. J Org Chem. 1976;41:3497. doi: 10.1021/jo00884a001. [DOI] [PubMed] [Google Scholar]; (b) Denmark SE, Jones TK. J Org Chem. 1982;47:4595. [Google Scholar]
- 15.Cowell A, Stille JK. J Am Chem Soc. 1980;102:4193. [Google Scholar]
- 16.Hoye TR, Humpal PE, Jimenez JI, Mayer MJ, Tan L, Ye Z. Tetrahedron Lett. 1994;35:7517. [Google Scholar]
- 17.Ponpipom MM, Hanessian S. Carbohydr Res. 1971;18:342. [Google Scholar]
- 18.For other examples of the use of this reaction in total synthesis see: Das S, Abraham S, Sinha SC. Org Lett. 2007;9:2273. doi: 10.1021/ol070517g.Keaton KA, Phillips AJ. Org Lett. 2007;9:2717. doi: 10.1021/ol0710111.
- 19.Brown CA, Coleman RA. J Org Chem. 1979;44:2328. [Google Scholar]
- 20.As described and discussed by Rein and Takahashi in references 4a, 4b and 4c, there is a discrepancy between the optical rotations for synthetic and natural pyranicin: natural pyranicin [α]D −9.7 (c 0.008, CHCl3); synthetic pyranicin (Takahashi): [α]D +19.5 (c 0.55, CHCl3); synthetic pyranicin (Rein): [α]D +21.1 (c 0.24, CHCl3). Our material had optical rotation of [α]D +24.8 (c 0.20, CHCl3).
-
21.The possibility of the discrepancy in optical rotations being due to aggregation effects was seems unlikely as the rotation in chloroform for our synthetic material does not vary significantly with changing concentration:
Concentration (c)
α
[α]D
0.200 +0.049 +24.8 0.125 +0.022 +17.8 0.063 +0.016 +26.3 0.031 +0.007 +22.7 0.015 +0.003 +21.3
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.